
The Roles of Luteinizing Hormone, Follicle-Stimulating Hormone and Testosterone in Spermatogenesis and Folliculogenesis Revisited



Abstract
1. Introduction
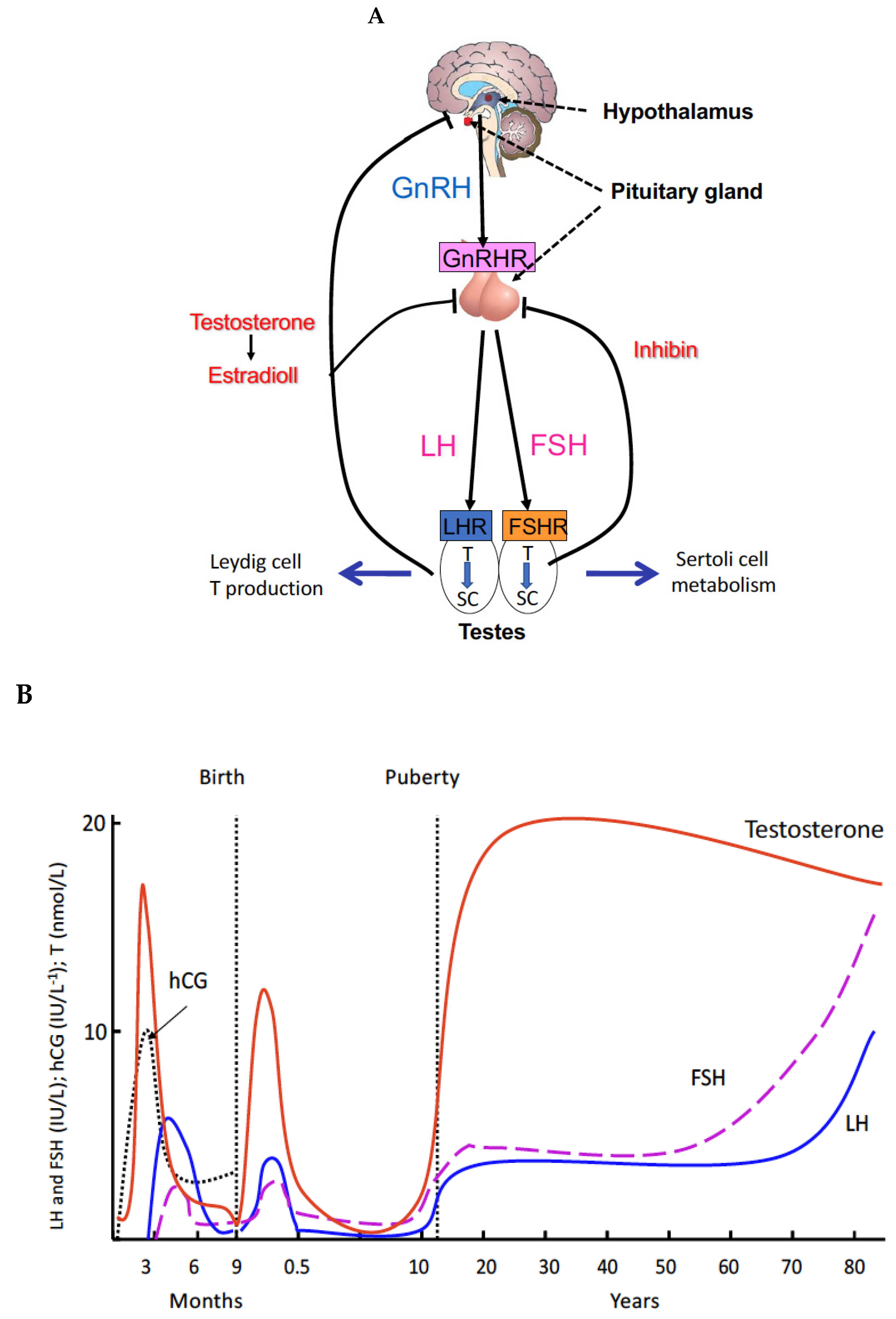
2. Principles and Regulations of Spermatogenesis
3. Genetic Defects of the Gonadal Axis Have Advanced the Understanding of the Role of Gonadotropins in Human Reproduction
4. Proliferation of Testosterone-Producing Leydig Cells in Different Waves
5. A New Concept of the Role of LH and T in Spermatogenesis
6. Testosterone and Spermatogenesis in Rodents: High Intratesticular Testosterone (ITT) Microenvironment Is Not Essential for Sperm Production
7. FSH Is an Important Stimulus for Sertoli Cells and Essential Regulator of Spermatid Maturation
8. FSH Is Essential for Quantitative Spermatogenesis in Rodents
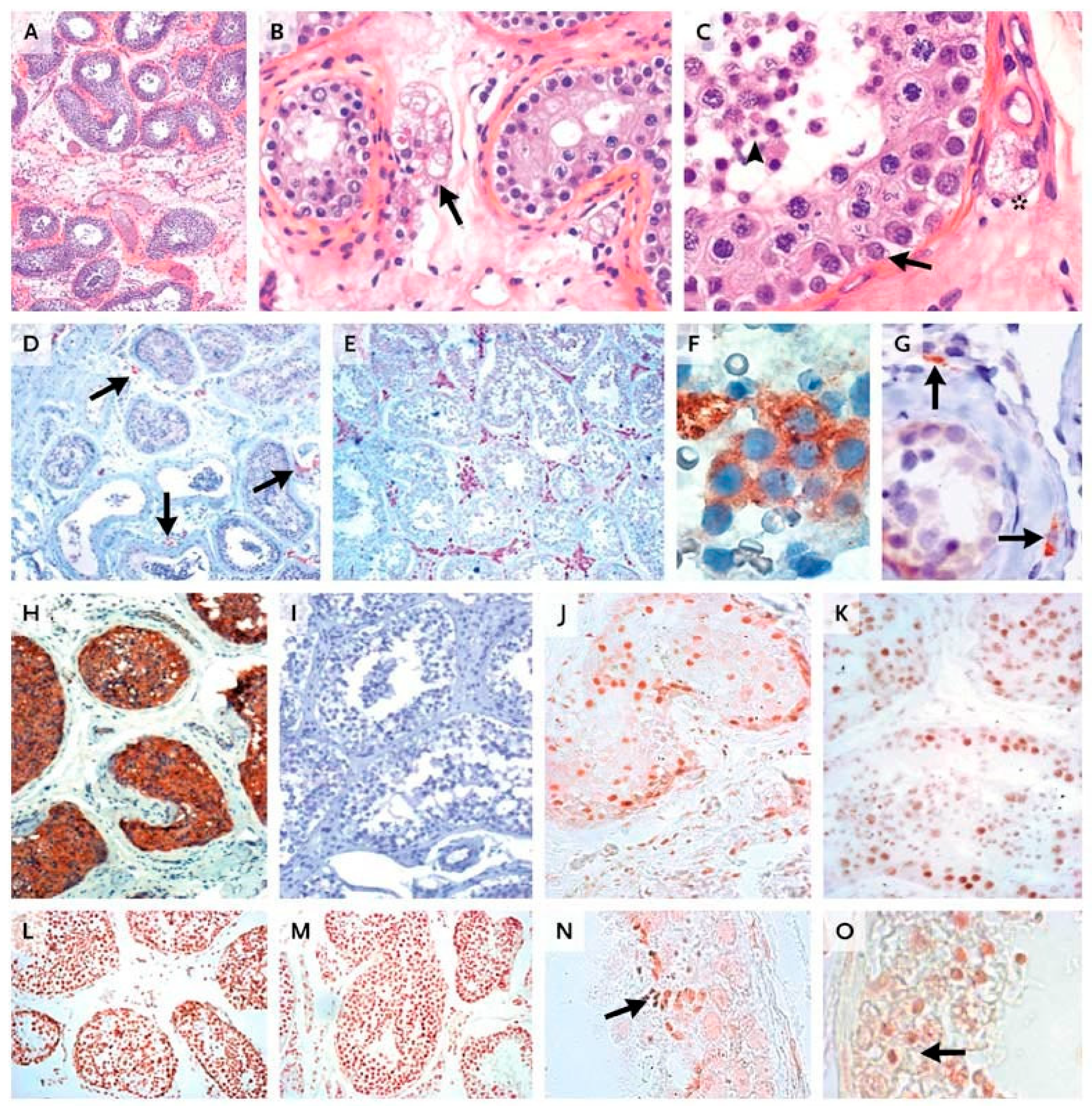
9. Relevance of LH and FSH in Human Spermatogenesis
10. The Role of FSH in the Human Ovary Revisited
10.1. Human In Vivo Model of Complete FSHR Inactivation
10.2. Lessons Derived from Studying a Complete Molecular Alteration of the Human FSHR in Human Primary Ovarian Insufficiency
10.3. Lessons Derived from Studying Partial Molecular Alteration of the Human FSHR In Vivo
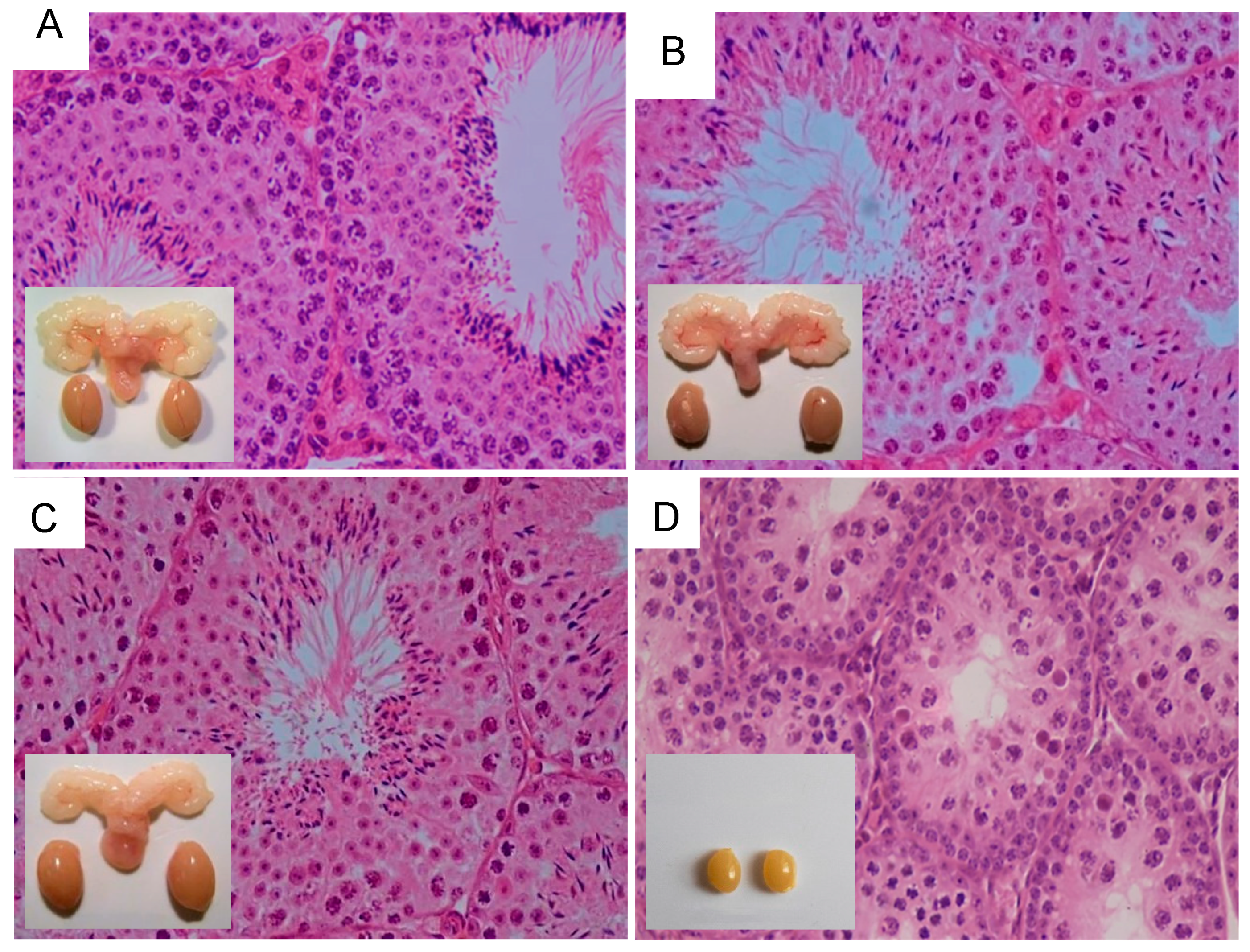
11. Influence of Excessive FSH Action on Testicular Function
12. LH/T Regulation of Spermatogenesis and Strong FSH Activation
13. Inference and Summary
Author Contributions
Funding
Institutional Review Board Statemen
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huhtaniemi, I.; Hovatta, O.; La Marca, A.; Livera, G.; Monniaux, D.; Persani, L.; Heddar, A.; Jarzabek, K.; Laisk-Podar, T.; Salumets, A.; et al. Advances in the Molecular Pathophysiology, Genetics, and Treatment of Primary Ovarian Insufficiency. Trends Endocrinol. Metab. 2018, 29, 400–419. [Google Scholar] [CrossRef] [PubMed]
- Krausz, C.; Riera-Escamilla, A. Genetics of Male Infertility. Nat. Rev. Urol. 2018, 15, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Levine, H.; Jørgensen, N.; Martino-Andrade, A.; Mendiola, J.; Weksler-Derri, D.; Mindlis, I.; Pinotti, R.; Swan, S.H. Temporal Trends in Sperm Count: A Systematic Review and Meta-Regression Analysis. Hum. Reprod. Update 2017, 23, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.A.; Baird, D.T. Male Contraception. Endocr. Rev. 2002, 23, 735–762. [Google Scholar] [CrossRef]
- McLachlan, R.I.; O’Donnell, L.; Meachem, S.J.; Stanton, P.G.; de Kretser, D.M.; Pratis, K.; Robertson, D.M. Identification of Specific Sites of Hormonal Regulation in Spermatogenesis in Rats, Monkeys, and Man. Recent Prog. Horm. Res. 2002, 57, 149–179. [Google Scholar] [CrossRef]
- Smith, L.; Walker, W. Hormonal signaling in the testis. In Knobil and Neill’s Physiology of Reproduction; Plant, T.M., Zeleznik, A.J., Eds.; Academic Press: Waltham, MA, USA, 2015; pp. 637–675. [Google Scholar]
- Saez, J.M. Leydig Cells: Endocrine, Paracrine, and Autocrine Regulation. Endocr. Rev. 1994, 15, 574–626. [Google Scholar] [CrossRef]
- Mruk, D.D.; Cheng, C.Y. The Mammalian Blood-Testis Barrier: Its Biology and Regulation. Endocr. Rev. 2015, 36, 564–591. [Google Scholar] [CrossRef]
- Steinberger, E.; Steinberger, A. Spermatogenic function of the testis. In Handbook of Physiology; Greep, R.O., Hamilton, D.W., Eds.; American Physiological Society: Washington, DC, USA, 1975; pp. 1–10. [Google Scholar]
- Ehmcke, J.; Schlatt, S. A Revised Model for Spermatogonial Expansion in Man: Lessons from Non-Human Primates. Reproduction 2006, 132, 673–680. [Google Scholar] [CrossRef]
- Dubé, E.; Cyr, D.G. The Blood-Epididymis Barrier and Human Male Fertility. Adv. Exp. Med. Biol. 2012, 763, 218–236. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Weinbauer, G.F. Endocrine Control of Spermatogenesis: Role of FSH and LH/Testosterone. Spermatogenesis 2014, 4, e996025. [Google Scholar] [CrossRef]
- Huhtaniemi, I. Mechanisms in Endocrinology: Hormonal Regulation of Spermatogenesis: Mutant Mice Challenging Old Paradigms. Eur. J. Endocrinol. 2018, 179, R143–R150. [Google Scholar] [CrossRef]
- Oduwole, O.O.; Huhtaniemi, I.T. Feasibility of Male Hormonal Contraception: Lessons from Clinical Trials and Animal Experiments. Curr. Mol. Pharmacol. 2014, 7, 109–118. [Google Scholar] [CrossRef]
- Hayes, F.J.; DeCruz, S.; Seminara, S.B.; Boepple, P.A.; Crowley, W.F. Differential Regulation of Gonadotropin Secretion by Testosterone in the Human Male: Absence of a Negative Feedback Effect of Testosterone on Follicle-Stimulating Hormone Secretion. J. Clin. Endocrinol. Metab. 2001, 86, 53–58. [Google Scholar] [CrossRef]
- Manetti, G.J.; Honig, S.C. Update on Male Hormonal Contraception: Is the Vasectomy in Jeopardy? Int. J. Impot. Res. 2010, 22, 159–170. [Google Scholar] [CrossRef]
- Sprengel, R.; Braun, T.; Nikolics, K.; Segaloff, D.L.; Seeburg, P.H. The Testicular Receptor for Follicle Stimulating Hormone: Structure and Functional Expression of Cloned CDNA. Mol. Endocrinol. 1990, 4, 525–530. [Google Scholar] [CrossRef]
- Loosfelt, H.; Misrahi, M.; Atger, M.; Salesse, R.; Vu Hai-Luu Thi, M.T.; Jolivet, A.; Guiochon-Mantel, A.; Sar, S.; Jallal, B.; Garnier, J. Cloning and Sequencing of Porcine LH-HCG Receptor CDNA: Variants Lacking Transmembrane Domain. Science 1989, 245, 525–528. [Google Scholar] [CrossRef]
- Atger, M.; Misrahi, M.; Sar, S.; Le Flem, L.; Dessen, P.; Milgrom, E. Structure of the Human Luteinizing Hormone-Choriogonadotropin Receptor Gene: Unusual Promoter and 5′ Non-Coding Regions. Mol. Cell. Endocrinol. 1995, 111, 113–123. [Google Scholar] [CrossRef]
- McFarland, K.C.; Sprengel, R.; Phillips, H.S.; Köhler, M.; Rosemblit, N.; Nikolics, K.; Segaloff, D.L.; Seeburg, P.H. Lutropin-Choriogonadotropin Receptor: An Unusual Member of the G Protein-Coupled Receptor Family. Science 1989, 245, 494–499. [Google Scholar] [CrossRef]
- Orth, J.; Christensen, A.K. Localization of 125I-Labeled FSH in the Testes of Hypophy-Sectomized Rats by Autoradiography at the Light and Electron Microscope Levels. Endocrinology 1977, 101, 262–278. [Google Scholar] [CrossRef]
- Kangasniemi, M.; Kaipia, A.; Toppari, J.; Perheentupa, A.; Huhtaniemi, I.; Parvinen, M. Cellular Regulation of Follicle-Stimulating Hormone (FSH) Binding in Rat Seminiferous Tubules. J. Androl. 1990, 11, 336–343. [Google Scholar]
- Simoni, M.; Gromoll, J.; Nieschlag, E. The Follicle-Stimulating Hormone Receptor: Biochemistry, Molecular Biology, Physiology, and Pathophysiology. Endocr. Rev. 1997, 18, 739–773. [Google Scholar] [CrossRef]
- Welsh, M.; Saunders, P.T.K.; Atanassova, N.; Sharpe, R.M.; Smith, L.B. Androgen Action via Testicular Peritubular Myoid Cells Is Essential for Male Fertility. FASEB J. 2009, 23, 4218–4230. [Google Scholar] [CrossRef]
- Rousseau-Merck, M.F.; Atger, M.; Loosfelt, H.; Milgrom, E.; Berger, R. The Chromosomal Localization of the Human Follicle-Stimulating Hormone Receptor Gene (FSHR) on 2p21-P16 Is Similar to That of the Luteinizing Hormone Receptor Gene. Genomics 1993, 15, 222–224. [Google Scholar] [CrossRef]
- Rousseau-Merck, M.F.; Misrahi, M.; Loosfelt, H.; Atger, M.; Milgrom, E.; Berger, R. Assignment of the Human Thyroid Stimulating Hormone Receptor (TSHR) Gene to Chromosome 14q31. Genomics 1990, 8, 233–236. [Google Scholar] [CrossRef]
- Gross, B.; Misrahi, M.; Sar, S.; Milgrom, E. Composite Structure of the Human Thyrotropin Receptor Gene. Biochem. Biophys. Res. Commun. 1991, 177, 679–687. [Google Scholar] [CrossRef]
- Heckert, L.L.; Daley, I.J.; Griswold, M.D. Structural Organization of the Follicle-Stimulating Hormone Receptor Gene. Mol. Endocrinol. 1992, 6, 70–80. [Google Scholar] [CrossRef][Green Version]
- Aittomäki, K.; Lucena, J.L.; Pakarinen, P.; Sistonen, P.; Tapanainen, J.; Gromoll, J.; Kaskikari, R.; Sankila, E.M.; Lehväslaiho, H.; Engel, A.R.; et al. Mutation in the Follicle-Stimulating Hormone Receptor Gene Causes Hereditary Hypergonadotropic Ovarian Failure. Cell 1995, 82, 959–968. [Google Scholar] [CrossRef]
- Gross, B.; Misrahi, M.; Loosfelt, H.; Vu Hai Luu Thi, M.; Atger, M.; Meduri, G.; Pichon, C.; Jolivet, A.; Milgrom, E. Receptors for Pituitary Glycoprotein Hormones. C R Acad. Sci. III 1992, 314, 21–22. [Google Scholar] [PubMed]
- Misrahi, M.; Beau, I.; Meduri, G.; Bouvattier, C.; Atger, M.; Loosfelt, H.; Ghinea, N.; Hai, M.V.; Bougnères, P.F.; Milgrom, E. Gonadotropin Receptors and the Control of Gonadal Steroidogenesis: Physiology and Pathology. Baillieres Clin. Endocrinol. Metab. 1998, 12, 35–66. [Google Scholar] [CrossRef]
- Kato, Y.; Shiraishi, K.; Matsuyama, H. Expression of Testicular Androgen Receptor in Non-Obstructive Azoospermia and Its Change after Hormonal Therapy. Andrology 2014, 2, 734–740. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, L.; Smith, L.B. Androgen Receptor Roles in Spermatogenesis and Infertility. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 595–605. [Google Scholar] [CrossRef]
- Themmen, A.P.N.; Huhtaniemi, I.T. Mutations of Gonadotropins and Gonadotropin Receptors: Elucidating the Physiology and Pathophysiology of Pituitary-Gonadal Function. Endocr. Rev. 2000, 21, 551–583. [Google Scholar] [CrossRef]
- Themmen, A.P.N. An Update of the Pathophysiology of Human Gonadotrophin Subunit and Receptor Gene Mutations and Polymorphisms. Reproduction 2005, 130, 263–274. [Google Scholar] [CrossRef]
- Meduri, G.; Bachelot, A.; Cocca, M.P.; Vasseur, C.; Rodien, P.; Kuttenn, F.; Touraine, P.; Misrahi, M. Molecular Pathology of the FSH Receptor: New Insights into FSH Physiology. Mol. Cell. Endocrinol. 2008, 282, 130–142. [Google Scholar] [CrossRef]
- Huhtaniemi, I.T.; Themmen, A.P.N. Mutations in Human Gonadotropin and Gonadotropin-Receptor Genes. Endocrine 2005, 26, 207–217. [Google Scholar] [CrossRef]
- Weiss, J.; Axelrod, L.; Whitcomb, R.W.; Harris, P.E.; Crowley, W.F.; Jameson, J.L. Hypogonadism Caused by a Single Amino Acid Substitution in the Beta Subunit of Luteinizing Hormone. N. Engl. J. Med. 1992, 326, 179–183. [Google Scholar] [CrossRef]
- Axelrod, L.; Neer, R.M.; Kliman, B. Hypogonadism in a Male with Immunologically Active, Biologically Inactive Luteinizing Hormone: An Exception to a Venerable Rule. J. Clin. Endocrinol. Metab. 1979, 48, 279–287. [Google Scholar] [CrossRef]
- Valdes-Socin, H.; Salvi, R.; Daly, A.F.; Gaillard, R.C.; Quatresooz, P.; Tebeu, P.-M.; Pralong, F.P.; Beckers, A. Hypogonadism in a Patient with a Mutation in the Luteinizing Hormone Beta-Subunit Gene. N. Engl. J. Med. 2004, 351, 2619–2625. [Google Scholar] [CrossRef]
- Lofrano-Porto, A.; Barra, G.B.; Giacomini, L.A.; Nascimento, P.P.; Latronico, A.C.; Casulari, L.A.; da Rocha Neves, F.D.A. Luteinizing Hormone Beta Mutation and Hypogonadism in Men and Women. N. Engl. J. Med. 2007, 357, 897–904. [Google Scholar] [CrossRef]
- Grumbach, M.M. A Window of Opportunity: The Diagnosis of Gonadotropin Deficiency in the Male Infant. J. Clin. Endocrinol. Metab. 2005, 90, 3122–3127. [Google Scholar] [CrossRef]
- Valdes-Socin, H.; Salvi, R.; Thiry, A.; Daly, A.F.; Pralong, F.P.; Gaillard, R.; Beckers, A. Testicular Effects of Isolated Luteinizing Hormone Deficiency and Reversal by Long-Term Human Chorionic Gonadotropin Treatment. J. Clin. Endocrinol. Metab. 2009, 94, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ochin, H.; Shu, L.; Liu, J.; Shen, J.; Liu, J.; Lin, C.; Cui, Y. Homozygous Nonsense Mutation Trp28X in the LHB Gene Causes Male Hypogonadism. J. Assist. Reprod. Genet. 2018, 35, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Tapanainen, J.; Kellokumpu-Lehtinen, P.; Pelliniemi, L.; Huhtaniemi, I. Age-Related Changes in Endogenous Steroids of Human Fetal Testis during Early and Midpregnancy. J. Clin. Endocrinol. Metab. 1981, 52, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Achard, C.; Courtillot, C.; Lahuna, O.; Méduri, G.; Soufir, J.-C.; Lière, P.; Bachelot, A.; Benyounes, H.; Schumacher, M.; Kuttenn, F.; et al. Normal Spermatogenesis in a Man with Mutant Luteinizing Hormone. N. Engl. J. Med. 2009, 361, 1856–1863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.P.; Poutanen, M.; Wilbertz, J.; Huhtaniemi, I. Normal Prenatal but Arrested Postnatal Sexual Development of Luteinizing Hormone Receptor Knockout (LuRKO) Mice. Mol. Endocrinol. 2001, 15, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.M.; Mishra, S.; Zou, W.; Xu, B.; Foltz, M.; Li, X.; Rao, C.V. Targeted Disruption of Luteinizing Hormone/Human Chorionic Gonadotropin Receptor Gene. Mol. Endocrinol. 2001, 15, 184–200. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, L.; Meachem, S.; Stanton, P. McLachlan Endocrine regulation of spermatogenesis. In Knobil and Neill’s Physiology of Reproduction, 3rd ed.; Neill, J.D., Ed.; Academic Press: Amsterdam, The Netherlands, 2006; pp. 1017–1069. [Google Scholar]
- Layman, L.C. Hypogonadotropic Hypogonadism. Endocrinol. Metab. Clin. N. Am. 2007, 36, 283–296. [Google Scholar] [CrossRef]
- Fraietta, R.; Zylberstejn, D.S.; Esteves, S.C. Hypogonadotropic Hypogonadism Revisited. Clinics 2013, 68 (Suppl. S1), 81–88. [Google Scholar] [CrossRef]
- Huhtaniemi, I. A Short Evolutionary History of FSH-Stimulated Spermatogenesis. Hormones 2015, 14, 468–478. [Google Scholar] [CrossRef]
- Kumar, T.R. Mouse Models for Gonadotropins: A 15-Year Saga. Mol. Cell Endocrinol. 2007, 260–262, 249–254. [Google Scholar] [CrossRef]
- Gilbert, S.B.; Roof, A.K.; Rajendra Kumar, T. Mouse Models for the Analysis of Gonadotropin Secretion and Action. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 219–239. [Google Scholar] [CrossRef]
- Ma, X.; Dong, Y.; Matzuk, M.M.; Kumar, T.R. Targeted Disruption of Luteinizing Hormone Beta-Subunit Leads to Hypogonadism, Defects in Gonadal Steroidogenesis, and Infertility. Proc. Natl. Acad. Sci. USA 2004, 101, 17294–17299. [Google Scholar] [CrossRef]
- Cattanach, B.M.; Iddon, C.A.; Charlton, H.M.; Chiappa, S.A.; Fink, G. Gonadotrophin-Releasing Hormone Deficiency in a Mutant Mouse with Hypogonadism. Nature 1977, 269, 338–340. [Google Scholar] [CrossRef]
- Ebling, F.J.P.; Nwagwu, M.O.; Baines, H.; Myers, M.; Kerr, J.B. The Hypogonadal (Hpg) Mouse as a Model to Investigate the Estrogenic Regulation of Spermatogenesis. Hum. Fertil. 2006, 9, 127–135. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J. Hormonal Control of Germ Cell Development and Spermatogenesis. Semin. Cell Dev. Biol. 2014, 29, 55–65. [Google Scholar] [CrossRef]
- Zhang, F.-P.; Pakarainen, T.; Poutanen, M.; Toppari, J.; Huhtaniemi, I. The Low Gonadotropin-Independent Constitutive Production of Testicular Testosterone Is Sufficient to Maintain Spermatogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 13692–13697. [Google Scholar] [CrossRef]
- Lei, Z.M.; Mishra, S.; Ponnuru, P.; Li, X.; Yang, Z.W.; Rao, C.V. Testicular Phenotype in Luteinizing Hormone Receptor Knockout Animals and the Effect of Testosterone Replacement Therapy. Biol. Reprod. 2004, 71, 1605–1613. [Google Scholar] [CrossRef]
- Hammond, G.L.; Ruokonen, A.; Kontturi, M.; Koskela, E.; Vihko, R. The Simultaneous Radioimmunoassay of Seven Steroids in Human Spermatic and Peripheral Venous Blood. J. Clin. Endocrinol. Metab. 1977, 45, 16–24. [Google Scholar] [CrossRef]
- Huhtaniemi, I.; Nikula, H.; Rannikko, S. Treatment of Prostatic Cancer with a Gonadotropin-Releasing Hormone Agonist Analog: Acute and Long Term Effects on Endocrine Functions of Testis Tissue. J. Clin. Endocrinol. Metab. 1985, 61, 698–704. [Google Scholar] [CrossRef]
- Huhtaniemi, I. A Hormonal Contraceptive for Men: How Close Are We? Prog. Brain Res. 2010, 181, 273–288. [Google Scholar] [CrossRef]
- Cunningham, G.R.; Huckins, C. Persistence of Complete Spermatogenesis in the Presence of Low Intratesticular Concentrations of Testosterone. Endocrinology 1979, 105, 177–186. [Google Scholar] [CrossRef]
- Spaliviero, J.A.; Jimenez, M.; Allan, C.M.; Handelsman, D.J. Luteinizing Hormone Receptor-Mediated Effects on Initiation of Spermatogenesis in Gonadotropin-Deficient (Hpg) Mice Are Replicated by Testosterone. Biol. Reprod. 2004, 70, 32–38. [Google Scholar] [CrossRef]
- Griffin, D.K.; Ellis, P.J.; Dunmore, B.; Bauer, J.; Abel, M.H.; Affara, N.A. Transcriptional Profiling of Luteinizing Hormone Receptor-Deficient Mice before and after Testosterone Treatment Provides Insight into the Hormonal Control of Postnatal Testicular Development and Leydig Cell Differentiation. Biol. Reprod. 2010, 82, 1139–1150. [Google Scholar] [CrossRef]
- Pakarainen, T.; Zhang, F.-P.; Mäkelä, S.; Poutanen, M.; Huhtaniemi, I. Testosterone Replacement Therapy Induces Spermatogenesis and Partially Restores Fertility in Luteinizing Hormone Receptor Knockout Mice. Endocrinology 2005, 146, 596–606. [Google Scholar] [CrossRef]
- Oduwole, O.O.; Vydra, N.; Wood, N.E.M.; Samanta, L.; Owen, L.; Keevil, B.; Donaldson, M.; Naresh, K.; Huhtaniemi, I.T. Overlapping Dose Responses of Spermatogenic and Extragonadal Testosterone Actions Jeopardize the Principle of Hormonal Male Contraception. FASEB J. 2014, 28, 2566–2576. [Google Scholar] [CrossRef]
- Roth, M.Y.; Lin, K.; Amory, J.K.; Matsumoto, A.M.; Anawalt, B.D.; Snyder, C.N.; Kalhorn, T.F.; Bremner, W.J.; Page, S.T. Serum LH Correlates Highly with Intratesticular Steroid Levels in Normal Men. J. Androl. 2010, 31, 138–145. [Google Scholar] [CrossRef]
- Roth, M.Y.; Lin, K.; Bay, K.; Amory, J.K.; Anawalt, B.D.; Matsumoto, A.M.; Marck, B.T.; Bremner, W.J.; Page, S.T. Serum Insulin-like Factor 3 Is Highly Correlated with Intratesticular Testosterone in Normal Men with Acute, Experimental Gonadotropin Deficiency Stimulated with Low-Dose Human Chorionic Gonadotropin: A Randomized, Controlled Trial. Fertil. Steril. 2013, 99, 132–139. [Google Scholar] [CrossRef] [PubMed]
- El-Gehani, F.; Zhang, F.P.; Pakarinen, P.; Rannikko, A.; Huhtaniemi, I. Gonadotropin-Independent Regulation of Steroidogenesis in the Fetal Rat Testis. Biol. Reprod. 1998, 58, 116–123. [Google Scholar] [CrossRef] [PubMed]
- De Gendt, K.; Swinnen, J.V.; Saunders, P.T.K.; Schoonjans, L.; Dewerchin, M.; Devos, A.; Tan, K.; Atanassova, N.; Claessens, F.; Lécureuil, C.; et al. A Sertoli Cell-Selective Knockout of the Androgen Receptor Causes Spermatogenic Arrest in Meiosis. Proc. Natl. Acad. Sci. USA 2004, 101, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Chen, Y.-T.; Yeh, S.-D.; Xu, Q.; Wang, R.-S.; Guillou, F.; Lardy, H.; Yeh, S. Infertility with Defective Spermatogenesis and Hypotestosteronemia in Male Mice Lacking the Androgen Receptor in Sertoli Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 6876–6881. [Google Scholar] [CrossRef] [PubMed]
- Quigley, C.A.; De Bellis, A.; Marschke, K.B.; el-Awady, M.K.; Wilson, E.M.; French, F.S. Androgen Receptor Defects: Historical, Clinical, and Molecular Perspectives. Endocr. Rev. 1995, 16, 271–321. [Google Scholar] [CrossRef]
- Gao, W.; Bohl, C.E.; Dalton, J.T. Chemistry and Structural Biology of Androgen Receptor. Chem. Rev. 2005, 105, 3352–3370. [Google Scholar] [CrossRef]
- Wang, R.-S.; Yeh, S.; Tzeng, C.-R.; Chang, C. Androgen Receptor Roles in Spermatogenesis and Fertility: Lessons from Testicular Cell-Specific Androgen Receptor Knockout Mice. Endocr. Rev. 2009, 30, 119–132. [Google Scholar] [CrossRef]
- Walters, K.A.; Simanainen, U.; Handelsman, D.J. Molecular Insights into Androgen Actions in Male and Female Reproductive Function from Androgen Receptor Knockout Models. Hum. Reprod. Update 2010, 16, 543–558. [Google Scholar] [CrossRef]
- Yeh, S.; Tsai, M.-Y.; Xu, Q.; Mu, X.-M.; Lardy, H.; Huang, K.-E.; Lin, H.; Yeh, S.-D.; Altuwaijri, S.; Zhou, X.; et al. Generation and Characterization of Androgen Receptor Knockout (ARKO) Mice: An in Vivo Model for the Study of Androgen Functions in Selective Tissues. Proc. Natl. Acad. Sci. USA 2002, 99, 13498–13503. [Google Scholar] [CrossRef]
- Huhtaniemi, I.T.; Yamamoto, M.; Ranta, T.; Jalkanen, J.; Jaffe, R.B. Follicle-Stimulating Hormone Receptors Appear Earlier in the Primate Fetal Testis than in the Ovary. J. Clin. Endocrinol. Metab. 1987, 65, 1210–1214. [Google Scholar] [CrossRef]
- Warren, D.W.; Huhtaniemi, I.T.; Tapanainen, J.; Dufau, M.L.; Catt, K.J. Ontogeny of Gonadotropin Receptors in the Fetal and Neonatal Rat Testis. Endocrinology 1984, 114, 470–476. [Google Scholar] [CrossRef]
- Sharpe, R.M.; McKinnell, C.; Kivlin, C.; Fisher, J.S. Proliferation and Functional Maturation of Sertoli Cells, and Their Relevance to Disorders of Testis Function in Adulthood. Reproduction 2003, 125, 769–784. [Google Scholar] [CrossRef]
- Orth, J.M. The Role of Follicle-Stimulating Hormone in Controlling Sertoli Cell Proliferation in Testes of Fetal Rats. Endocrinology 1984, 115, 1248–1255. [Google Scholar] [CrossRef]
- Tapanainen, J.S.; Aittomäki, K.; Min, J.; Vaskivuo, T.; Huhtaniemi, I.T. Men Homozygous for an Inactivating Mutation of the Follicle-Stimulating Hormone (FSH) Receptor Gene Present Variable Suppression of Spermatogenesis and Fertility. Nat. Genet. 1997, 15, 205–206. [Google Scholar] [CrossRef]
- Nieschlag, E.; Simoni, M.; Gromoll, J.; Weinbauer, G.F. Role of FSH in the Regulation of Spermatogenesis: Clinical Aspects. Clin. Endocrinol. 1999, 51, 139–146. [Google Scholar] [CrossRef]
- Allan, C.M.; Haywood, M.; Swaraj, S.; Spaliviero, J.; Koch, A.; Jimenez, M.; Poutanen, M.; Levallet, J.; Huhtaniemi, I.; Illingworth, P.; et al. A Novel Transgenic Model to Characterize the Specific Effects of Follicle-Stimulating Hormone on Gonadal Physiology in the Absence of Luteinizing Hormone Actions. Endocrinology 2001, 142, 2213–2220. [Google Scholar] [CrossRef]
- Allan, C.M.; Garcia, A.; Spaliviero, J.; Zhang, F.-P.; Jimenez, M.; Huhtaniemi, I.; Handelsman, D.J. Complete Sertoli Cell Proliferation Induced by Follicle-Stimulating Hormone (FSH) Independently of Luteinizing Hormone Activity: Evidence from Genetic Models of Isolated FSH Action. Endocrinology 2004, 145, 1587–1593. [Google Scholar] [CrossRef]
- De Kretser, D.M.; Loveland, K.L.; Meinhardt, A.; Simorangkir, D.; Wreford, N. Spermatogenesis. Hum. Reprod. 1998, 13 (Suppl. S1), 1–8. [Google Scholar] [CrossRef]
- Wreford, N.G.; Rajendra Kumar, T.; Matzuk, M.M.; de Kretser, D.M. Analysis of the Testicular Phenotype of the Follicle-Stimulating Hormone Beta-Subunit Knockout and the Activin Type II Receptor Knockout Mice by Stereological Analysis. Endocrinology 2001, 142, 2916–2920. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J.; Monteiro, A.; Abel, M. Testicular Development in Mice Lacking Receptors for Follicle Stimulating Hormone and Androgen. PLoS ONE 2012, 7, e35136. [Google Scholar] [CrossRef]
- Abel, M.H.; Baker, P.J.; Charlton, H.M.; Monteiro, A.; Verhoeven, G.; De Gendt, K.; Guillou, F.; O’Shaughnessy, P.J. Spermatogenesis and Sertoli Cell Activity in Mice Lacking Sertoli Cell Receptors for Follicle-Stimulating Hormone and Androgen. Endocrinology 2008, 149, 3279–3285. [Google Scholar] [CrossRef]
- Orth, J.M.; McGuinness, M.P.; Qiu, J.; Jester, W.F.; Li, L.H. Use of in Vitro Systems to Study Male Germ Cell Development in Neonatal Rats. Theriogenology 1998, 49, 431–439. [Google Scholar] [CrossRef]
- Meroni, S.B.; Galardo, M.N.; Rindone, G.; Gorga, A.; Riera, M.F.; Cigorraga, S.B. Molecular Mechanisms and Signaling Pathways Involved in Sertoli Cell Proliferation. Front. Endocrinol. 2019, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Griswold, M.D. The Central Role of Sertoli Cells in Spermatogenesis. Semin. Cell Dev. Biol. 1998, 9, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.R.; Wang, Y.; Lu, N.; Matzuk, M.M. Follicle Stimulating Hormone Is Required for Ovarian Follicle Maturation but Not Male Fertility. Nat. Genet. 1997, 15, 201–204. [Google Scholar] [CrossRef]
- Abel, M.H.; Wootton, A.N.; Wilkins, V.; Huhtaniemi, I.; Knight, P.G.; Charlton, H.M. The Effect of a Null Mutation in the Follicle-Stimulating Hormone Receptor Gene on Mouse Reproduction. Endocrinology 2000, 141, 1795–1803. [Google Scholar] [CrossRef]
- Dierich, A.; Sairam, M.R.; Monaco, L.; Fimia, G.M.; Gansmuller, A.; LeMeur, M.; Sassone-Corsi, P. Impairing Follicle-Stimulating Hormone (FSH) Signaling in Vivo: Targeted Disruption of the FSH Receptor Leads to Aberrant Gametogenesis and Hormonal Imbalance. Proc. Natl. Acad. Sci. USA 1998, 95, 13612–13617. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J.; Monteiro, A.; Verhoeven, G.; De Gendt, K.; Abel, M.H. Effect of FSH on Testicular Morphology and Spermatogenesis in Gonadotrophin-Deficient Hypogonadal Mice Lacking Androgen Receptors. Reproduction 2010, 139, 177–184. [Google Scholar] [CrossRef]
- Singh, J.; Handelsman, D.J. Neonatal Administration of FSH Increases Sertoli Cell Numbers and Spermatogenesis in Gonadotropin-Deficient (Hpg) Mice. J. Endocrinol. 1996, 151, 37–48. [Google Scholar] [CrossRef]
- McLachlan, R.I.; Wreford, N.G.; Meachem, S.J.; De Kretser, D.M.; Robertson, D.M. Effects of Testosterone on Spermatogenic Cell Populations in the Adult Rat. Biol. Reprod. 1994, 51, 945–955. [Google Scholar] [CrossRef][Green Version]
- McLachlan, R.I.; Wreford, N.G.; Tsonis, C.; De Kretser, D.M.; Robertson, D.M. Testosterone Effects on Spermatogenesis in the Gonadotropin-Releasing Hormone-Immunized Rat. Biol. Reprod. 1994, 50, 271–280. [Google Scholar] [CrossRef][Green Version]
- Meachem, S.J.; Mclachlan, R.I.; Stanton, P.G.; Robertson, D.M.; Wreford, N.G. FSH Immunoneutralization Acutely Impairs Spermatogonial Development in Normal Adult Rats. J. Androl. 1999, 20, 756–762, discussion 755. [Google Scholar]
- Plant, T.M.; Marshall, G.R. The Functional Significance of FSH in Spermatogenesis and the Control of Its Secretion in Male Primates. Endocr. Rev. 2001, 22, 764–786. [Google Scholar] [CrossRef]
- Siegel, E.T.; Kim, H.-G.; Nishimoto, H.K.; Layman, L.C. The Molecular Basis of Impaired Follicle-Stimulating Hormone Action: Evidence from Human Mutations and Mouse Models. Reprod. Sci. 2013, 20, 211–233. [Google Scholar] [CrossRef]
- Rannikko, A.; Pakarinen, P.; Manna, P.R.; Beau, I.; Misrahi, M.; Aittomäki, K.; Huhtaniemi, I. Functional Characterization of the Human FSH Receptor with an Inactivating Ala189Val Mutation. Mol. Hum. Reprod. 2002, 8, 311–317. [Google Scholar] [CrossRef]
- Tranchant, T.; Durand, G.; Gauthier, C.; Crépieux, P.; Ulloa-Aguirre, A.; Royère, D.; Reiter, E. Preferential β-Arrestin Signalling at Low Receptor Density Revealed by Functional Characterization of the Human FSH Receptor A189 V Mutation. Mol. Cell. Endocrinol. 2011, 331, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yan, Z.; Cai, R.; Khor, S.; Wu, L.; Sun, L.; Wang, Y.; Xu, Y.; Tian, H.; Chen, Q.; et al. Pregnancy and Live Birth In Women With Pathogenic LHCGR Variants Using Their Own Oocytes. J. Clin. Endocrinol. Metab. 2019, 104, 5877–5892. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Mao, J.; Cui, M.; Liu, Z.; Wang, X.; Xiong, S.; Nie, M.; Wu, X. Novel FSHβ Mutation in a Male Patient with Isolated FSH Deficiency and Infertility. Eur. J. Med. Genet. 2017, 60, 335–339. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Vaskivuo, T.E.; Aittomäki, K.; Anttonen, M.; Ruokonen, A.; Herva, R.; Osawa, Y.; Heikinheimo, M.; Huhtaniemi, I.; Tapanainen, J.S. Effects of Follicle-Stimulating Hormone (FSH) and Human Chorionic Gonadotropin in Individuals with an Inactivating Mutation of the FSH Receptor. Fertil. Steril. 2002, 78, 108–113. [Google Scholar] [CrossRef]
- Meduri, G.; Touraine, P.; Beau, I.; Lahuna, O.; Desroches, A.; Vacher-Lavenu, M.C.; Kuttenn, F.; Misrahi, M. Delayed Puberty and Primary Amenorrhea Associated with a Novel Mutation of the Human Follicle-Stimulating Hormone Receptor: Clinical, Histological, and Molecular Studies. J. Clin. Endocrinol. Metab. 2003, 88, 3491–3498. [Google Scholar] [CrossRef]
- Beau, I.; Touraine, P.; Meduri, G.; Gougeon, A.; Desroches, A.; Matuchansky, C.; Milgrom, E.; Kuttenn, F.; Misrahi, M. A Novel Phenotype Related to Partial Loss of Function Mutations of the Follicle Stimulating Hormone Receptor. J. Clin. Investig. 1998, 102, 1352–1359. [Google Scholar] [CrossRef]
- Touraine, P.; Beau, I.; Gougeon, A.; Meduri, G.; Desroches, A.; Pichard, C.; Detoeuf, M.; Paniel, B.; Prieur, M.; Zorn, J.R.; et al. New Natural Inactivating Mutations of the Follicle-Stimulating Hormone Receptor: Correlations between Receptor Function and Phenotype. Mol. Endocrinol. 1999, 13, 1844–1854. [Google Scholar] [CrossRef]
- Bachelot, A.; Rouxel, A.; Massin, N.; Dulon, J.; Courtillot, C.; Matuchansky, C.; Badachi, Y.; Fortin, A.; Paniel, B.; Lecuru, F.; et al. Phenotyping and Genetic Studies of 357 Consecutive Patients Presenting with Premature Ovarian Failure. Eur. J. Endocrinol. 2009, 161, 179–187. [Google Scholar] [CrossRef]
- Massin, N.; Méduri, G.; Bachelot, A.; Misrahi, M.; Kuttenn, F.; Touraine, P. Evaluation of Different Markers of the Ovarian Reserve in Patients Presenting with Premature Ovarian Failure. Mol. Cell. Endocrinol. 2008, 282, 95–100. [Google Scholar] [CrossRef]
- Méduri, G.; Massin, N.; Guibourdenche, J.; Bachelot, A.; Fiori, O.; Kuttenn, F.; Misrahi, M.; Touraine, P. Serum Anti-Müllerian Hormone Expression in Women with Premature Ovarian Failure. Hum. Reprod. 2007, 22, 117–123. [Google Scholar] [CrossRef]
- Campbell, B.K.; Souza, C.; Gong, J.; Webb, R.; Kendall, N.; Marsters, P.; Robinson, G.; Mitchell, A.; Telfer, E.E.; Baird, D.T. Domestic Ruminants as Models for the Elucidation of the Mechanisms Controlling Ovarian Follicle Development in Humans. Reprod. Suppl. 2003, 61, 429–443. [Google Scholar] [CrossRef]
- Erickson, G.F.; Shimasaki, S. The Physiology of Folliculogenesis: The Role of Novel Growth Factors. Fertil. Steril. 2001, 76, 943–949. [Google Scholar] [CrossRef]
- Zeleznik, A.J. The Physiology of Follicle Selection. Reprod. Biol. Endocrinol. 2004, 2, 31. [Google Scholar] [CrossRef]
- Vo, K.C.T.; Kawamura, K. In Vitro Activation Early Follicles: From the Basic Science to the Clinical Perspectives. Int. J. Mol. Sci. 2021, 22, 3785. [Google Scholar] [CrossRef]
- Misrahi, M.; Meduri, G.; Pissard, S.; Bouvattier, C.; Beau, I.; Loosfelt, H.; Jolivet, A.; Rappaport, R.; Milgrom, E.; Bougneres, P. Comparison of Immunocytochemical and Molecular Features with the Phenotype in a Case of Incomplete Male Pseudohermaphroditism Associated with a Mutation of the Luteinizing Hormone Receptor. J. Clin. Endocrinol. Metab. 1997, 82, 2159–2165. [Google Scholar] [CrossRef][Green Version]
- De Roux, N.; Young, J.; Misrahi, M.; Genet, R.; Chanson, P.; Schaison, G.; Milgrom, E. A Family with Hypogonadotropic Hypogonadism and Mutations in the Gonadotropin-Releasing Hormone Receptor. N. Engl. J. Med. 1997, 337, 1597–1602. [Google Scholar] [CrossRef]
- Grynberg, M.; Peltoketo, H.; Christin-Maître, S.; Poulain, M.; Bouchard, P.; Fanchin, R. First Birth Achieved after in Vitro Maturation of Oocytes from a Woman Endowed with Multiple Antral Follicles Unresponsive to Follicle-Stimulating Hormone. J. Clin. Endocrinol. Metab. 2013, 98, 4493–4498. [Google Scholar] [CrossRef]
- Meachem, S.J.; McLachlan, R.I.; de Kretser, D.M.; Robertson, D.M.; Wreford, N.G. Neonatal Exposure of Rats to Recombinant Follicle Stimulating Hormone Increases Adult Sertoli and Spermatogenic Cell Numbers. Biol. Reprod. 1996, 54, 36–44. [Google Scholar] [CrossRef]
- Galway, A.B.; Hsueh, A.J.; Daneshdoost, L.; Zhou, M.H.; Pavlou, S.N.; Snyder, P.J. Gonadotroph Adenomas in Men Produce Biologically Active Follicle-Stimulating Hormone. J. Clin. Endocrinol. Metab. 1990, 71, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Snyder, P.J. Gonadotroph Cell Pituitary Adenomas. Endocrinol. Metab. Clin. N. Am. 1987, 16, 755–764. [Google Scholar] [CrossRef]
- Vasseur, C.; Rodien, P.; Beau, I.; Desroches, A.; Gérard, C.; de Poncheville, L.; Chaplot, S.; Savagner, F.; Croué, A.; Mathieu, E.; et al. A Chorionic Gonadotropin-Sensitive Mutation in the Follicle-Stimulating Hormone Receptor as a Cause of Familial Gestational Spontaneous Ovarian Hyperstimulation Syndrome. N. Engl. J. Med. 2003, 349, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Lussiana, C.; Guani, B.; Mari, C.; Restagno, G.; Massobrio, M.; Revelli, A. Mutations and Polymorphisms of the FSH Receptor (FSHR) Gene: Clinical Implications in Female Fecundity and Molecular Biology of FSHR Protein and Gene. Obstet. Gynecol. Surv. 2008, 63, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Smits, G.; Olatunbosun, O.; Delbaere, A.; Pierson, R.; Vassart, G.; Costagliola, S. Ovarian Hyperstimulation Syndrome Due to a Mutation in the Follicle-Stimulating Hormone Receptor. N. Engl. J. Med. 2003, 349, 760–766. [Google Scholar] [CrossRef]
- Gromoll, J.; Simoni, M.; Nieschlag, E. An Activating Mutation of the Follicle-Stimulating Hormone Receptor Autonomously Sustains Spermatogenesis in a Hypophysectomized Man. J. Clin. Endocrinol. Metab. 1996, 81, 1367–1370. [Google Scholar] [CrossRef]
- Casas-González, P.; Scaglia, H.E.; Pérez-Solís, M.A.; Durand, G.; Scaglia, J.; Zariñán, T.; Dias, J.A.; Reiter, E.; Ulloa-Aguirre, A. Normal Testicular Function without Detectable Follicle-Stimulating Hormone. A Novel Mutation in the Follicle-Stimulating Hormone Receptor Gene Leading to Apparent Constitutive Activity and Impaired Agonist-Induced Desensitization and Internalization. Mol. Cell. Endocrinol. 2012, 364, 71–82. [Google Scholar] [CrossRef]
- Jeha, G.S.; Lowenthal, E.D.; Chan, W.-Y.; Wu, S.-M.; Karaviti, L.P. Variable Presentation of Precocious Puberty Associated with the D564G Mutation of the LHCGR Gene in Children with Testotoxicosis. J. Pediatr. 2006, 149, 271–274. [Google Scholar] [CrossRef]
- Macedo, D.B.; Silveira, L.F.G.; Bessa, D.S.; Brito, V.N.; Latronico, A.C. Sexual Precocity-Genetic Bases of Central Precocious Puberty and Autonomous Gonadal Activation. Endocr. Dev. 2016, 29, 50–71. [Google Scholar] [CrossRef]
- Oduwole, O.O.; Peltoketo, H.; Poliandri, A.; Vengadabady, L.; Chrusciel, M.; Doroszko, M.; Samanta, L.; Owen, L.; Keevil, B.; Rahman, N.A.; et al. Constitutively Active Follicle-Stimulating Hormone Receptor Enables Androgen-Independent Spermatogenesis. J. Clin. Investig. 2018, 128, 1787–1792. [Google Scholar] [CrossRef]
- Peltoketo, H.; Strauss, L.; Karjalainen, R.; Zhang, M.; Stamp, G.W.; Segaloff, D.L.; Poutanen, M.; Huhtaniemi, I.T. Female Mice Expressing Constitutively Active Mutants of FSH Receptor Present with a Phenotype of Premature Follicle Depletion and Estrogen Excess. Endocrinology 2010, 151, 1872–1883. [Google Scholar] [CrossRef][Green Version]
- Lottini, T.; Iorio, J.; Lastraioli, E.; Carraresi, L.; Duranti, C.; Sala, C.; Armenio, M.; Noci, I.; Pillozzi, S.; Arcangeli, A. Transgenic Mice Overexpressing the LH Receptor in the Female Reproductive System Spontaneously Develop Endometrial Tumour Masses. Sci. Rep. 2021, 11, 8847. [Google Scholar] [CrossRef]
- Hai, L.; McGee, S.R.; Rabideau, A.C.; Paquet, M.; Narayan, P. Infertility in Female Mice with a Gain-of-Function Mutation in the Luteinizing Hormone Receptor Is Due to Irregular Estrous Cyclicity, Anovulation, Hormonal Alterations, and Polycystic Ovaries. Biol. Reprod. 2015, 93, 16. [Google Scholar] [CrossRef][Green Version]
- Büchter, D.; Behre, H.M.; Kliesch, S.; Nieschlag, E. Pulsatile GnRH or Human Chorionic Gonadotropin/Human Menopausal Gonadotropin as Effective Treatment for Men with Hypogonadotropic Hypogonadism: A Review of 42 Cases. Eur. J. Endocrinol. 1998, 139, 298–303. [Google Scholar] [CrossRef]
- Burgués, S.; Calderón, M.D. Subcutaneous Self-Administration of Highly Purified Follicle Stimulating Hormone and Human Chorionic Gonadotrophin for the Treatment of Male Hypogonadotrophic Hypogonadism. Spanish Collaborative Group on Male Hypogonadotropic Hypogonadism. Hum. Reprod. 1997, 12, 980–986. [Google Scholar] [CrossRef]
- Haywood, M.; Tymchenko, N.; Spaliviero, J.; Koch, A.; Jimenez, M.; Gromoll, J.; Simoni, M.; Nordhoff, V.; Handelsman, D.J.; Allan, C.M. An Activated Human Follicle-Stimulating Hormone (FSH) Receptor Stimulates FSH-like Activity in Gonadotropin-Deficient Transgenic Mice. Mol. Endocrinol. 2002, 16, 2582–2591. [Google Scholar] [CrossRef][Green Version]
- Walker, W.H.; Cheng, J. FSH and Testosterone Signaling in Sertoli Cells. Reproduction 2005, 130, 15–28. [Google Scholar] [CrossRef]
- Toocheck, C.; Clister, T.; Shupe, J.; Crum, C.; Ravindranathan, P.; Lee, T.-K.; Ahn, J.-M.; Raj, G.V.; Sukhwani, M.; Orwig, K.E.; et al. Mouse Spermatogenesis Requires Classical and Nonclassical Testosterone Signaling. Biol. Reprod. 2016, 94, 11. [Google Scholar] [CrossRef]
- Gorczynska, E.; Handelsman, D.J. Androgens Rapidly Increase the Cytosolic Calcium Concentration in Sertoli Cells. Endocrinology 1995, 136, 2052–2059. [Google Scholar] [CrossRef]
- Lyng, F.M.; Jones, G.R.; Rommerts, F.F. Rapid Androgen Actions on Calcium Signaling in Rat Sertoli Cells and Two Human Prostatic Cell Lines: Similar Biphasic Responses between 1 Picomolar and 100 Nanomolar Concentrations. Biol. Reprod. 2000, 63, 736–747. [Google Scholar] [CrossRef][Green Version]
- Bougnères, P.; François, M.; Pantalone, L.; Rodrigue, D.; Bouvattier, C.; Demesteere, E.; Roger, D.; Lahlou, N. Effects of an Early Postnatal Treatment of Hypogonadotropic Hypogonadism with a Continuous Subcutaneous Infusion of Recombinant Follicle-Stimulating Hormone and Luteinizing Hormone. J. Clin. Endocrinol. Metab. 2008, 93, 2202–2205. [Google Scholar] [CrossRef]
- Rastrelli, G.; Corona, G.; Mannucci, E.; Maggi, M. Factors Affecting Spermatogenesis upon Gonadotropin-Replacement Therapy: A Meta-Analytic Study. Andrology 2014, 2, 794–808. [Google Scholar] [CrossRef]
- Attia, A.M.; Abou-Setta, A.M.; Al-Inany, H.G. Gonadotrophins for Idiopathic Male Factor Subfertility. Cochrane Database Syst. Rev. 2013, CD005071. [Google Scholar] [CrossRef]
- Valenti, D.; La Vignera, S.; Condorelli, R.A.; Rago, R.; Barone, N.; Vicari, E.; Calogero, A.E. Follicle-Stimulating Hormone Treatment in Normogonadotropic Infertile Men. Nat. Rev. Urol. 2013, 10, 55–62. [Google Scholar] [CrossRef]
- Simoni, M.; Brigante, G.; Rochira, V.; Santi, D.; Casarini, L. Prospects for FSH Treatment of Male Infertility. J. Clin. Endocrinol. Metab. 2020, 105, dgaa243. [Google Scholar] [CrossRef]
- Colacurci, N.; De Leo, V.; Ruvolo, G.; Piomboni, P.; Caprio, F.; Pivonello, R.; Papaleo, E.; La Verde, E.; Depalo, R.; Lispi, M.; et al. Recombinant FSH Improves Sperm DNA Damage in Male Infertility: A Phase II Clinical Trial. Front. Endocrinol. 2018, 9, 383. [Google Scholar] [CrossRef]
- Simoni, M.; Santi, D.; Negri, L.; Hoffmann, I.; Muratori, M.; Baldi, E.; Cambi, M.; Marcou, M.; Greither, T.; Baraldi, E.; et al. Treatment with Human, Recombinant FSH Improves Sperm DNA Fragmentation in Idiopathic Infertile Men Depending on the FSH Receptor Polymorphism p.N680S: A Pharmacogenetic Study. Hum. Reprod. 2016, 31, 1960–1969. [Google Scholar] [CrossRef]
- Paradisi, R.; Busacchi, P.; Seracchioli, R.; Porcu, E.; Venturoli, S. Effects of High Doses of Recombinant Human Follicle-Stimulating Hormone in the Treatment of Male Factor Infertility: Results of a Pilot Study. Fertil. Steril. 2006, 86, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhang, X.; Li, J.-P.; Chen, S.; Zhang, R.; Tan, W.; Shi, X.-J. Treatment of Idiopathic Oligozoospermia with Recombinant Human Follicle-Stimulating Hormone: A Prospective, Randomized, Double-Blind, Placebo-Controlled Clinical Study in Chinese Population. Clin. Endocrinol. 2015, 83, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.S.; Kliesch, S.; Tüttelmann, F.; Gromoll, J. FSHB-211G>T Stratification for Follicle-Stimulating Hormone Treatment of Male Infertility Patients: Making the Case for a Pharmacogenetic Approach in Genetic Functional Secondary Hypogonadism. Andrology 2015, 3, 1050–1053. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject No. † | Sex | Age | LH | FSH | Testosterone | Androstene | 17β-Estradiol | Estrone | Inhibin B | AMH | α-Subunit | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | Peak | Baseline | Peak | ||||||||||
| Yr | IU/L | ng/mL | μg/L | pg/mL | pg/mL | pg/mL | ng/mL | IU/L | |||||
| IV-3 (proband) | M | 43 | ND | ND | 20.7 | 29.8 | 0.48 * | 0.62 | <10 | 29 | 71.6 | 9.3 | 1.28 |
| IV-5 (affected sister) | F | 46 | ND | ND | 53.3 | 13 | |||||||
| IV-4 (unaffected) | F | 32 | 4.5 | 4.6 | 66 | ||||||||
| IV-2 (unaffected) | M | 40 | 2.5 | 5.4 | 4.00 | ||||||||
| III-2 (unaffected) | F | 71 | 24.5 | 42.3 | <10 | ||||||||
| Normal range | |||||||||||||
| Male | 0.8–7.6 | 0.7–11.1 | 3.30–10.00 | 0.2–2.90 | <35 | 10–60 | 60.0–330.0 | 3.0–5.4 | <0.70 | ||||
| Female | 1.1–11.6 | 3.3–46.4 | 3.0–14.4 | 4.5–28.8 | 30–50 for follicular phase, 150–230 for luteal phase | ||||||||
| Patient/FSHR Mutation * | Puberty | Amenorrhea | Ovary | Ovarian Histology | Receptor Function In Vitro |
|---|---|---|---|---|---|
| Paris 1 a/Pro519Thr | Delayed | Primary | Hypoplasic | ↑Primordial and primary follicles | Total loss of function |
| Finnish 1 b/Ala189Val | Delayed | Primary | Hypoplasic | ↓Primordial; ↓secondary rare mature follicles | c-AMP: ∼29% of WT FSHR; IP3: almost totally lost |
| Paris 2 c/Asp224Val, Leu601Val | Normal | Primary | Normal | Primordial, primary, secondary and antral follicles up to 3 mm diameter | c-AMP: ∼4% and 12% of WT FSHR |
| Paris 3 d/Ile160Thr, Arg573Cys | Normal | Secondary | Normal | Primordial, primary, secondary and antral follicles up to 5 mm diameter | c-AMP: ∼9% and 24% of WT FSHR |
| Animal model | |||||
| FSHR KO mice e | Hypoplasic | Primordial, primary, secondary follicles | Total inactivation | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oduwole, O.O.; Huhtaniemi, I.T.; Misrahi, M. The Roles of Luteinizing Hormone, Follicle-Stimulating Hormone and Testosterone in Spermatogenesis and Folliculogenesis Revisited. Int. J. Mol. Sci. 2021, 22, 12735. https://doi.org/10.3390/ijms222312735
Oduwole OO, Huhtaniemi IT, Misrahi M. The Roles of Luteinizing Hormone, Follicle-Stimulating Hormone and Testosterone in Spermatogenesis and Folliculogenesis Revisited. International Journal of Molecular Sciences. 2021; 22(23):12735. https://doi.org/10.3390/ijms222312735
Chicago/Turabian StyleOduwole, Olayiwola O., Ilpo T. Huhtaniemi, and Micheline Misrahi. 2021. "The Roles of Luteinizing Hormone, Follicle-Stimulating Hormone and Testosterone in Spermatogenesis and Folliculogenesis Revisited" International Journal of Molecular Sciences 22, no. 23: 12735. https://doi.org/10.3390/ijms222312735
APA StyleOduwole, O. O., Huhtaniemi, I. T., & Misrahi, M. (2021). The Roles of Luteinizing Hormone, Follicle-Stimulating Hormone and Testosterone in Spermatogenesis and Folliculogenesis Revisited. International Journal of Molecular Sciences, 22(23), 12735. https://doi.org/10.3390/ijms222312735