
Regulation of the Cell-Intrinsic DNA Damage Response by the Innate Immune Machinery


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. RLRs and the Cell-Intrinsic DNA Damage Response
3. cGAS–STING and Cellular Senescence
4. STING-Independent Nuclear Functions of cGAS
5. STING and Autophagy
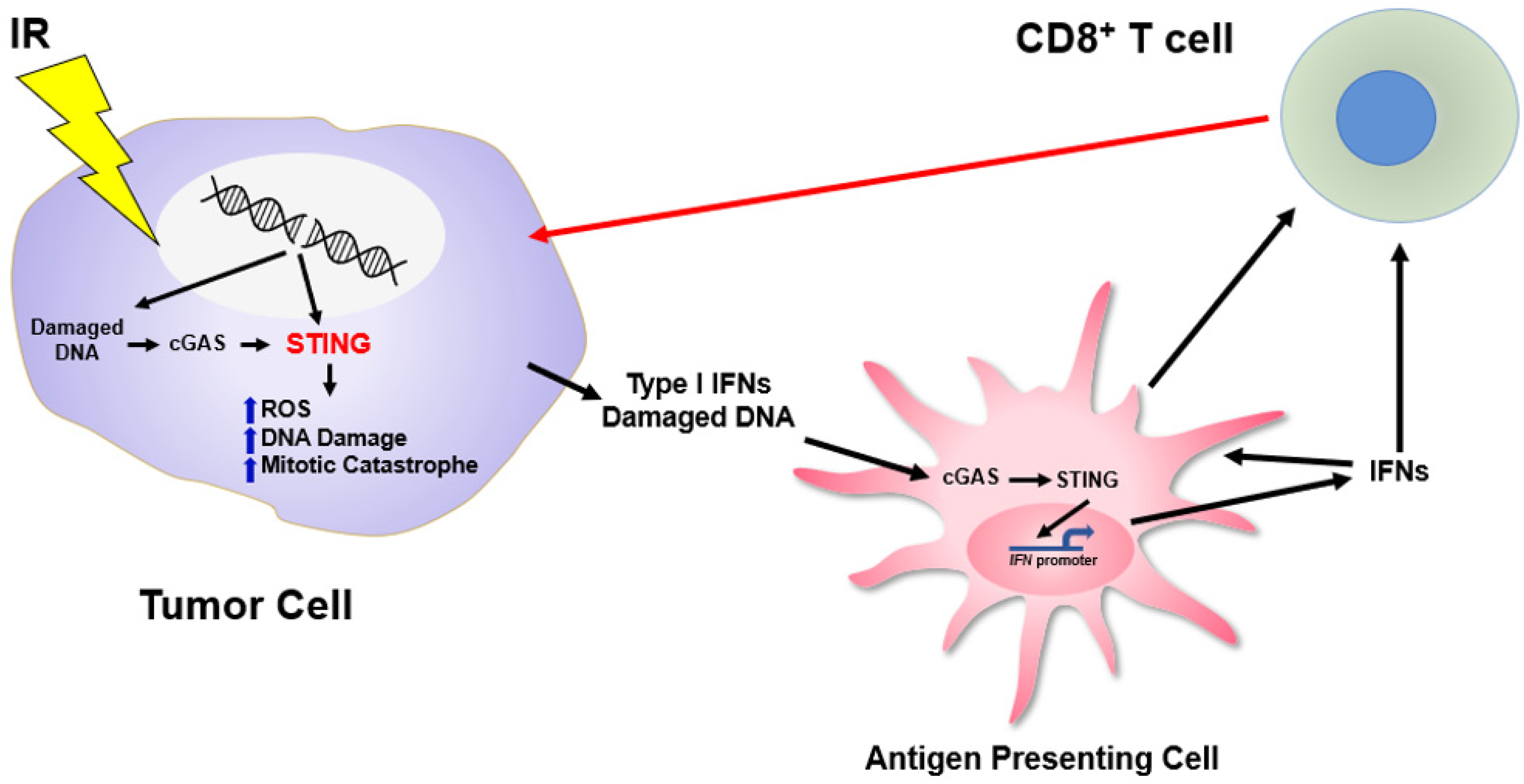
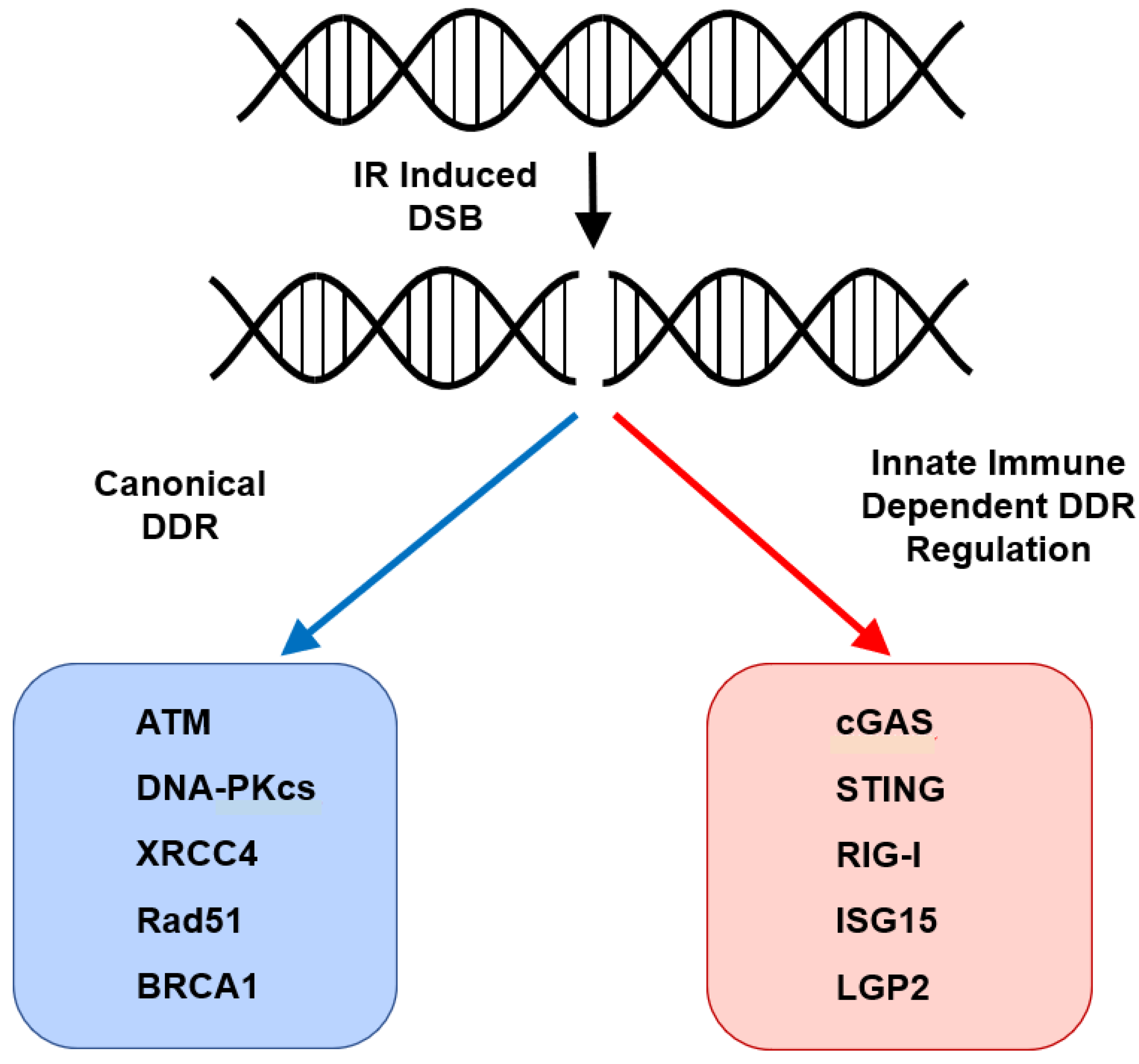
6. STING and Regulation of the Cell-Intrinsic Response to DNA Damage
7. ISG15 and DNA Replication
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Takata, M.; Sasaki, M.S.; Sonoda, E.; Morrison, C.; Hashimoto, M.; Utsumi, H.; Yamaguchi-Iwai, Y.; Shinohara, A.; Takeda, S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998, 17, 5497–5508. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.J.; Yu, V.P.; Lee, H.; Corcoran, A.; Thistlethwaite, F.C.; Evans, M.J.; Colledge, W.H.; Friedman, L.S.; Ponder, B.A.; Venkitaraman, A.R. Involvement of Brca2 in DNA repair. Mol. Cell 1998, 1, 347–357. [Google Scholar] [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Reislander, T.; Groelly, F.J.; Tarsounas, M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol. Cell 2020, 80, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Bednarski, J.J.; Sleckman, B.P. At the intersection of DNA damage and immune responses. Nat. Rev. Immunol. 2019, 19, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Sleckman, B.P. The response to and repair of RAG-mediated DNA double-strand breaks. Annu. Rev. Immunol. 2012, 30, 175–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desiderio, S.; Lin, W.C.; Li, Z. The cell cycle and V(D)J recombination. Curr. Top. MicroBiol. Immunol. 1996, 217, 45–59. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Alt, F.W. Class-switch recombination: Interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 2004, 4, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Basu, U.; Meng, F.L.; Keim, C.; Grinstein, V.; Pefanis, E.; Eccleston, J.; Zhang, T.; Myers, D.; Wasserman, C.R.; Wesemann, D.R.; et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell 2011, 144, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boboila, C.; Alt, F.W.; Schwer, B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 2012, 116, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Cox, R.A.; Rotem, Z. Foreign nucleic acids as the stimulus to make interferon. Lancet 1963, 2, 113–116. [Google Scholar] [CrossRef]
- Turnell, A.S.; Grand, R.J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef]
- Zur Hausen, H. Induction of specific chromosomal aberrations by adenovirus type 12 in human embryonic kidney cells. J. Virol. 1967, 1, 1174–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDougall, J.K. Adenovirus-induced chromosome aberrations in human cells. J. Gen. Virol. 1971, 12, 43–51. [Google Scholar] [CrossRef] [PubMed]
- McDougall, J.K. Effects of adenoviruses on the chromosomes of normal human cells and cells trisomic for an E chromosome. Nature 1970, 225, 456–458. [Google Scholar] [CrossRef]
- Wilson, C.B.; Leopard, J.; Cheresh, D.A.; Nakamura, R.M. Extracellular matrix and integrin composition of the normal bladder wall. World J. Urol. 1996, 14 (Suppl. 1), S30–S37. [Google Scholar] [CrossRef]
- Daniel, R.; Katz, R.A.; Skalka, A.M. A role for DNA-PK in retroviral DNA integration. Science 1999, 284, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.A.; Jackson, S.P. Involvement of DNA end-binding protein Ku in Ty element retrotransposition. Mol. Cell Biol. 1999, 19, 6260–6268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, R.; Katz, R.A.; Merkel, G.; Hittle, J.C.; Yen, T.J.; Skalka, A.M. Wortmannin potentiates integrase-mediated killing of lymphocytes and reduces the efficiency of stable transduction by retroviruses. Mol. Cell Biol. 2001, 21, 1164–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, R.; Kao, G.; Taganov, K.; Greger, J.G.; Favorova, O.; Merkel, G.; Yen, T.J.; Katz, R.A.; Skalka, A.M. Evidence that the retroviral DNA integration process triggers an ATR-dependent DNA damage response. Proc. Natl. Acad. Sci. USA 2003, 100, 4778–4783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalka, A.M.; Katz, R.A. Retroviral DNA integration and the DNA damage response. Cell Death Differ. 2005, 12 (Suppl. 1), 971–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Parrish, M.; Chan, T.K.; Yin, L.; Rai, P.; Yoshiyuki, Y.; Abolhassani, N.; Tan, K.B.; Kiraly, O.; Chow, V.T.; et al. Influenza infection induces host DNA damage and dynamic DNA damage responses during tissue regeneration. Cell Mol. Life Sci. 2015, 72, 2973–2988. [Google Scholar] [CrossRef] [Green Version]
- Querido, E.; Blanchette, P.; Yan, Q.; Kamura, T.; Morrison, M.; Boivin, D.; Kaelin, W.G.; Conaway, R.C.; Conaway, J.W.; Branton, P.E. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001, 15, 3104–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Madden-Fuentes, R.J.; Lou, B.X.; Pipas, J.M.; Gerhardt, J.; Rigell, C.J.; Fanning, E. Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J. Virol. 2008, 82, 5316–5328. [Google Scholar] [CrossRef] [Green Version]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakad, R.; Schumacher, B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front. Genet. 2016, 7, 147. [Google Scholar] [CrossRef] [Green Version]
- McGee, H.M.; Marciscano, A.E.; Campbell, A.M.; Monjazeb, A.M.; Kaech, S.M.; Teijaro, J.R. Parallels Between the Antiviral State and the Irradiated State. J. Natl. Cancer Inst. 2021, 113, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal. Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Ascano, M.; Wu, Y.; Barchet, W.; Gaffney, B.L.; Zillinger, T.; Serganov, A.A.; Liu, Y.; Jones, R.A.; Hartmann, G.; et al. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 2013, 153, 1094–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T.; Barber, G.N.; Arai, H.; Taguchi, T. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- De Oliveira Mann, C.C.; Orzalli, M.H.; King, D.S.; Kagan, J.C.; Lee, A.S.Y.; Kranzusch, P.J. Modular Architecture of the STING C-Terminal Tail Allows Interferon and NF-kappaB Signaling Adaptation. Cell Rep. 2019, 27, 1165–1175 e1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonugunta, V.K.; Sakai, T.; Pokatayev, V.; Yang, K.; Wu, J.; Dobbs, N.; Yan, N. Trafficking-Mediated STING Degradation Requires Sorting to Acidified Endolysosomes and Can Be Targeted to Enhance Anti-tumor Response. Cell Rep. 2017, 21, 3234–3242. [Google Scholar] [CrossRef] [Green Version]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from Irradiated Cancer Cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef] [Green Version]
- Grabosch, S.; Bulatovic, M.; Zeng, F.; Ma, T.; Zhang, L.; Ross, M.; Brozick, J.; Fang, Y.; Tseng, G.; Kim, E.; et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 2019, 38, 2380–2393. [Google Scholar] [CrossRef]
- Maekawa, H.; Inoue, T.; Ouchi, H.; Jao, T.M.; Inoue, R.; Nishi, H.; Fujii, R.; Ishidate, F.; Tanaka, T.; Tanaka, Y.; et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 2019, 29, 1261–1273 e1266. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980 e2975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Ranoa, D.R.E.; Huang, X.; Hou, Y.; Yang, K.; Poli, E.C.; Beckett, M.A.; Fu, Y.X.; Weichselbaum, R.R. RIG-I-Like Receptor LGP2 Is Required for Tumor Control by Radiotherapy. Cancer Res. 2020, 80, 5633–5641. [Google Scholar] [CrossRef] [PubMed]
- Ranoa, D.R.; Parekh, A.D.; Pitroda, S.P.; Huang, X.; Darga, T.; Wong, A.C.; Huang, L.; Andrade, J.; Staley, J.P.; Satoh, T.; et al. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 2016, 7, 26496–26515. [Google Scholar] [CrossRef] [PubMed]
- Carozza, J.A.; Böhnert, V.; Nguyen, K.C.; Skariah, G.; Shaw, K.E.; Brown, J.A.; Rafat, M.; von Eyben, R.; Graves, E.E.; Glenn, J.S.; et al. Extracellular cGAMP is a cancer-cell-produced immunotransmitter involved in radiation-induced anticancer immunity. Nat. Cancer 2020, 1, 184–196. [Google Scholar] [CrossRef]
- Ramanjulu, J.M.; Pesiridis, G.S.; Yang, J.; Concha, N.; Singhaus, R.; Zhang, S.Y.; Tran, J.L.; Moore, P.; Lehmann, S.; Eberl, H.C.; et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018, 564, 439–443. [Google Scholar] [CrossRef]
- Villanueva, M.T. Cancer immunotherapy: STINGing systemically. Nat. Rev. Drug Discov. 2018, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Crowe, W.N.; Wang, L.; Lu, Y.; Petty, W.J.; Habib, A.A.; Zhao, D. An inhalable nanoparticulate STING agonist synergizes with radiotherapy to confer long-term control of lung metastases. Nat. Commun. 2019, 10, 5108. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, C. Drug developers switch gears to inhibit STING. Nat. Biotechnol. 2019, 37, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.R.; Friedman, D.; Cottam, B.; Dubensky, T.W., Jr.; Kanne, D.B.; Bambina, S.; Bahjat, K.; Crittenden, M.R.; Gough, M.J. Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res. 2016, 76, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Green, M.D.; Lang, X.; Lazarus, J.; Parsels, J.D.; Wei, S.; Parsels, L.A.; Shi, J.; Ramnath, N.; Wahl, D.R.; et al. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res. 2019, 79, 3940–3951. [Google Scholar] [CrossRef] [PubMed]
- Reislander, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10, 3143. [Google Scholar] [CrossRef] [Green Version]
- Wolf, C.; Rapp, A.; Berndt, N.; Staroske, W.; Schuster, M.; Dobrick-Mattheuer, M.; Kretschmer, S.; Konig, N.; Kurth, T.; Wieczorek, D.; et al. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat. Commun. 2016, 7, 11752. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Lu, C.; Jin, Q.; Lu, H.; Chen, X.; Tian, L.; Zhang, Y.; Ortega, J.; Zhang, J.; Siteni, S.; et al. MLH1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the cGAS-STING Pathway. Cancer Cell 2021, 39, 109–121 e105. [Google Scholar] [CrossRef] [PubMed]
- Sivick, K.E.; Desbien, A.L.; Glickman, L.H.; Reiner, G.L.; Corrales, L.; Surh, N.H.; Hudson, T.E.; Vu, U.T.; Francica, B.J.; Banda, T.; et al. Magnitude of Therapeutic STING Activation Determines CD8(+) T Cell-Mediated Anti-tumor Immunity. Cell Rep. 2018, 25, 3074–3085 e3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez David, R.Y.; Combredet, C.; Najburg, V.; Millot, G.A.; Beauclair, G.; Schwikowski, B.; Leger, T.; Camadro, J.M.; Jacob, Y.; Bellalou, J.; et al. LGP2 binds to PACT to regulate RIG-I- and MDA5-mediated antiviral responses. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Widau, R.C.; Parekh, A.D.; Ranck, M.C.; Golden, D.W.; Kumar, K.A.; Sood, R.F.; Pitroda, S.P.; Liao, Z.; Huang, X.; Darga, T.E.; et al. RIG-I-like receptor LGP2 protects tumor cells from ionizing radiation. Proc. Natl. Acad. Sci. USA 2014, 111, E484–E491. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.; Gao, M.; Gao, X.; Zhu, B.; Huang, J.; Tu, X.; Kim, W.; Zhao, F.; Zhou, Q.; Zhu, S.; et al. Reciprocal regulation of RIG-I and XRCC4 connects DNA repair with RIG-I immune signaling. Nat. Commun. 2021, 12, 2187. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Olvera, J.M.; Yoder, K.E.; Mitchell, R.S.; Butler, S.L.; Lieber, M.; Martin, S.L.; Bushman, F.D. Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J. 2001, 20, 3272–3281. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluck, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front. Cell Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; Broekema, N.M.; Diner, B.A.; Hancks, D.C.; Elde, N.C.; Cristea, I.M.; Knipe, D.M. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E1773–E1781. [Google Scholar] [CrossRef] [Green Version]
- Lahaye, X.; Gentili, M.; Silvin, A.; Conrad, C.; Picard, L.; Jouve, M.; Zueva, E.; Maurin, M.; Nadalin, F.; Knott, G.J.; et al. NONO Detects the Nuclear HIV Capsid to Promote cGAS-Mediated Innate Immune Activation. Cell 2018, 175, 488–501 e422. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]
- Jiang, H.; Xue, X.; Panda, S.; Kawale, A.; Hooy, R.M.; Liang, F.; Sohn, J.; Sung, P.; Gekara, N.O. Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J. 2019, 38, e102718. [Google Scholar] [CrossRef] [PubMed]
- Volkman, H.E.; Cambier, S.; Gray, E.E.; Stetson, D.B. Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife 2019, 8, e47491. [Google Scholar] [CrossRef]
- Zierhut, C.; Yamaguchi, N.; Paredes, M.; Luo, J.D.; Carroll, T.; Funabiki, H. The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 2019, 178, 302–315 e323. [Google Scholar] [CrossRef]
- De Oliveira Mann, C.C.; Hopfner, K.P. Nuclear cGAS: Guard or prisoner? EMBO J. 2021, 40, e108293. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, H.; Zhang, J.; Wang, Y.; Simoneau, A.; Yang, H.; Levine, A.S.; Zou, L.; Chen, Z.; Lan, L. cGAS suppresses genomic instability as a decelerator of replication forks. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef]
- Nassour, J.; Radford, R.; Correia, A.; Fuste, J.M.; Schoell, B.; Jauch, A.; Shaw, R.J.; Karlseder, J. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 2019, 565, 659–663. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.T.; Cesare, A.J.; Rivera, T.; Karlseder, J. Cell death during crisis is mediated by mitotic telomere deprotection. Nature 2015, 522, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jonsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappaB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760 e745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayman, T.J.; Baro, M.; MacNeil, T.; Phoomak, C.; Aung, T.N.; Cui, W.; Leach, K.; Iyer, R.; Challa, S.; Sandoval-Schaefer, T.; et al. STING enhances cell death through regulation of reactive oxygen species and DNA damage. Nat. Commun. 2021, 12, 2327. [Google Scholar] [CrossRef]
- Ranoa, D.R.E.; Widau, R.C.; Mallon, S.; Parekh, A.D.; Nicolae, C.M.; Huang, X.; Bolt, M.J.; Arina, A.; Parry, R.; Kron, S.J.; et al. STING Promotes Homeostasis via Regulation of Cell Proliferation and Chromosomal Stability. Cancer Res. 2019, 79, 1465–1479. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.; Becares, M.; Falqui, M.; Fernandez-Lozano, C.; Guerra, S. ISG15, a Small Molecule with Huge Implications: Regulation of Mitochondrial Homeostasis. Viruses 2018, 10, 629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.B.; Miyauchi-Ishida, S.; Arimoto, K.; Liu, D.; Yan, M.; Liu, C.W.; Gyorffy, B.; Zhang, D.E. Type I IFN induces protein ISGylation to enhance cytokine expression and augments colonic inflammation. Proc. Natl. Acad. Sci. USA 2015, 112, 14313–14318. [Google Scholar] [CrossRef] [Green Version]
- Huo, Y.; Zong, Z.; Wang, Q.; Zhang, Z.; Deng, H. ISG15 silencing increases cisplatin resistance via activating p53-mediated cell DNA repair. Oncotarget 2017, 8, 107452–107461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. MicroBiol. 2018, 16, 423–439. [Google Scholar] [CrossRef]
- Lou, Z.; Wei, J.; Riethman, H.; Baur, J.A.; Voglauer, R.; Shay, J.W.; Wright, W.E. Telomere length regulates ISG15 expression in human cells. Aging 2009, 1, 608–621. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Yang, S.W.; Park, J.M.; Ka, S.H.; Kim, J.H.; Kong, Y.Y.; Jeon, Y.J.; Seol, J.H.; Chung, C.H. Positive feedback regulation of p53 transactivity by DNA damage-induced ISG15 modification. Nat. Commun. 2016, 7, 12513. [Google Scholar] [CrossRef] [Green Version]
- Park, J.M.; Yang, S.W.; Yu, K.R.; Ka, S.H.; Lee, S.W.; Seol, J.H.; Jeon, Y.J.; Chung, C.H. Modification of PCNA by ISG15 plays a crucial role in termination of error-prone translesion DNA synthesis. Mol. Cell 2014, 54, 626–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raso, M.C.; Djoric, N.; Walser, F.; Hess, S.; Schmid, F.M.; Burger, S.; Knobeloch, K.P.; Penengo, L. Interferon-stimulated gene 15 accelerates replication fork progression inducing chromosomal breakage. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayman, T.J.; Glazer, P.M. Regulation of the Cell-Intrinsic DNA Damage Response by the Innate Immune Machinery. Int. J. Mol. Sci. 2021, 22, 12761. https://doi.org/10.3390/ijms222312761
Hayman TJ, Glazer PM. Regulation of the Cell-Intrinsic DNA Damage Response by the Innate Immune Machinery. International Journal of Molecular Sciences. 2021; 22(23):12761. https://doi.org/10.3390/ijms222312761
Chicago/Turabian StyleHayman, Thomas J., and Peter M. Glazer. 2021. "Regulation of the Cell-Intrinsic DNA Damage Response by the Innate Immune Machinery" International Journal of Molecular Sciences 22, no. 23: 12761. https://doi.org/10.3390/ijms222312761
APA StyleHayman, T. J., & Glazer, P. M. (2021). Regulation of the Cell-Intrinsic DNA Damage Response by the Innate Immune Machinery. International Journal of Molecular Sciences, 22(23), 12761. https://doi.org/10.3390/ijms222312761