
A Comparative Transcriptomic Meta-Analysis Revealed Conserved Key Genes and Regulatory Networks Involved in Drought Tolerance in Cereal Crops

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
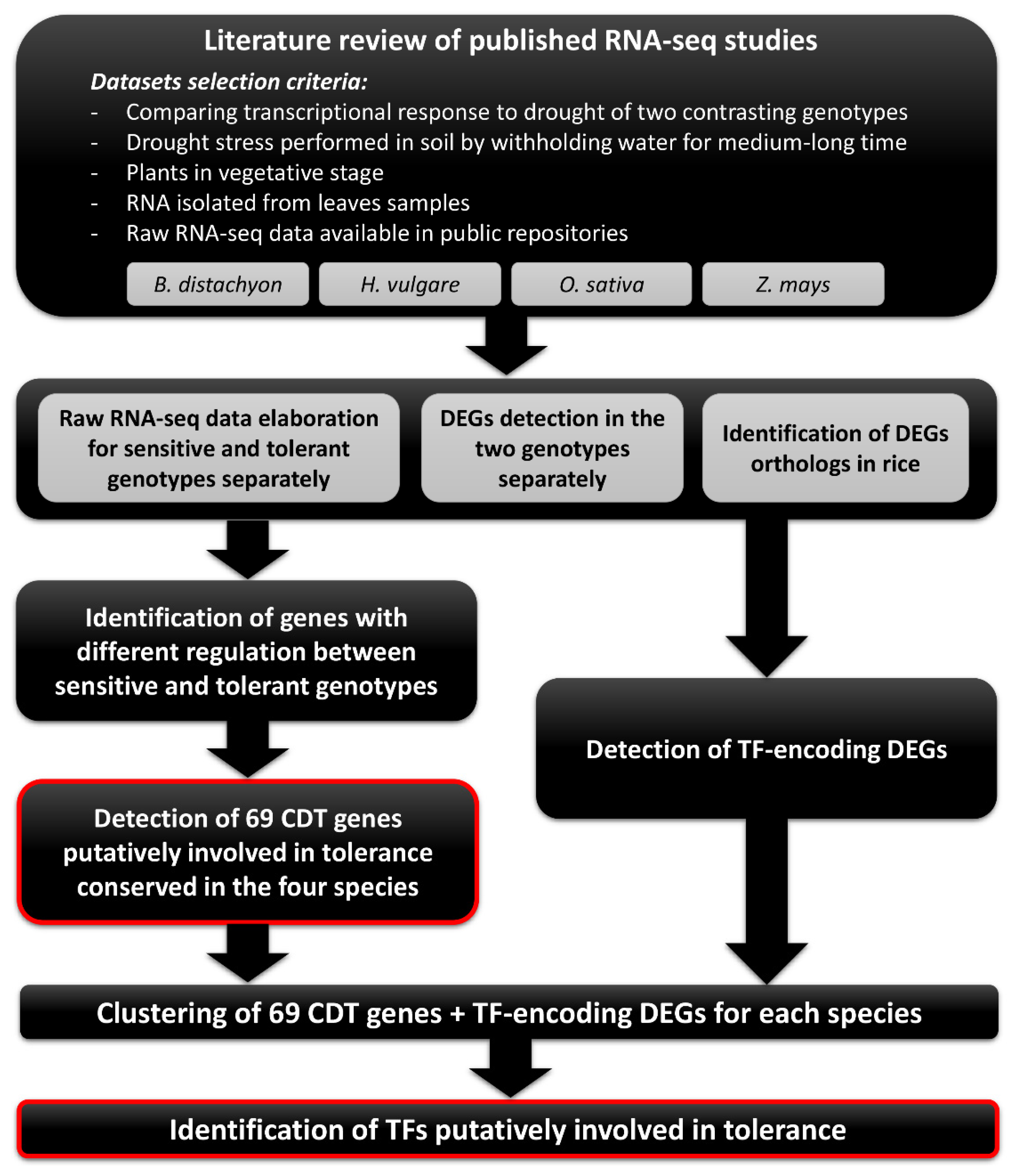
2.1. Selection of RNA-Seq Datasets for Meta-Analysis
2.2. RNA-Seq Data Processing and Differential Expression Analyses
2.3. Comparison of the Level of Drought Stress among the Four Datasets
2.4. Identification of Genes Involved in Tolerance to Drought Stress in each Species
2.5. Identification of Genes Involved in Tolerance to Drought Stress across Multiple Species
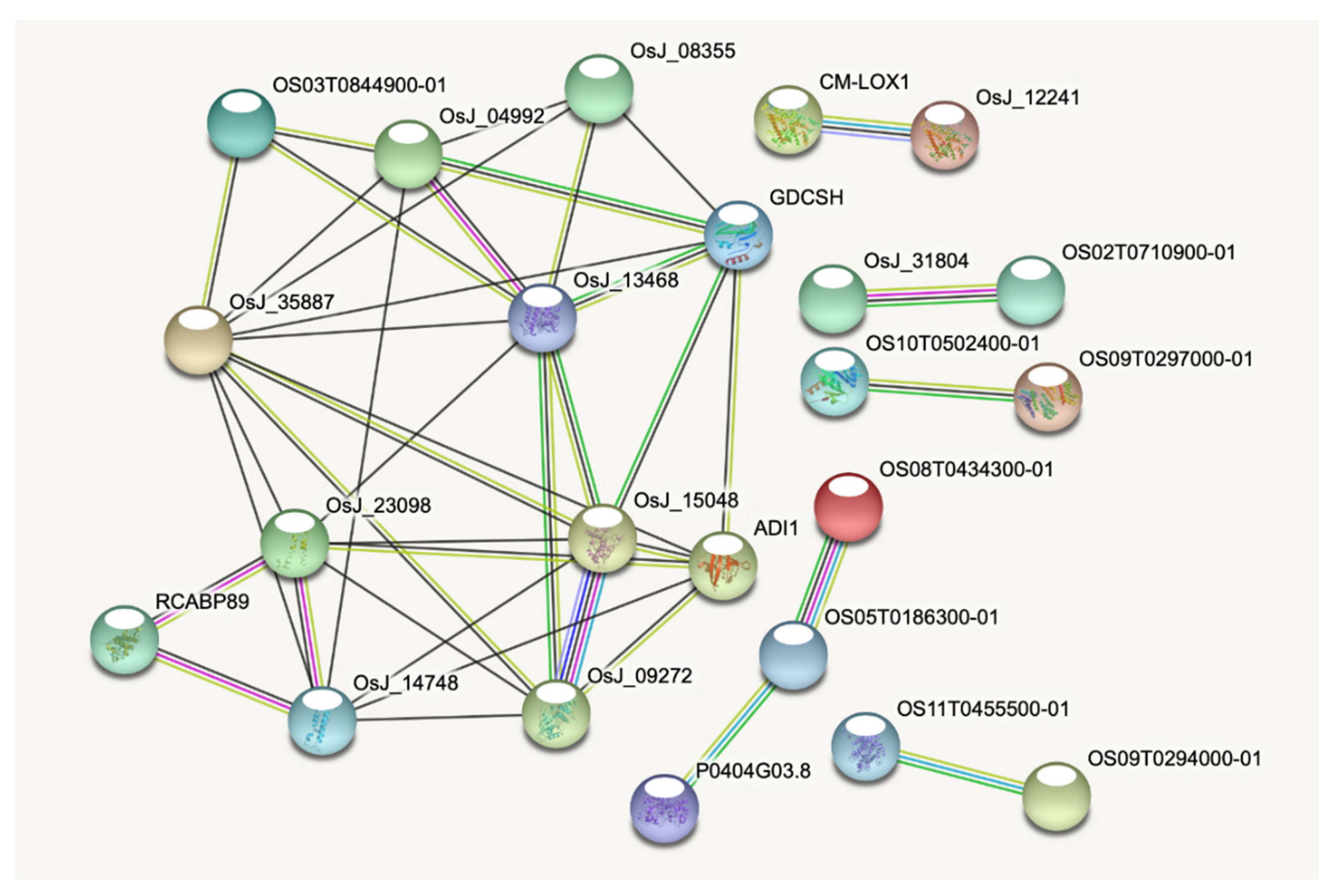
2.6. Principal Component Analysis and Protein–Protein Interaction of CDT Genes
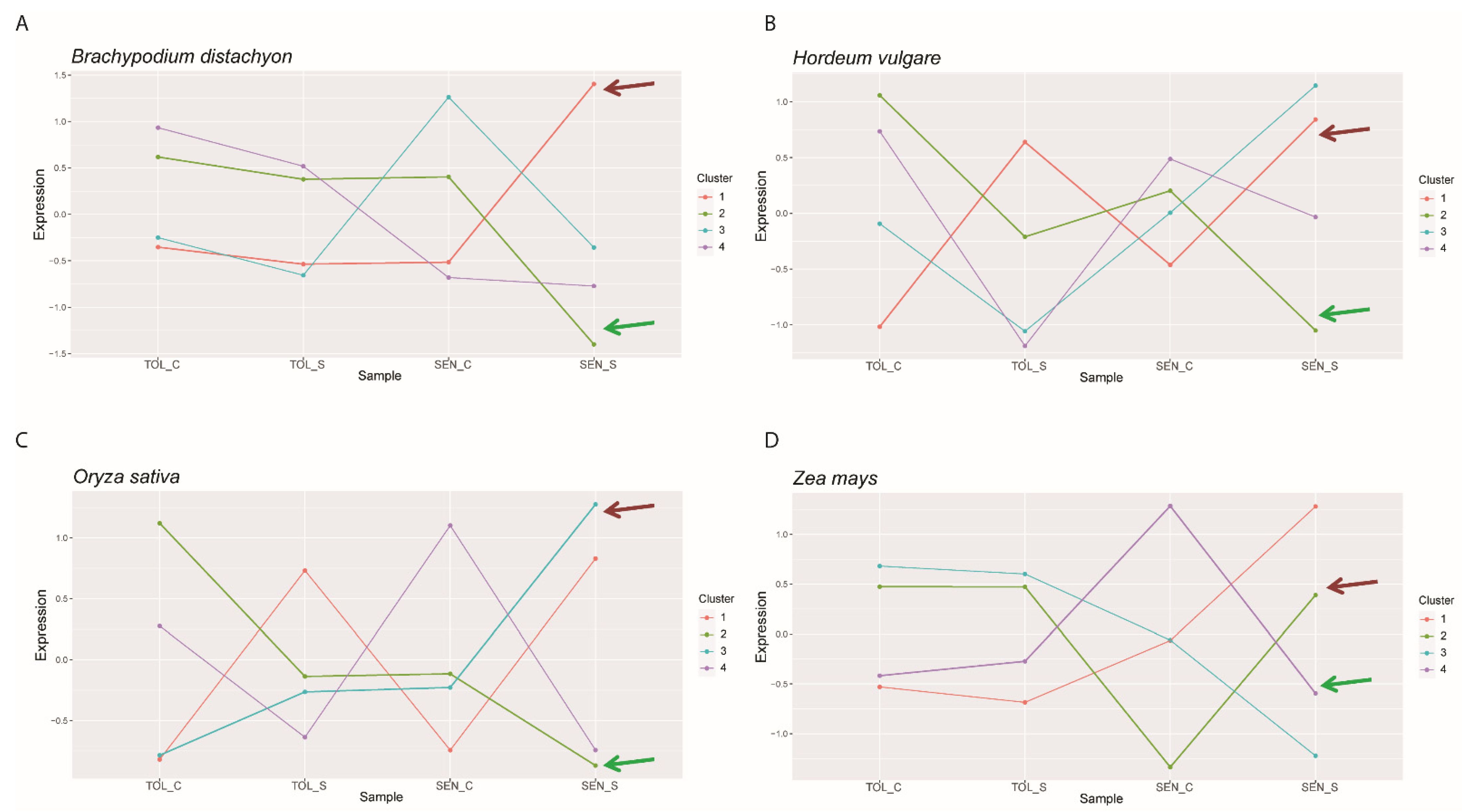
2.7. Clustering Gene Expression to Find Common Patterns and Associated Transcription Factors
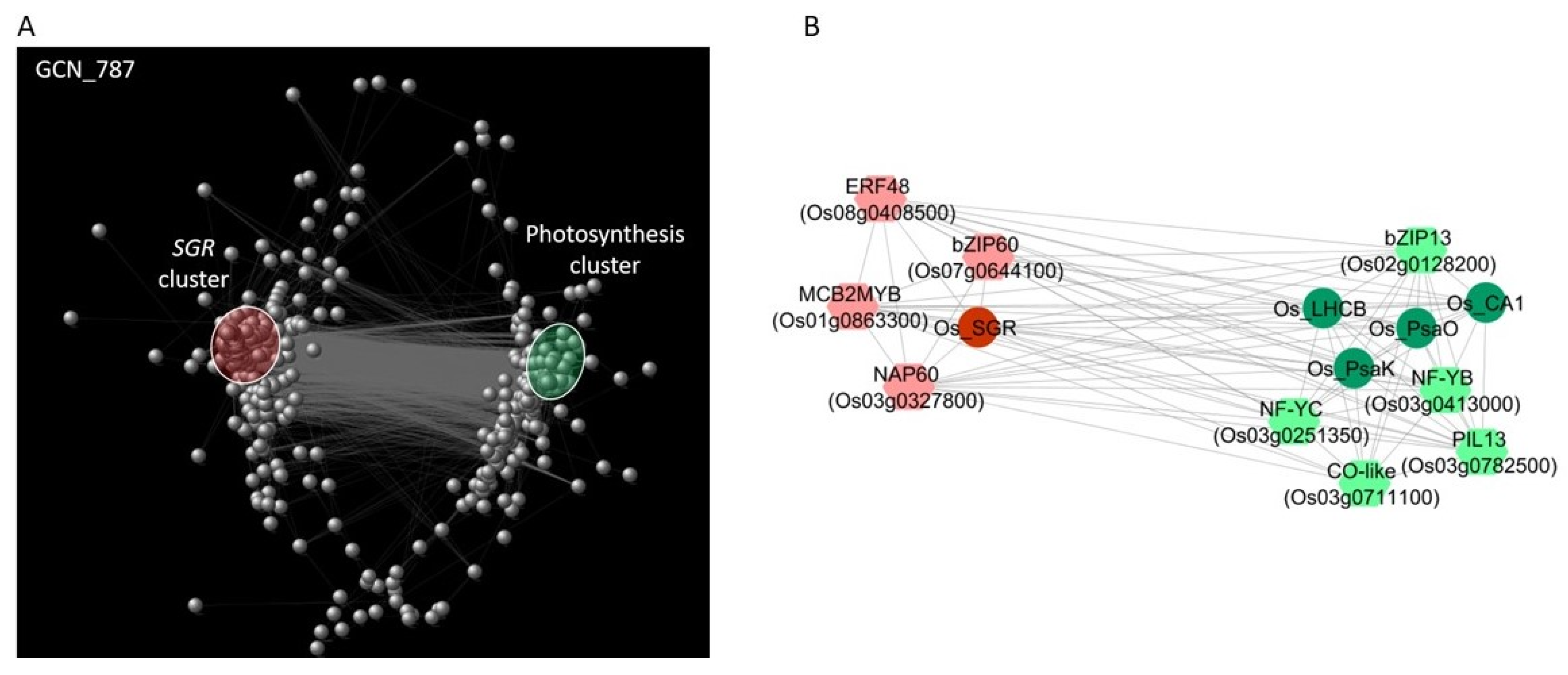
2.8. Gene Co-Expression Network Analysis of CDT Genes and TF DEGs on an Independent Water Stress Experiment in Rice
2.9. Analysis of TF Binding Sites on the Promoter Sequences of the Rice CDT Genes
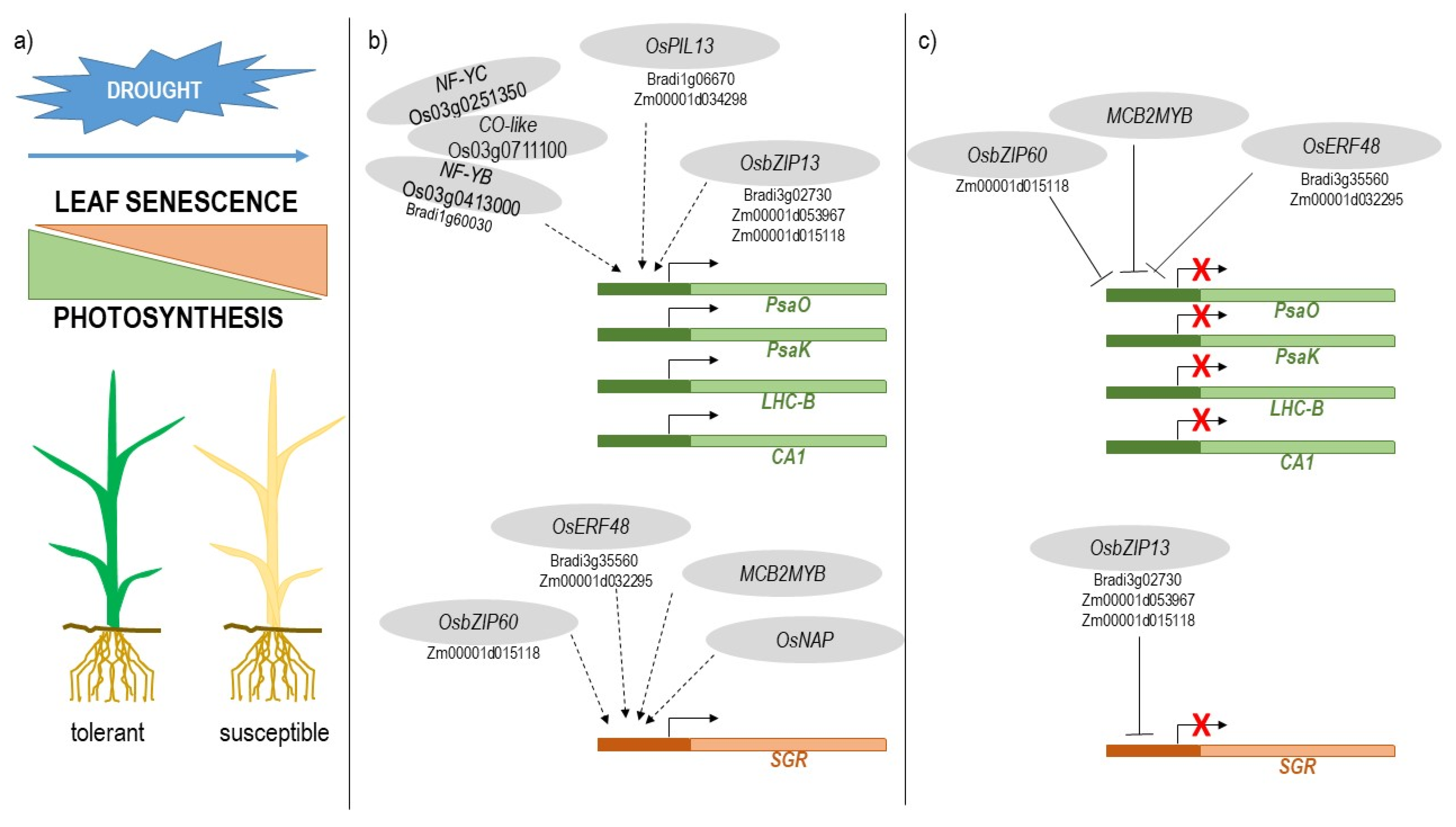
3. Discussion
3.1. CDT Genes Characterized the Drought Response of Sensitive and Tolerant Genotypes in the Four Cereal Species
3.2. The Balance between the Induction of Leaf Senescence and the Maintenance of Photosynthesis Plays a Major Role in Drought Tolerance
3.3. Identification of Transcription Factors Involved in the Balance between Leaf Senescence and Photosynthesis
3.4. Rice Transcription Factors Involved in the Positive Regulation of CDT Photosynthesis Genes
3.5. Rice Transcription Factors Involved in the Positive Regulation of Leaf Senescence
3.6. Putative Orthologs of Rice Transcription Factors Are Involved in the Regulation of CDT Genes in Brachypodium, Barley, and Maize
4. Materials and Methods
4.1. Selection of Published RNA-Seq Studies
4.2. RNA-Seq Data Handling and Gene Expression Quantification
4.3. Differential Expression Analysis and Orthology Study
4.4. Transcription Factors Annotation and Orthology
4.5. Protein–Protein Interaction Network Analysis
4.6. K-Means Cluster Analysis
4.7. Gene Co-Expression Network (GCN) Construction and Analysis
4.8. Analysis of the Cis-Regulatory Elements in the CDT Promoters
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Lesk, C.; Rowhani, P.; Ramankutty, N. Influence of extreme weather disasters on global crop production. Nature 2016, 529, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Daryanto, S.; Wang, L.; Jacinthe, P.A. Global synthesis of drought effects on maize and wheat production. PLoS ONE 2016, 11, e0156362. [Google Scholar] [CrossRef]
- Tardieu, F.; Simonneau, T.; Muller, B. The Physiological Basis of Drought Tolerance in Crop Plants: A Scenario-Dependent Probabilistic Approach. Annu. Rev. Plant Biol. 2018, 69, 733–759. [Google Scholar] [CrossRef] [Green Version]
- Bailey-Serres, J.; Parker, J.E.; Ainsworth, E.A.; Oldroyd, G.E.D.; Schroeder, J.I. Genetic strategies for improving crop yields. Nature 2019, 575, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Trenberth, K.E.; Dai, A.; Van Der Schrier, G.; Jones, P.D.; Barichivich, J.; Briffa, K.R.; Sheffield, J. Global warming and changes in drought. Nat. Clim. Chang. 2014, 4, 17–22. [Google Scholar] [CrossRef]
- Fàbregas, N.; Fernie, A.R. The metabolic response to drought. J. Exp. Bot. 2019, 70, 1077–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, A. Osmotic adjustment is a prime drought stress adaptive engine in support of plant production. Plant Cell Environ. 2017, 40, 4–10. [Google Scholar] [CrossRef]
- Per, T.S.; Khan, N.A.; Reddy, P.S.; Masood, A.; Hasanuzzaman, M.; Khan, M.I.R.; Anjum, N.A. Approaches in modulating proline metabolism in plants for salt and drought stress tolerance: Phytohormones, mineral nutrients and transgenics. Plant Physiol. Biochem. 2017, 115, 126–140. [Google Scholar] [CrossRef]
- Claeys, H.; Inzé, D. The agony of choice: How plants balance growth and survival under water-limiting conditions. Plant Physiol. 2013, 162, 1768–1779. [Google Scholar] [CrossRef]
- Estravis-Barcala, M.; Mattera, M.G.; Soliani, C.; Bellora, N.; Opgenoorth, L.; Heer, K.; Arana, M.V.; Gutiérrez, R. Molecular bases of responses to abiotic stress in trees. J. Exp. Bot. 2020, 71, 3765–3779. [Google Scholar] [CrossRef] [PubMed]
- Laxa, M.; Liebthal, M.; Telman, W.; Chibani, K.; Dietz, K.J. The role of the plant antioxidant system in drought tolerance. Antioxidants 2019, 8, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peleg, Z.; Blumwald, E. Hormone balance and abiotic stress tolerance in crop plants. Curr. Opin. Plant Biol. 2011, 14, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Kazan, K. Diverse roles of jasmonates and ethylene in abiotic stress tolerance. Trends Plant Sci. 2015, 20, 219–229. [Google Scholar] [CrossRef]
- de Ollas, C.; Dodd, I.C. Physiological impacts of ABA–JA interactions under water-limitation. Plant Mol. Biol. 2016, 91, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Mogami, J.; Yamaguchi-Shinozaki, K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 2014, 21, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Todaka, D.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Recent advances in the dissection of drought-stress regulatory networks and strategies for development of drought-tolerant transgenic rice plants. Front. Plant Sci. 2015, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Takasaki, H.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. NAC transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta—Gene Regul. Mech. 2012, 1819, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Dietz, K.J.; Vogel, M.O.; Viehhauser, A. AP2/EREBP transcription factors are part of gene regulatory networks and integrate metabolic, hormonal and environmental signals in stress acclimation and retrograde signalling. Protoplasma 2010, 245, 3–14. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Sui, N. Transcriptional regulation of bHLH during plant response to stress. Biochem. Biophys. Res. Commun. 2018, 503, 397–401. [Google Scholar] [CrossRef]
- Baldoni, E.; Genga, A.; Cominelli, E. Plant MYB transcription factors: Their role in drought response mechanisms. Int. J. Mol. Sci. 2015, 16, 15811–15851. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Pang, S.; Lu, Z.; Jin, B. Function and mechanism of WRKY transcription factors in abiotic stress responses of plants. Plants 2020, 9, 1515. [Google Scholar] [CrossRef]
- Han, G.; Lu, C.; Guo, J.; Qiao, Z.; Sui, N.; Qiu, N.; Wang, B. C2H2 Zinc Finger Proteins: Master Regulators of Abiotic Stress Responses in Plants. Front. Plant Sci. 2020, 11, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Front. Plant Sci. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Chandra, B.R. Breeding for drought resistance in rice: An integrated view from physiology. Babu R. Chandra 2010, 1, 1133–1141. [Google Scholar]
- Fukai, S.; Cooper, M. Development of drought-resistant cultivars using physiomorphological traits in rice. Field Crops Res. 1995, 40, 67–86. [Google Scholar] [CrossRef]
- Zhang, Z.F.; Li, Y.Y.; Xiao, B.Z. Comparative transcriptome analysis highlights the crucial roles of photosynthetic system in drought stress adaptation in upland rice. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Baldoni, E.; Bagnaresi, P.; Locatelli, F.; Mattana, M.; Genga, A. Comparative Leaf and Root Transcriptomic Analysis of two Rice Japonica Cultivars Reveals Major Differences in the Root Early Response to Osmotic Stress. Rice 2016, 9, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Cantalapiedra, C.P.; García-Pereira, M.J.; Gracia, M.P.; Igartua, E.; Casas, A.M.; Contreras-Moreira, B. Large differences in gene expression responses to drought and heat stress between elite barley cultivar scarlett and a Spanish landrace. Front. Plant Sci. 2017, 8, 647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenk, I.; Fisher, L.H.C.; Vickers, M.; Akinyemi, A.; Didion, T.; Swain, M.; Jensen, C.S.; Mur, L.A.J.; Bosch, M. Transcriptional and metabolomic analyses indicate that cell wall properties are associated with drought tolerance in brachypodium distachyon. Int. J. Mol. Sci. 2019, 20, 1758. [Google Scholar] [CrossRef] [Green Version]
- Zenda, T.; Liu, S.; Wang, X.; Liu, G.; Jin, H.; Dong, A.; Yang, Y.; Duan, H. Key maize drought-responsive genes and pathways revealed by comparative transcriptome and physiological analyses of contrasting inbred lines. Int. J. Mol. Sci. 2019, 20, 1268. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Lou, Q.; Xu, K.; Yan, M.; Xia, H.; Ma, X.; Yu, X.; Luo, L. Alternative splicing complexity contributes to genetic improvement of drought resistance in the rice maintainer HuHan2B. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Lenka, S.K.; Katiyar, A.; Chinnusamy, V.; Bansal, K.C. Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotechnol. J. 2011, 9, 315–327. [Google Scholar] [CrossRef]
- Muthuramalingam, P.; Krishnan, S.R.; Pothiraj, R.; Ramesh, M. Global transcriptome analysis of combined abiotic stress signaling genes unravels key players in Oryza sativa L.: An in silico approach. Front. Plant Sci. 2017, 8, 759. [Google Scholar] [CrossRef] [Green Version]
- Buti, M.; Baldoni, E.; Formentin, E.; Milc, J.; Frugis, G.; Lo Schiavo, F.; Genga, A.; Francia, E. A meta-analysis of comparative transcriptomic data reveals a set of key genes involved in the tolerance to abiotic stresses in rice. Int. J. Mol. Sci. 2019, 20, 5662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sircar, S.; Parekh, N. Meta-analysis of drought-tolerant genotypes in Oryza sativa: A network-based approach. PLoS ONE 2019, 14, e0216068. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.P.; Leach, J.E. Abiotic and biotic stresses induce a core transcriptome response in rice. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Todaka, D.; Zhao, Y.; Yoshida, T.; Kudo, M.; Kidokoro, S.; Mizoi, J.; Kodaira, K.S.; Takebayashi, Y.; Kojima, M.; Sakakibara, H.; et al. Temporal and spatial changes in gene expression, metabolite accumulation and phytohormone content in rice seedlings grown under drought stress conditions. Plant J. 2017, 90, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. The plant hormone abscisic acid mediates the drought-induced expression but not the seed-specific expression of rd22, a gene responsive to dehydration stress in Arabidopsis thaliana. MGG Mol. Gen. Genet. 1993, 238, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Martínez, I.M.; Chrispeels, M.J. Genomic analysis of the unfolded protein response in Arabidopsis shows its connection to important cellular processes. Plant Cell 2003, 15, 561–576. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.E.; Bassi, R.; Boekema, E.J.; Dekker, J.P.; Jansson, S.; Leister, D.; Robinson, C.; Scheller, H.V. Structure, function and regulation of plant photosystem I. Biochim. Biophys. Acta—Bioenerg. 2007, 1767, 335–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Wu, H.; Wu, J.; Fan, X.; Li, X.; Lin, Y. Absence of OsβCA1 causes a CO2 deficit and affects leaf photosynthesis and the stomatal response to CO2 in rice. Plant J. 2017, 90, 344–357. [Google Scholar] [CrossRef] [Green Version]
- Muthusamy, S.K.; Lenka, S.K.; Katiyar, A.; Chinnusamy, V.; Singh, A.K.; Bansal, K.C. Genome-Wide Identification and Analysis of Biotic and Abiotic Stress Regulation of C 4 Photosynthetic Pathway Genes in Rice. Appl. Biochem. Biotechnol. 2019, 187, 221–238. [Google Scholar] [CrossRef]
- Jiang, H.; Li, M.; Liang, N.; Yan, H.; Wei, Y.; Xu, X.; Liu, J.; Xu, Z.; Chen, F.; Wu, G. Molecular cloning and function analysis of the stay green gene in rice. Plant J. 2007, 52, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Yu, J.W.; Park, J.S.; Li, J.; Yoo, S.C.; Lee, N.Y.; Lee, S.K.; Jeong, S.W.; Hak, S.S.; Koh, H.J.; et al. The senescence-induced staygreen protein regulates chlorophyll degradation. Plant Cell 2007, 19, 1649–1664. [Google Scholar] [CrossRef] [Green Version]
- Rong, H.; Tang, Y.; Zhang, H.; Wu, P.; Chen, Y.; Li, M.; Wu, G.; Jiang, H. The Stay-Green Rice like (SGRL) gene regulates chlorophyll degradation in rice. J. Plant Physiol. 2013, 170, 1367–1373. [Google Scholar] [CrossRef]
- Zeng, Z.; Lin, T.; Zhao, J.; Zheng, T.; Xu, L.; Wang, Y.; Liu, L.; Jiang, L.; Chen, S.; Wan, J. OsHemA gene, encoding glutamyl-tRNA reductase (GluTR) is essential for chlorophyll biosynthesis in rice (Oryza sativa). J. Integr. Agric. 2020, 19, 612–623. [Google Scholar] [CrossRef]
- Zhou, Q.; Yu, Q.; Wang, Z.; Pan, Y.; Lv, W.; Zhu, L.; Chen, R.; He, G. Knockdown of GDCH gene reveals reactive oxygen species-induced leaf senescence in rice. Plant Cell Environ. 2013, 36, 1476–1489. [Google Scholar] [CrossRef]
- Kolton, M.; Keren, I.; Shevtsov, S.; Shaya, F.; Peled-Zehavi, H.; Danon, A.; Ostersetzer-Biran, O. Plastidic redox switches: Ferredoxins as novel RNA-binding proteins. J. Endocytobiosis Cell Res. 2011, 5, 1–18. [Google Scholar]
- Mei, J.; Li, F.; Liu, X.; Hu, G.; Fu, Y.; Liu, W. Newly identified CSP41b gene localized in chloroplasts affects leaf color in rice. Plant Sci. 2017, 256, 39–45. [Google Scholar] [CrossRef]
- Wang, G.F.; Li, W.Q.; Li, W.Y.; Wu, G.L.; Zhou, C.Y.; Chen, K.M. Characterization of rice NADPH oxidase genes and their expression under various environmental conditions. Int. J. Mol. Sci. 2013, 14, 9440–9458. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Li, L.; Zhang, P.; Wang, D.; Yang, L.; Reza, B.M.; Lin, W. Lsi1 modulates the antioxidant capacity of rice and protects against ultraviolet-B radiation. Plant Sci. 2019, 278, 96–106. [Google Scholar] [CrossRef]
- Bai, Z.; Qi, T.; Liu, Y.; Wu, Z.; Ma, L.; Liu, W.; Cao, Y.; Bao, Y.; Fu, C. Alteration of S-adenosylhomocysteine levels affects lignin biosynthesis in switchgrass. Plant Biotechnol. J. 2018, 16, 2016–2026. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Tao, Y.; Ali, A.; Zhuang, Z.; Guo, D.; Guo, Q.; Riaz, A.; Zhang, H.; Xu, P.; Liao, Y.; et al. Transcriptome and Proteome Profiling of Different Colored Rice Reveals Physiological Dynamics Involved in the Flavonoid Pathway. Int. J. Mol. Sci. 2019, 20, 2463. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Cho, K.; Jang, S.; Rakwal, R.; Iwahashi, H.; Agrawal, G.K.; Shim, J.; Han, O. Inverse correlation between jasmonic acid and salicylic acid during early wound response in rice. Biochem. Biophys. Res. Commun. 2004, 318, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Shim, Y.; Kang, K.; An, G.; Paek, N.C. Rice DNA-Binding One Zinc Finger 24 (OsDOF24) Delays Leaf Senescence in a Jasmonate-Mediated Pathway. Plant Cell Physiol. 2019, 60, 2065–2076. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhao, J.; Yang, B.; Sun, S.; Peng, L.; Wang, Z. Indole-3-acetate beta-glucosyltransferase OsIAGLU regulates seed vigour through mediating crosstalk between auxin and abscisic acid in rice. Plant Biotechnol. J. 2020, 18, 1933–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testone, G.; Baldoni, E.; Iannelli, M.A.; Nicolodi, C.; Di Giacomo, E.; Pietrini, F.; Mele, G.; Giannino, D.; Frugis, G. Transcription factor networks in leaves of cichorium endivia: New insights into the relationship between photosynthesis and leaf development. Plants 2019, 8, 531. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Hu, J.X.; Thomas, C.E.; Brunak, S. Network biology concepts in complex disease comorbidities. Nat. Rev. Genet. 2016, 17, 615–629. [Google Scholar] [CrossRef]
- Urao, T.; Yamaguchi-Shinozaki, K.; Urao, S.; Shinozaki, K. An Arabidopsis myb homolog is induced by dehydration stress and its gene product binds to the conserved MYB recognition sequence. Plant Cell 1993, 5, 1529–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Xiong, L. Genetic engineering and breeding of drought-resistant crops. Annu. Rev. Plant Biol. 2014, 65, 715–741. [Google Scholar] [CrossRef] [PubMed]
- Buti, M.; Pasquariello, M.; Ronga, D.; Milc, J.A.; Pecchioni, N.; Ho, V.T.; Pucciariello, C.; Perata, P.; Francia, E. Transcriptome profiling of short-term response to chilling stress in tolerant and sensitive Oryza sativa ssp. Japonica seedlings. Funct. Integr. Genom. 2018, 18, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Bjornson, M.; Dandekar, A.; Dehesh, K. Determinants of timing and amplitude in the plant general stress response. J. Integr. Plant Biol. 2016, 58, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Tahmasebi, A.; Niazi, A. Comparison of Transcriptional Response of C3 and C4 Plants to Drought Stress Using Meta-Analysis and Systems Biology Approach. Front. Plant Sci. 2021, 12, 668736. [Google Scholar] [CrossRef] [PubMed]
- Dal Bosco, C.; Lezhneva, L.; Bieh, A.; Leister, D.; Strotmann, H.; Wanner, G.; Meurer, J. Inactivation of the Chloroplast ATP Synthase γ Subunit Results in High Non-photochemical Fluorescence Quenching and Altered Nuclear Gene Expression in Arabidopsis thaliana. J. Biol. Chem. 2004, 279, 1060–1069. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.Y.; Kuai, B.K.; Jia, J.Z.; Jing, H.C. Regulation of Leaf Senescence and Crop Genetic Improvement. J. Integr. Plant Biol. 2012, 54, 936–952. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, H.; Lee, S.; Lee, J.; Kim, S.J.; Park, J.W.; Woo, H.R.; Lim, P.O.; An, G.; Nam, H.G.; et al. Molecular bases for differential aging programs between flag and second leaves during grain-filling in rice. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Leng, Y.; Ye, G.; Zeng, D. Genetic dissection of leaf senescence in rice. Int. J. Mol. Sci. 2017, 18, 2686. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.; Lu, S.; Lv, B.; Zhang, B.; Shen, J.; He, J.; Luo, L.; Xi, D.; Chen, X.; Ming, F. A rice nac transcription factor promotes leaf senescence via ABA biosynthesis. Plant Physiol. 2017, 174, 1747–1763. [Google Scholar] [CrossRef] [Green Version]
- Takasaki, H.; Maruyama, K.; Takahashi, F.; Fujita, M.; Yoshida, T.; Nakashima, K.; Myouga, F.; Toyooka, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. SNAC-As, stress-responsive NAC transcription factors, mediate ABA-inducible leaf senescence. Plant J. 2015, 84, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Rivero, R.M.; Kojima, M.; Gepstein, A.; Sakakibara, H.; Mittler, R.; Gepstein, S.; Blumwald, E. Delayed leaf senescence induces extreme drought tolerance in a flowering plant. Proc. Natl. Acad. Sci. USA 2007, 104, 19631–19636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, N.M.; Gorafi, Y.S.A.; Abdelrahman, M.; Abdellatef, E.; Tsujimoto, H. Stay-green trait: A prospective approach for yield potential, and drought and heat stress adaptation in globally important cereals. Int. J. Mol. Sci. 2019, 20, 5837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan, S.; Abbas, N.; Ashraf, M.; Ahmad, P. Roles of potential plant hormones and transcription factors in controlling leaf senescence and drought tolerance. Protoplasma 2019, 256, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Serin, E.A.R.; Nijveen, H.; Hilhorst, H.W.M.; Ligterink, W. Learning from Co-expression Networks: Possibilities and Challenges. Front. Plant Sci. 2016, 7, 444. [Google Scholar] [CrossRef] [Green Version]
- Zanetti, M.E.; Rípodas, C.; Niebel, A. Plant NF-Y transcription factors: Key players in plant-microbe interactions, root development and adaptation to stress. Biochim. Biophys. Acta—Gene Regul. Mech. 2017, 1860, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Leyva-González, M.A.; Ibarra-Laclette, E.; Cruz-Ramírez, A.; Herrera-Estrella, L. Functional and Transcriptome Analysis Reveals an Acclimatization Strategy for Abiotic Stress Tolerance Mediated by Arabidopsis NF-YA Family Members. PLoS ONE 2012, 7, e0048138. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, K.; Ito, Y.; Serizawa, A.; Kurata, N. OsHAP3 genes regulate chloroplast biogenesis in rice. Plant J. 2003, 36, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Gnesutta, N.; Kumimoto, R.W.; Swain, S.; Chiara, M.; Siriwardana, C.; Horner, D.S.; Holt, B.F.; Mantovani, R. CONSTANS imparts DNA sequence specificity to the histone fold NF-YB/NF-YC Dimer. Plant Cell 2017, 29, 1516–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, W.; Kim, E.Y.; Han, S.H.; Sakuraba, Y.; Paek, N.C. Rice phytochrome B (OsPhyB) negatively regulates dark- and starvation-induced leaf senescence. Plants 2015, 4, 644–663. [Google Scholar] [CrossRef] [Green Version]
- Todaka, D.; Nakashima, K.; Maruyama, K.; Kidokoro, S.; Osakabe, Y.; Ito, Y.; Matsukura, S.; Fujita, Y.; Yoshiwara, K.; Ohme-Takagi, M.; et al. Rice phytochrome-interacting factor-like protein OsPIL1 functions as a key regulator of internode elongation and induces a morphological response to drought stress. Proc. Natl. Acad. Sci. USA 2012, 109, 15947–15952. [Google Scholar] [CrossRef] [Green Version]
- Sakuraba, Y.; Kim, E.Y.; Han, S.H.; Piao, W.; An, G.; Todaka, D.; Yamaguchi-Shinozaki, K.; Paek, N.C. Rice Phytochrome-Interacting Factor-Like1 (OsPIL1) is involved in the promotion of chlorophyll biosynthesis through feed-forward regulatory loops. J. Exp. Bot. 2017, 68, 4103–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, B.; Kitazumi, A.; Cheung, C.Y.M.; Lakshmanan, M.; de los Reyes, B.G.; Jang, I.C.; Lee, D.Y. Identification of candidate network hubs involved in metabolic adjustments of rice under drought stress by integrating transcriptome data and genome-scale metabolic network. Plant Sci. 2016, 242, 224–239. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; Lv, B.; Li, J.; Luo, L.; Lu, S.; Zhang, X.; Ma, H.; Ming, F. The NAC family transcription factor OsNAP confers abiotic stress response through the ABA pathway. Plant Cell Physiol. 2014, 55, 604–619. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Wang, Y.; Zhu, Y.; Tang, J.; Hu, B.; Liu, L.; Ou, S.; Wu, H.; Sun, X.; Chu, J.; et al. OsNAP connects abscisic acid and leaf senescence by fine-tuning abscisic acid biosynthesis and directly targeting senescence-associated genes in rice. Proc. Natl. Acad. Sci. USA 2014, 111, 10013–10018. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Huang, W.; Liu, L.; Chen, T.; Zhou, F.; Lin, Y. Identification and functional characterization of a rice NAC gene involved in the regulation of leaf senescence. BMC Plant Biol. 2013, 13, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Chung, P.J.; Park, S.H.; Redillas, M.C.F.R.; Kim, Y.S.; Suh, J.W.; Kim, J.K. Overexpression of OsERF48 causes regulation of OsCML16, a calmodulin-like protein gene that enhances root growth and drought tolerance. Plant Biotechnol. J. 2017, 15, 1295–1308. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Chen, J.; Luo, N.; Qu, R.; Guo, Z.; Lu, S. Cholesterol accumulation by suppression of SMT1 leads to dwarfism and improved drought tolerance in herbaceous plants. Plant Cell Environ. 2018, 41, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Baisakh, N.; Yabes, J.; Gutierrez, A.; Mangu, V.; Ma, P.; Famoso, A.; Pereira, A. Genetic mapping identifies consistent quantitative trait loci for yield traits of rice under greenhouse drought conditions. Genes 2020, 11, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.; Yu, J.; Miao, J.; Li, J.; Zhang, H.; Wang, X.; Liu, P.; Zhao, Y.; Jiang, C.; Yin, Z.; et al. Natural variation in OsLG3 increases drought tolerance in rice by inducing ROS scavenging. Plant Physiol. 2018, 178, 451–467. [Google Scholar] [CrossRef] [Green Version]
- Baldoni, E.; Genga, A.; Medici, A.; Coraggio, I.; Locatelli, F. The OsMyb4 gene family: Stress response and transcriptional auto-regulation mechanisms. Biol. Plant. 2013, 57, 691–700. [Google Scholar] [CrossRef]
- Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta—Gene Regul. Mech. 2012, 1819, 86–96. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, Y.; Cai, H.; Guo, M.; Chai, M.; She, Z.; Ye, L.; Cheng, Y.; Wang, B.; Qin, Y. The bzip transcription factor gmbzip15 negatively regulates salt-and drought-stress responses in soybean. Int. J. Mol. Sci. 2020, 21, 7778. [Google Scholar] [CrossRef]
- Sekhon, R.S.; Saski, C.; Kumar, R.; Flinn, B.S.; Luo, F.; Beissinger, T.M.; Ackerman, A.J.; Breitzman, M.W.; Bridges, W.C.; de Leon, N.; et al. Integrated genome-scale analysis identifies novel genes and networks underlying senescence in maize. Plant Cell 2019, 31, 1968–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Haider, I.; Kohlen, W.; Jiang, L.; Bouwmeester, H.; Meijer, A.H.; Schluepmann, H.; Liu, C.M.; Ouwerkerk, P.B.F. Function of the HD-Zip I gene Oshox22 in ABA-mediated drought and salt tolerances in rice. Plant Mol. Biol. 2012, 80, 571–585. [Google Scholar] [CrossRef]
- Agalou, A.; Purwantomo, S.; Övernäs, E.; Johannesson, H.; Zhu, X.; Estiati, A.; De Kam, R.J.; Engström, P.; Slamet-Loedin, I.H.; Zhu, Z.; et al. A genome-wide survey of HD-Zip genes in rice and analysis of drought-responsive family members. Plant Mol. Biol. 2008, 66, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, X.; Wang, W.; Pan, Y.; Huang, L.; Liu, X.; Zong, Y.; Zhu, L.; Yang, D.; Fu, B. Comparative transcriptome profiling of chilling stress responsiveness in two contrasting rice genotypes. PLoS ONE 2012, 7, e0043274. [Google Scholar] [CrossRef] [Green Version]
- Baldoni, E.; Mattana, M.; Locatelli, F.; Consonni, R.; Cagliani, L.R.; Picchi, V.; Abbruscato, P.; Genga, A. Analysis of transcript and metabolite levels in Italian rice (Oryzasativa L.) cultivars subjected to osmotic stress or benzothiadiazole treatment. Plant Physiol. Biochem. 2013, 70, 492–503. [Google Scholar] [CrossRef]
- Andrews, S. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 September 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.P.; Garvin, D.F.; Mockler, T.C.; Schmutz, J.; Rokhsar, D.; Bevan, M.W.; Barry, K.; Lucas, S.; Harmon-Smith, M.; Lail, K.; et al. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [Google Scholar] [CrossRef]
- Mayer, K.F.X.; Waugh, R.; Langridge, P.; Close, T.J.; Wise, R.P.; Graner, A.; Matsumoto, T.; Sato, K.; Schulman, A.; Ariyadasa, R.; et al. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Wu, J.; Kanamori, H.; Katayose, Y.; Fujisawa, M.; Namiki, N.; Mizuno, H.; Yamamoto, K.; Antonio, B.A.; Baba, T.; et al. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014, 42, D1182–D1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinsella, R.J.; Ka, A.; Spudich, G.; Almeida-king, J.; Staines, D.; Derwent, P.; Kerhornou, A.; Kersey, P.; Flicek, P. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.; Obenchain, V.; Hester, J.; Pagès, H. Summarized Experiment: Summarized Experiment Container. R Package Version 1.24.0. 2021. Available online: https://bioconductor.org/packages/SummarizedExperiment (accessed on 30 September 2021).
- Wei, T.; Simko, V. R Package “Corrplot”: Visualization of a Correlation Matrix (Version 0.84). 2017. Available online: https://github.com/taiyun/corrplot (accessed on 30 September 2021).
- Nguyen, N.T.T.; Contreras-Moreira, B.; Castro-Mondragon, J.A.; Santana-Garcia, W.; Ossio, R.; Robles-Espinoza, C.D.; Bahin, M.; Collombet, S.; Vincens, P.; Thieffry, D.; et al. RSAT 2018: Regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res. 2018, 46, W209–W214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999, 27, 297–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemose, S.; Jensen, M.K.; De Velde, J.; O’Shea, C.; Heyndrickx, K.S.; Workman, C.T.; Vandepoele, K.; Skriver, K.; De Masi, F. A DNA-binding-site landscape and regulatory network analysis for NAC transcription factors in Arabidopsis thaliana. Nucleic Acids Res. 2014, 42, 7681–7693. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B. distachyon | H. vulgare | O. sativa | Z. mays | |
|---|---|---|---|---|
| Reference | [30] | [29] | [32] | [31] |
| Sensitive accession | KOZ1 | Scarlett | HanFengB | MO17 |
| Tolerant accession | ABR8 | SBCC073 | HuHuan2B | YE8112 |
| Biological replicates | 3 | 3 | Pooled | 2–3 |
| Drought time | 8 days | 30 days | 20 days | 7 days |
| Sequencer | HiSeq 2000 | HiSeq 2000 | HiSeq 2500 | Hiseq Xten |
| Reads length | 100 nt paired-end | 101 nt paired-end | 126 nt paired-end | 150 nt single-end |
| BioProject accession | PRJNA524106 | PRJEB12540 | PRJNA306542 | PRJNA397964 |
| B. distachyon | H. vulgare | O. sativa | Z. mays | |||||
|---|---|---|---|---|---|---|---|---|
| Sensitive | Tolerant | Sensitive | Tolerant | Sensitive | Tolerant | Sensitive | Tolerant | |
| Predicted genes | 35,118 | 35,118 | 43,050 | 43,050 | 39,978 | 39,978 | 45,953 | 45,953 |
| Active genes | 19,404 | 18,411 | 16,092 | 15,773 | 18,449 | 18,285 | 20,094 | 21,048 |
| Down-reg DEGs | 3551 | 61 | 287 | 1261 | 1634 | 1360 | 2915 | 583 |
| Up-reg DEGs | 3145 | 234 | 448 | 653 | 1650 | 1134 | 2129 | 348 |
| Total DEGs | 6696 | 295 | 735 | 1914 | 3284 | 2494 | 5044 | 931 |
| Class | B. distachyon | H. vulgare | O. sativa | Z. mays |
|---|---|---|---|---|
| sen+ | 3125 | 313 | 942 | 2026 |
| sen− | 3340 | 168 | 901 | 2722 |
| tol+ | 38 | 543 | 421 | 251 |
| tol− | 26 | 1117 | 632 | 384 |
| ∆LFC > 1 | 16 | 12 | 88 | 56 |
| ∆LFC < −1 | 4 | 13 | 40 | 9 |
| sen−/tol+ | 185 | 1 | 19 | 77 |
| sen+/tol− | 9 | 26 | 14 | 83 |
| TOTAL | 6743 | 2193 | 3057 | 5608 |
| n. | RAP ID | Gene Name | Related Pathway |
|---|---|---|---|
| Os_1 | Os01g0111100 | uncharacterized gene | |
| Os_2 | Os01g0117300 | OsRLCK17 | signalling |
| Os_3 | Os01g0227800 | uncharacterized gene | |
| Os_4 | Os01g0551000 | uncharacterized gene | |
| Os_5 | Os01g0639900 | OsCA1 | PHOTOSYNTHESIS (CO2 fixation) |
| Os_6 | Os01g0733500 | RD22 | ABIOTIC STRESS RESPONSE |
| Os_7 | Os01g0803200 | OsCYS1 | cysteine proteinase inhibitors |
| Os_8 | Os02g0101500 | Peroxisomal hydroxypyruvate reductase | |
| Os_9 | Os02g0465900 | ChaC-like | TRANSPORT |
| Os_10 | Os02g0626400 | PAL2-2 | SECONDARY METABOLISM |
| Os_11 | Os02g0710900 | HSP70—OsBiP | ABIOTIC STRESS RESPONSE |
| Os_12 | Os02g0744000 | uncharacterized gene | |
| Os_13 | Os02g0783625 | lysine ketoglutarate reductase | |
| Os_14 | Os02g0815700 | uncharacterized gene | |
| Os_15 | Os03g0114800 | OsCd1 | TRANSPORT |
| Os_16 | Os03g0129300 | G3PDH | PHOTOSYNTHESIS (Calvin Cycle) |
| Os_17 | Os03g0192700 | Inositol-3-phosphate synthase 1 | |
| Os_18 | Os03g0238300 | inositol-1,4,5-trisphosphate 5-phosphatase | |
| Os_19 | Os03g0336000 | uncharacterized gene | |
| Os_20 | Os03g0592500 | LHCB | PHOTOSYNTHESIS (photosystem assembly) |
| Os_21 | Os03g0700700 | OsLOX8 | JA SIGNALLING |
| Os_22 | Os03g0744700 | uncharacterized gene | |
| Os_23 | Os03g0844900 | uncharacterized gene | |
| Os_24 | Os03g0849800 | Glycosyltransferase | |
| Os_25 | Os03g0850400 | aspartate kinase | |
| Os_26 | Os04g0234600 | SBPase | PHOTOSYNTHESIS (Calvin Cycle) |
| Os_27 | Os04g0390100 | OsHIPP29 | TRANSPORT |
| Os_28 | Os04g0414700 | PsaO | PHOTOSYNTHESIS (photosystem assembly) |
| Os_29 | Os04g0459500 | G3PDH | PHOTOSYNTHESIS (Calvin Cycle) |
| Os_30 | Os04g0496000 | uncharacterized gene | |
| Os_31 | Os04g0508800 | uncharacterized gene | |
| Os_32 | Os04g0618200 | UDP-arabinose 4-epimerase 2 | |
| Os_33 | Os05g0186300 | OsNADP-ME3 | PHOTOSYNTHESIS (Calvin Cycle) |
| Os_34 | Os05g0429500 | Dienelactone hydrolase | |
| Os_35 | Os05g0439400 | OsPUB44 | E3 ubiquitin ligase |
| Os_36 | Os05g0453300 | uncharacterized gene | |
| Os_37 | Os05g0468900 | uncharacterized gene | |
| Os_38 | Os05g0501700 | uncharacterized gene | |
| Os_39 | Os05g0535900 | IQ calmodulin-binding motif family protein | |
| Os_40 | Os06g0129400 | OsSPX-MFS3 | CELLULAR TRANSPORT |
| Os_41 | Os06g0486800 | FDH (Formate dehydrogenase) | CELLULAR TRANSPORT |
| Os_42 | Os06g0528600 | Aminopropyl transferase | |
| Os_43 | Os06g0716000 | uncharacterized gene | |
| Os_44 | Os07g0148900 | Psak | PHOTOSYNTHESIS (photosystem assembly) |
| Os_45 | Os07g0524900 | uncharacterized gene | |
| Os_46 | Os08g0104600 | OsFd1 | OXIDATIVE STRESS RESPONSE |
| Os_47 | Os08g0141300 | OsVST1 | root system architecture |
| Os_48 | Os08g0141400 | NADH-ubiquinone oxidoreductase | |
| Os_49 | Os08g0364900 | uncharacterized gene | |
| Os_50 | Os08g0434300 | malate dehydrogenase | |
| Os_51 | Os08g0485900 | uncharacterized gene | |
| Os_52 | Os08g0508800 | OsHI-LOX | JA SIGNALLING |
| Os_53 | Os09g0294000 | Aspartate kinase-homoserine dehydrogenase | |
| Os_54 | Os09g0297000 | Ferrochelatase-1 | |
| Os_55 | Os09g0426800 | OsGL1-1 | leaf wax synthesis |
| Os_56 | Os09g0438000 | OsNOX7 | OXIDATIVE STRESS RESPONSE |
| Os_57 | Os09g0438100 | uncharacterized gene | |
| Os_58 | Os09g0532000 | SGR | LEAF SENESCENCE |
| Os_59 | Os10g0462900 | OsHsp60-1 | ABIOTIC STRESS RESPONSE |
| Os_60 | Os10g0502400 | OsHEMA | LEAF SENESCENCE |
| Os_61 | Os10g0516100 | OsGDCH | LEAF SENESCENCE |
| Os_62 | Os10g0530500 | glutathione S-transferase | OXIDATIVE STRESS RESPONSE |
| Os_63 | Os11g0145200 | Indole-3-acetate beta-glucosyltransferase | AUXIN SIGNALLING |
| Os_64 | Os11g0455500 | OsSAHH | SECONDARY METABOLISM |
| Os_65 | Os11g0520500 | uncharacterized gene | |
| Os_66 | Os12g0115700 | CHI | SECONDARY METABOLISM |
| Os_67 | Os12g0420200 | CSP41b | OXIDATIVE STRESS RESPONSE |
| Os_68 | Os12g0428000 | glycosyl hydrolase | |
| Os_69 | Os12g0534100 | uncharacterized gene | |
| B. distachyon Gene ID | H. vulgare Gene ID | O. sativa Gene ID | Z. mays Gene ID | Corresponding Clusters | TF Class |
|---|---|---|---|---|---|
| Bradi1g06670 | - | Os03g0782500 | Zm00001d034298 | photosynthesis | bHLH |
| Bradi2g00730 | HORVU3Hr1G000170 | Os01g0108400 | - | SGR | bHLH |
| Bradi2g60970 | - | Os01g0952800 | - | photosynthesis | bHLH |
| Bradi3g02730 | - | Os02g0128200 | Zm00001d053967 Zm00001d015118 | photosynthesis | bZIP |
| Bradi3g57960 | - | - | Zm00001d018178 | SGR | bZIP |
| - | Os07g0644100 | Zm00001d007042 | SGR | bZIP | |
| Bradi4g02570 | - | Os12g0601800 | - | photosynthesis | bZIP |
| Bradi1g11310 | - | - | Zm00001d033719 Zm00001d013443 | photosynthesis | CO-like |
| Bradi3g35560 | - | Os08g0408500 | Zm00001d032295 | SGR | ERF |
| Bradi4g29380 | - | Os09g0379600 | SGR | HD-ZIP | |
| Bradi5g17170 | HORVU2Hr1G092710 | - | Zm00001d002799Zm00001d025964 | SGR | HD-ZIP |
| Bradi3g16400 | - | Os08g0159500 | - | photosynthesis | LBD |
| Bradi2g16120 | - | Os05g0567600 | - | photosynthesis | MYB |
| Bradi3g16515 | HORVU7Hr1G070870 | - | - | photosynthesis | MYB |
| Bradi3g52807 | - | - | Zm00001d017782 | photosynthesis | MYB |
| - | HORVU5Hr1G099390 | - | Zm00001d013151 | SGR | NAC |
| Bradi1g60030 | - | Os03g0413000 | - | photosynthesis | NF-YB |
| - | - | Os06g0145800 | Zm00001d036148 | photosynthesis | Whirly |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldoni, E.; Frugis, G.; Martinelli, F.; Benny, J.; Paffetti, D.; Buti, M. A Comparative Transcriptomic Meta-Analysis Revealed Conserved Key Genes and Regulatory Networks Involved in Drought Tolerance in Cereal Crops. Int. J. Mol. Sci. 2021, 22, 13062. https://doi.org/10.3390/ijms222313062
Baldoni E, Frugis G, Martinelli F, Benny J, Paffetti D, Buti M. A Comparative Transcriptomic Meta-Analysis Revealed Conserved Key Genes and Regulatory Networks Involved in Drought Tolerance in Cereal Crops. International Journal of Molecular Sciences. 2021; 22(23):13062. https://doi.org/10.3390/ijms222313062
Chicago/Turabian StyleBaldoni, Elena, Giovanna Frugis, Federico Martinelli, Jubina Benny, Donatella Paffetti, and Matteo Buti. 2021. "A Comparative Transcriptomic Meta-Analysis Revealed Conserved Key Genes and Regulatory Networks Involved in Drought Tolerance in Cereal Crops" International Journal of Molecular Sciences 22, no. 23: 13062. https://doi.org/10.3390/ijms222313062
APA StyleBaldoni, E., Frugis, G., Martinelli, F., Benny, J., Paffetti, D., & Buti, M. (2021). A Comparative Transcriptomic Meta-Analysis Revealed Conserved Key Genes and Regulatory Networks Involved in Drought Tolerance in Cereal Crops. International Journal of Molecular Sciences, 22(23), 13062. https://doi.org/10.3390/ijms222313062