
Multiparameter Evaluation of the Platelet-Inhibitory Effects of Tyrosine Kinase Inhibitors Used for Cancer Treatment

 ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Characteristics of Selected TKIs
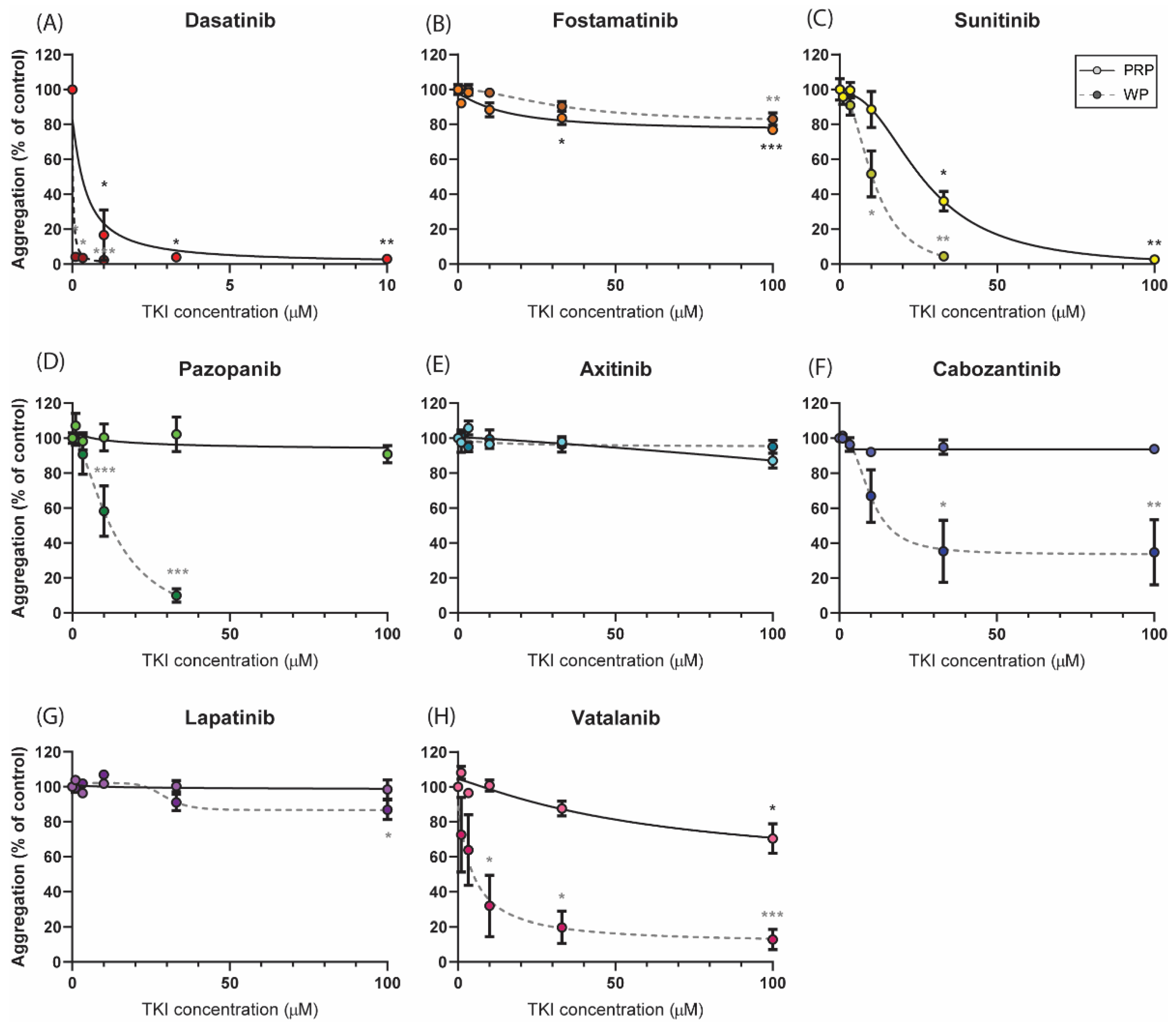
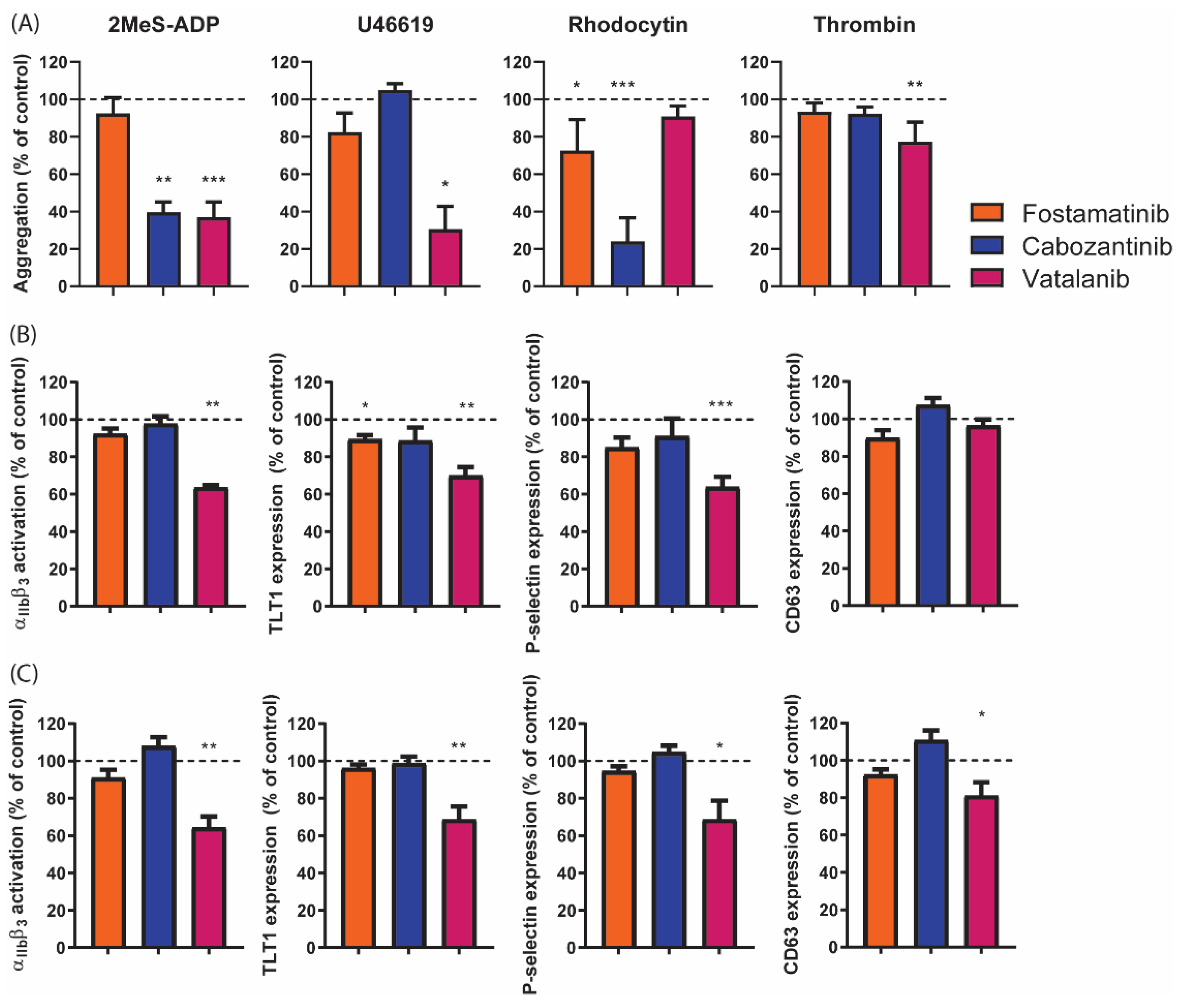
2.2. Distinct Effects of Selected TKIs on Collagen-Induced Platelet Aggregation in the Presence or Absence of Plasma
2.3. Whole-Blood Thrombus Formation over (Non-)Collagen Surfaces under Flow Is Most Strongly Affected by High-Affinity TKIs
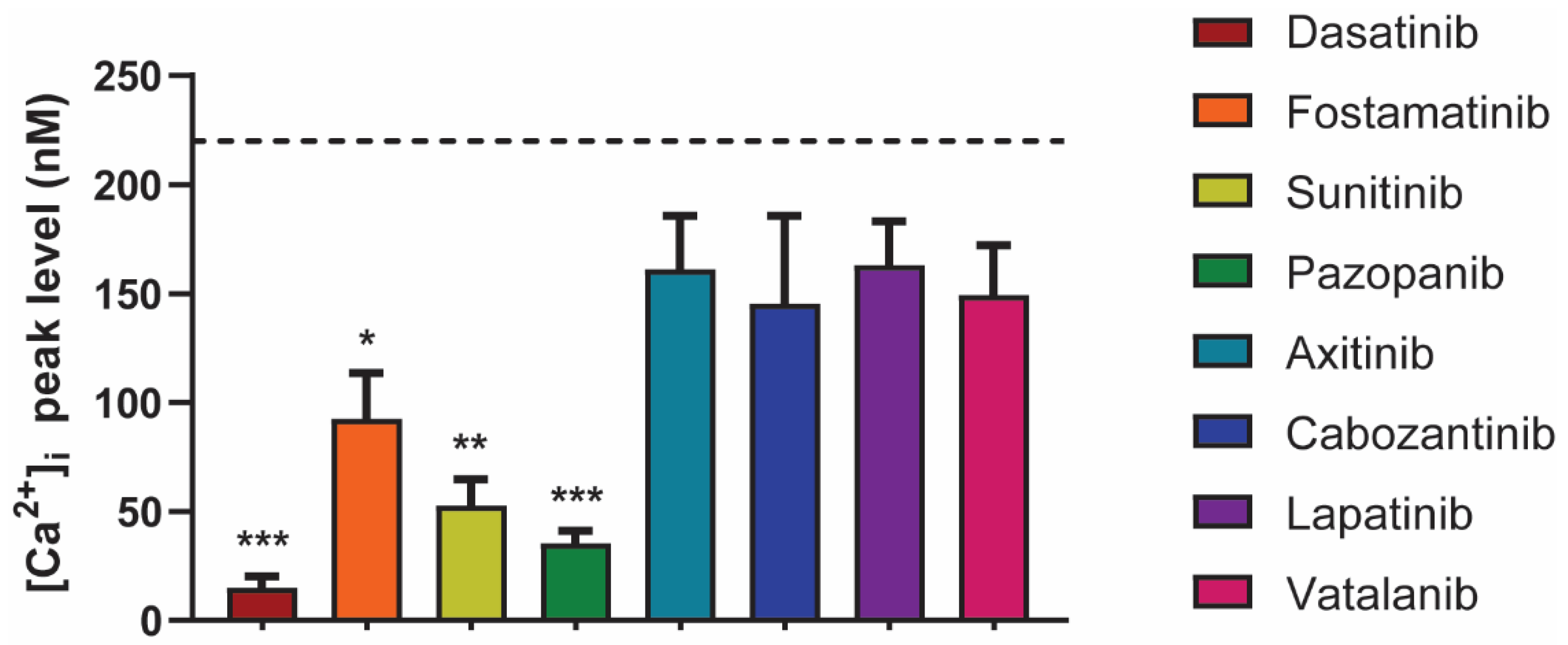
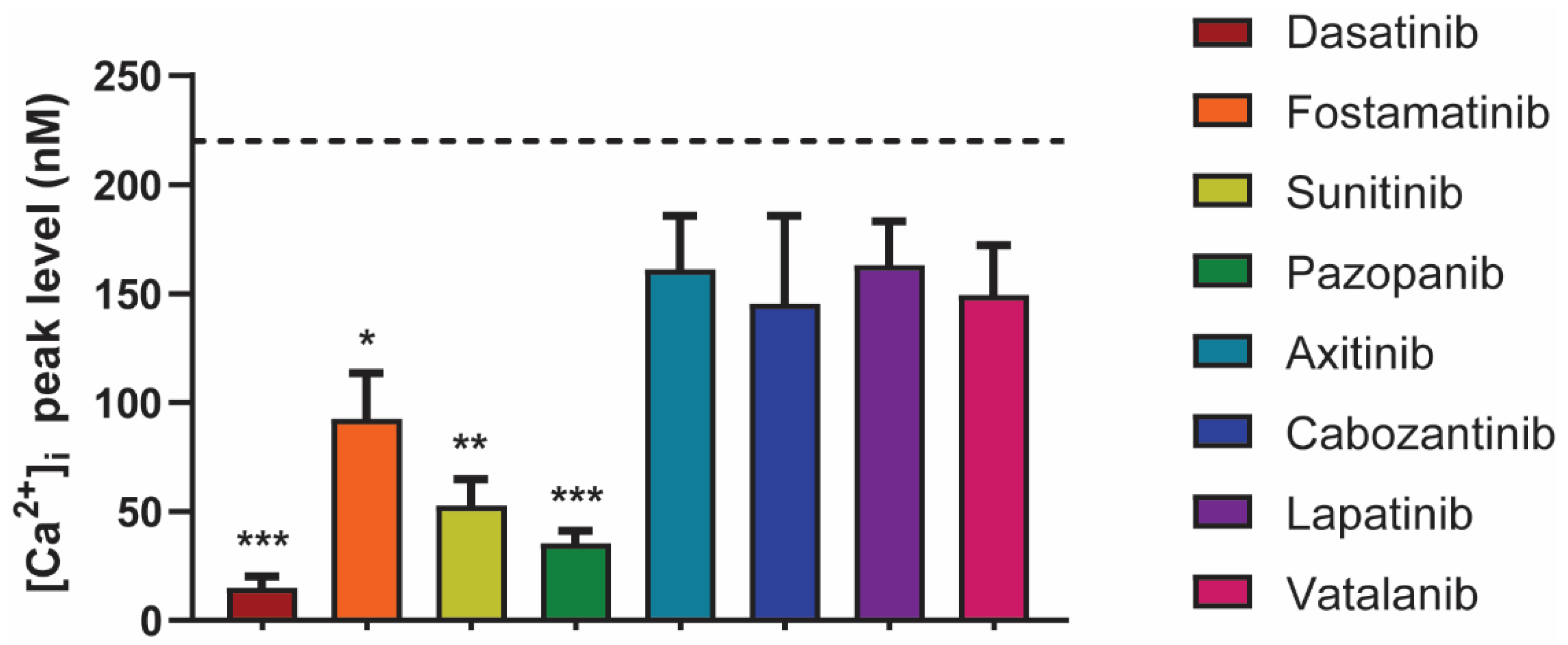
2.4. Differential Effects of TKIs on GPVI-Induced Calcium-Signaling
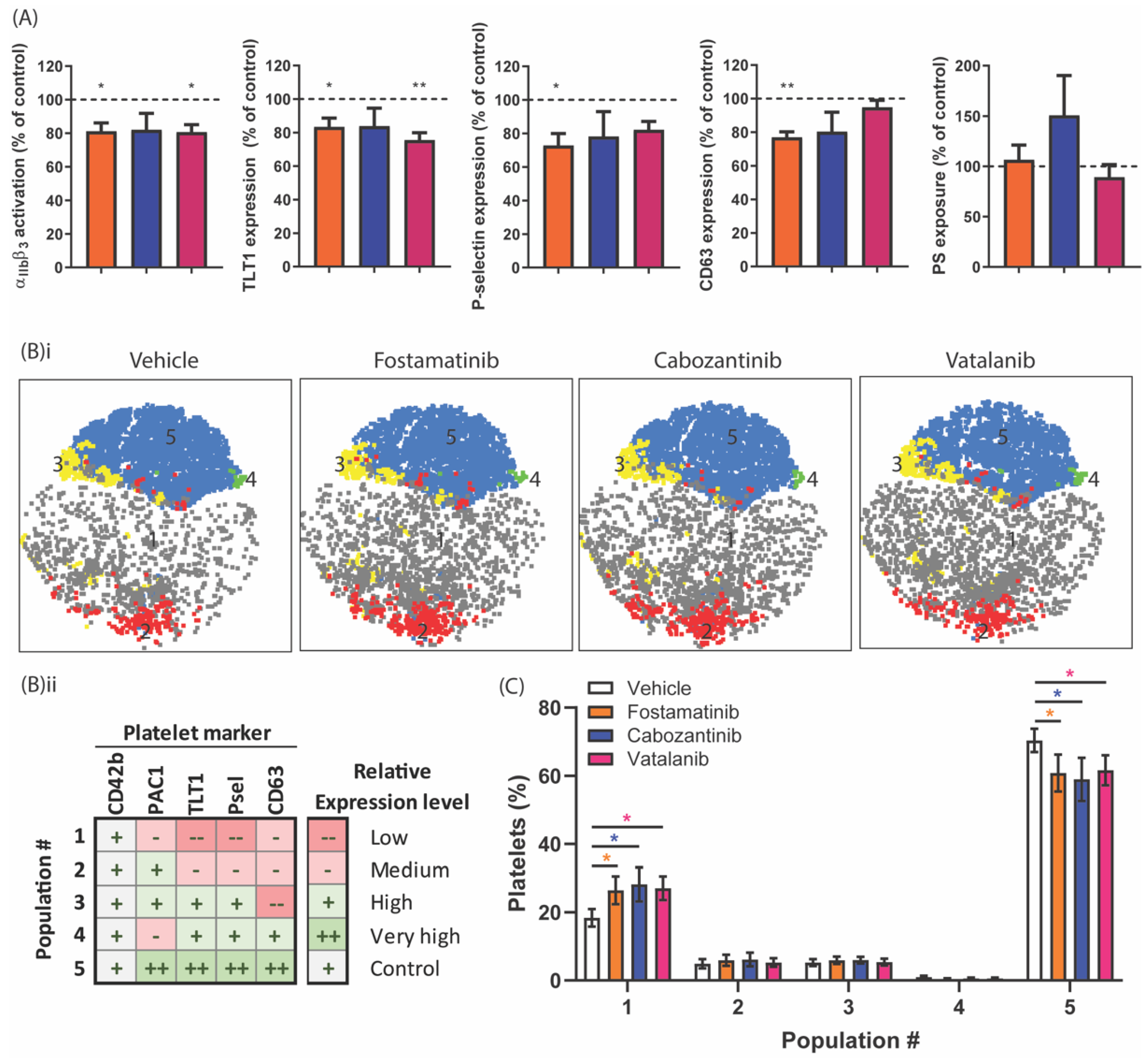
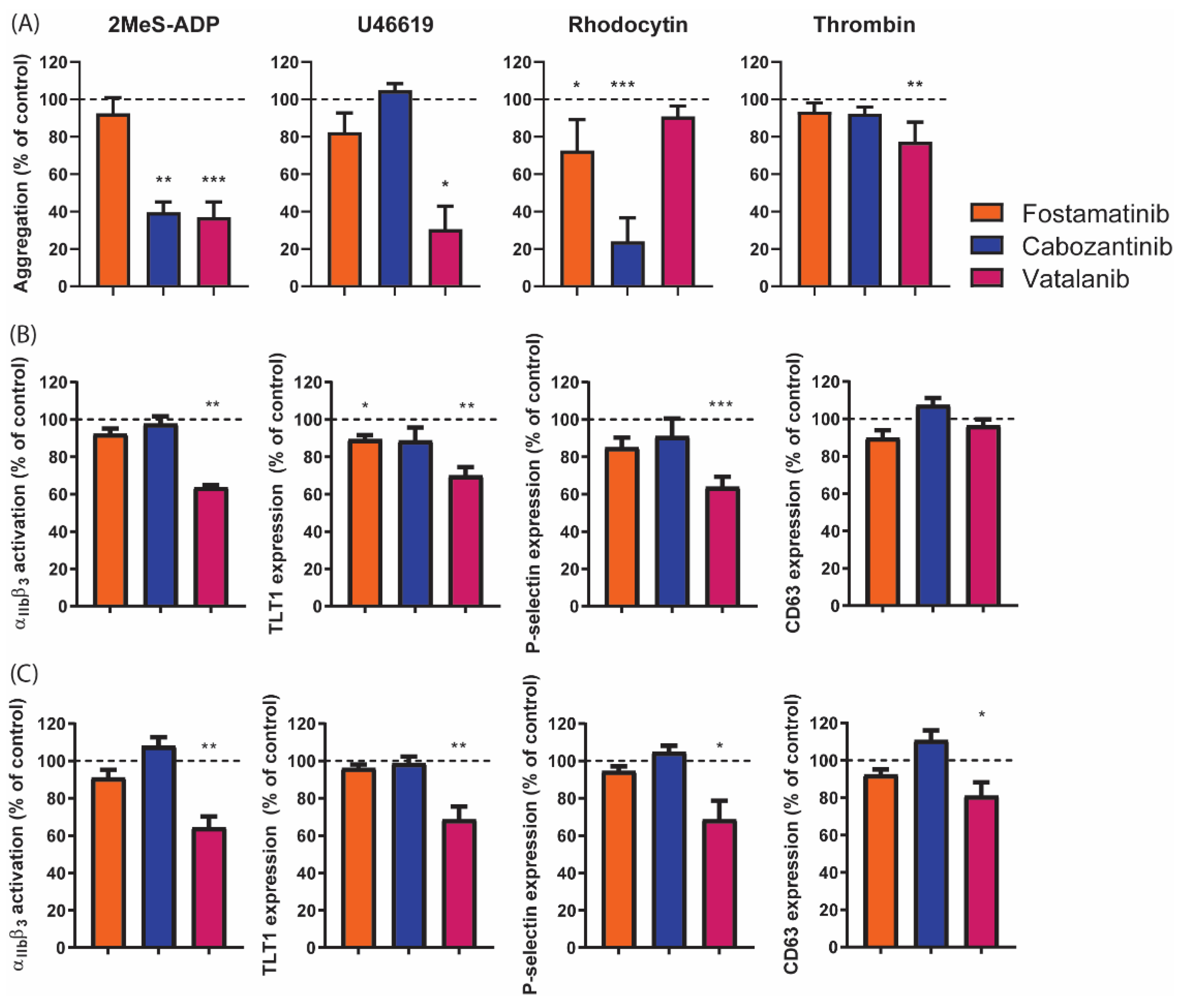
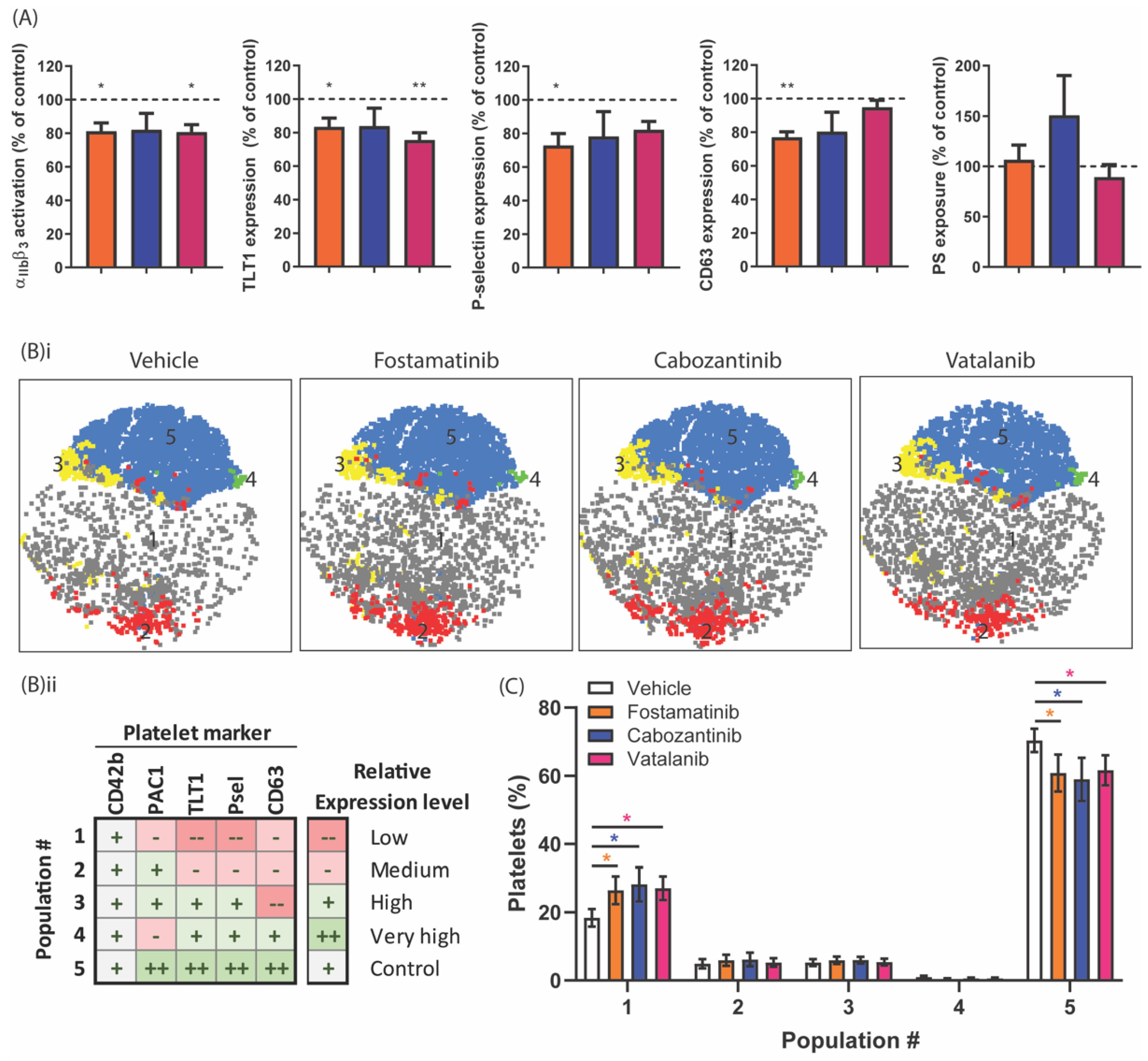
2.5. Diverse Effects of Fostamatinib, Cabozantinib, and Vatalanib on Platelet Activation Markers and Platelet Populations
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Blood Collection and Platelet Isolation
4.3. Thrombus Formation
4.4. Analysis of the Microscopic Images
4.5. Platelet Aggregation by Light Transmission Aggregometry
4.6. Cytosolic Ca2+ Measurements
4.7. Platelet Activation by Flow Cytometry
4.8. Analysis of Flow Cytometry Data
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 9 December 2020).
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.K. Reassessing the mechanisms of acute coronary syndromes. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Roffi, M.; Patrono, C.; Collet, J.P.; Mueller, C.; Valgimigli, M.; Andreotti, F.; Bax, J.J.; Borger, M.A.; Brotons, C.; Chew, D.P.; et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task force for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation of the European society of cardiology (ESC). Eur. Heart J. 2016, 37, 267–315. [Google Scholar]
- McFadyen, J.D.; Schaff, M.; Peter, K. Current and future antiplatelet therapies: Emphasis on preserving haemostasis. Nat. Rev. Cardiol. 2018, 15, 181–191. [Google Scholar] [CrossRef]
- Harbi, M.H.; Smith, C.W.; Nicolson, P.L.R.; Watson, S.P.; Thomas, M.R. Novel antiplatelet strategies targeting GPVI, CLEC-2 and tyrosine kinases. Platelets 2021, 32, 29–41. [Google Scholar] [CrossRef]
- Nieswandt, B.; Brakebusch, C.; Bergmeier, W.; Schulte, V.; Bouvard, D.; Mokhtari-Nejad, R.; Lindhout, T.; Heemskerk, J.W.M.; Zirngibl, H.; Fässler, R. Glycoprotein VI but not alpha2beta1 integrin is essential for platelet interaction with collagen. EMBO J. 2001, 20, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, O.M.; Hughes, C.E.; Montague, S.; Watson, S.K.; Frampton, J.; Bender, M.; Watson, S.P. Fibrin activates GPVI in human and mouse platelets. Blood 2015, 126, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Mangin, P.H.; Onselaer, M.B.; Receveur, N.; Le Lay, N.; Hardy, A.T.; Wilson, C.; Sanchez, X.; Loyau, S.; Dupuis, A.; Babar, A.K.; et al. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica 2018, 103, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Schulte, V.; Bergmeier, W.; Mokhtari-Nejad, R.; Rackebrandt, K.; Cazenave, J.P.; Ohlmann, P.; Gachet, C.; Zirngibl, H. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J. Exp. Med. 2001, 193, 459–469. [Google Scholar] [CrossRef]
- Lockyer, S.; Okuyama, K.; Begum, S.; Le, S.; Sun, B.; Watanabe, T.; Matsumoto, Y.; Yoshitake, M.; Kambayashi, J.; Tandon, N.N. GPVI-deficient mice lack collagen responses and are protected against experimentally induced pulmonary thromboembolism. Thromb. Res. 2006, 118, 371–380. [Google Scholar] [CrossRef]
- Schönberger, T.; Siegel-Axel, D.; Bussl, R.; Richter, S.; Judenhofer, M.S.; Haubner, R.; Reischl, G.; Klingel, K.; Münch, G.; Seizer, P.; et al. The immunoadhesin glycoprotein VI-Fc regulates arterial remodelling after mechanical injury in ApoE-/- mice. Cardiovasc. Res. 2008, 80, 131–137. [Google Scholar] [CrossRef]
- Kuijpers, M.J.E.; Gilio, K.; Reitsma, S.; Nergiz-Unal, R.; Prinzen, L.; Heeneman, S.; Lutgens, E.; van Zandvoort, M.A.; Nieswandt, B.; Egbrink, M.G.; et al. Complementary roles of platelets and coagulation in thrombus formation on plaques acutely ruptured by targeted ultrasound treatment: A novel intravital model. J. Thromb. Haemost. 2009, 7, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Moroi, M.; Jung, S.M.; Okuma, M.; Shinmyozu, K. A patient with platelets deficient in glycoprotein VI that lack both collagen-induced aggregation and adhesion. J. Clin. Investig. 1989, 84, 1440–1445. [Google Scholar] [CrossRef]
- Nagy, M.; Perrella, G.; Dalby, A.; Becerra, M.F.; Garcia Quintanilla, L.; Pike, J.A.; Morgan, N.V.; Gardiner, E.E.; Heemskerk, J.W.M.; Azócar, L.; et al. Flow studies on human GPVI-deficient blood under coagulating and noncoagulating conditions. Blood Adv. 2020, 4, 2953–2961. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Inoue, K.; Kato, Y.; Inoue, O.; Kaneko, M.K.; Mishima, K.; Yatomi, Y.; Yamazaki, Y.; Narimatsu, H.; Ozaki, Y. Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J. Biol. Chem. 2007, 282, 25993–26001. [Google Scholar] [CrossRef]
- Suzuki-Inoue, K.; Inoue, O.; Ding, G.; Nishimura, S.; Hokamura, K.; Eto, K.; Kashiwagi, H.; Tomiyama, Y.; Yatomi, Y.; Umemura, K.; et al. Essential in vivo roles of the C-type lectin receptor CLEC-2: Embryonic/neonatal lethality of CLEC-2-deficient mice by blood/lymphatic misconnections and impaired thrombus formation of CLEC-2-deficient platelets. J. Biol. Chem. 2010, 285, 24494–24507. [Google Scholar] [CrossRef] [PubMed]
- May, F.; Hagedorn, I.; Pleines, I.; Bender, M.; Vögtle, T.; Eble, J.; Elvers, M.; Nieswandt, B. CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood 2009, 114, 3464–3472. [Google Scholar] [CrossRef] [PubMed]
- Bender, M.; May, F.; Lorenz, V.; Thielmann, I.; Hagedorn, I.; Finney, B.A.; Vogtle, T.; Remer, K.; Braun, A.; Bosl, M.; et al. Combined in vivo depletion of glycoprotein VI and C-type lectin-like receptor 2 severely compromises hemostasis and abrogates arterial thrombosis in mice. Arterioscl. Thromb. Vasc Biol. 2013, 33, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Gitz, E.; Pollitt, A.Y.; Gitz-Francois, J.J.; Alshehri, O.; Mori, J.; Montague, S.; Nash, G.B.; Douglas, M.R.; Gardiner, E.E.; Andrews, R.K.; et al. CLEC-2 expression is maintained on activated platelets and on platelet microparticles. Blood 2014, 124, 2262–2270. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Heemskerk, J.W.M.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef]
- Rivera, J.; Lozano, M.L.; Navarro-Núñez, L.; Vicente, V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica 2009, 94, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Tsiamis, A.C.; Hayes, P.; Box, H.; Goodall, A.H.; Bell, P.R.; Brindle, N.P. Characterization and regulation of the receptor tyrosine kinase Tie-1 in platelets. J. Vasc. Res. 2000, 37, 437–442. [Google Scholar] [CrossRef]
- Tullemans, B.M.E.; Heemskerk, J.W.M.; Kuijpers, M.J.E. Acquired platelet antagonism: Off-target antiplatelet effects of malignancy treatment with tyrosine kinase inhibitors. J. Thromb. Haemost. 2018, 16, 1686–1699. [Google Scholar] [CrossRef] [PubMed]
- Levade, M.; Severin, S.; Gratacap, M.P.; Ysebaert, L.; Payrastre, B. Targeting kinases in cancer therapies: Adverse effects on blood platelets. Curr. Pharm. Des. 2016, 22, 2315–2322. [Google Scholar] [CrossRef] [PubMed]
- Spalton, J.C.; Mori, J.; Pollitt, A.Y.; Hughes, C.E.; Eble, J.A.; Watson, S.P. The novel Syk inhibitor R406 reveals mechanistic differences in the initiation of GPVI and CLEC-2 signaling in platelets. J. Thromb. Haemost. 2009, 7, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Braselmann, S.; Taylor, V.; Zhao, H.; Wang, S.; Sylvain, C.; Baluom, M.; Qu, K.; Herlaar, E.; Lau, A.; Young, C.; et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J. Pharmacol. Exp. Ther. 2006, 319, 998–1008. [Google Scholar] [CrossRef]
- Tullemans, B.M.E.; Nagy, M.; Sabrkhany, S.; Griffioen, A.W.; Oude Egbrink, M.G.A.; Aarts, M.; Heemskerk, J.W.M.; Kuijpers, M.J.E. Tyrosine kinase inhibitor pazopanib inhibits platelet procoagulant activity in renal cell carcinoma patients. Front. Cardiovasc. Med. 2018, 5, 142. [Google Scholar] [CrossRef] [PubMed]
- Sabrkhany, S.; Griffioen, A.W.; Pineda, S.; Sanders, L.; Mattheij, N.J.A.; van Geffen, J.P.; Aarts, M.J.E.; Heemskerk, J.W.M.; Oude Egbrink, M.G.; Kuijpers, M.J.E. Sunitinib uptake inhibits platelet function in cancer patients. Eur. J. Cancer 2016, 66, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Tullemans, B.M.E.; Fernández, D.I.; Veninga, A.; Baaten, C.C.F.M.J.; Peters, L.J.F.; Aarts, M.J.B.; Eble, J.A.; Campello, E.; Spiezia, L.; Simioni, P.; et al. Tyrosine Kinase Inhibitor Sunitinib Delays Platelet-Induced Coagulation: Additive Effects of Aspirin. Thromb. Haemost. 2021. Online ahead of print. [Google Scholar]
- Crist, M.; Hansen, E.; Chablani, L.; Guancial, E. Examining the bleeding incidences associated with targeted therapies used in metastatic renal cell carcinoma. Crit. Rev. Oncol. Hematol. 2017, 120, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Steeghs, N.; Nijenhuis, C.M.; Schellens, J.H.; Beijnen, J.H.; Huitema, A.D. Practical guidelines for therapeutic drug monitoring of anticancer tyrosine kinase inhibitors: Focus on the pharmacokinetic targets. Clin. Pharm. 2014, 53, 305–325. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, D.G.; Tsimberidou, A.M. Dasatinib in chronic myeloid leukemia: A review. Ther. Clin. Risk Manag. 2009, 5, 281–289. [Google Scholar] [PubMed]
- McAdoo, S.P.; Tam, F.W. Fostamatinib Disodium. Drugs Future 2011, 36, 273. [Google Scholar] [PubMed]
- Je, Y.; Schutz, F.A.; Choueiri, T.K. Risk of bleeding with vascular endothelial growth factor receptor tyrosine-kinase inhibitors sunitinib and sorafenib: A systematic review and meta-analysis of clinical trials. Lancet Oncol. 2009, 10, 967–974. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Mainwaring, P.N.; Rini, B.I.; Donskov, F.; Hammers, H.; Hutson, T.E.; Lee, J.L.; Peltola, K.; et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1814–1823. [Google Scholar] [CrossRef]
- Blackwell, K.L.; Burstein, H.J.; Storniolo, A.M.; Rugo, H.; Sledge, G.; Koehler, M.; Ellis, C.; Casey, M.; Vukelja, S.; Bischoff, J.; et al. Randomized study of Lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J. Clin. Oncol. 2010, 28, 1124–1130. [Google Scholar] [CrossRef]
- Hecht, J.R.; Trarbach, T.; Hainsworth, J.D.; Major, P.; Jäger, E.; Wolff, R.A.; Lloyd-Salvant, K.; Bodoky, G.; Pendergrass, K.; Berg, W.; et al. Randomized, placebo-controlled, phase III study of first-line oxaliplatin-based chemotherapy plus PTK787/ZK 222584, an oral vascular endothelial growth factor receptor inhibitor, in patients with metastatic colorectal adenocarcinoma. J. Clin. Oncol. 2011, 29, 1997–2003. [Google Scholar] [CrossRef]
- Fabian, M.A.; Biggs, W.H., 3rd; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef]
- EMA. Sprycel, INN-Dasatinib Scientific Discussion. Available online: https://www.ema.europa.eu/en/documents/scientific-discussion/sprycel-epar-scientific-discussion_en.pdf (accessed on 9 December 2020).
- de Witt, S.M.; Swieringa, F.; Heemskerk, J.W.M.; Cosemans, J.M.E.M. Multi-parameter assessment of thrombus formation on microspotted arrays of thrombogenic surfaces. Nat. Protoc. 2014, 5, 4257. [Google Scholar]
- Jooss, N.J.; De Simone, I.; Provenzale, I.; Fernández, D.I.; Brouns, S.L.N.; Farndale, R.W.; Henskens, Y.M.C.; Kuijpers, M.J.E.; Ten Cate, H.; van der Meijden, P.E.J.; et al. Role of platelet glycoprotein VI and tyrosine kinase Syk in thrombus formation on collagen-like surfaces. Int. J. Mol. Sci. 2019, 20, 2788. [Google Scholar] [CrossRef] [PubMed]
- Lhermusier, T.; van Rottem, J.; Garcia, C.; Xuereb, J.M.; Ragab, A.; Martin, V.; Gratacap, M.P.; Sié, P.; Payrastre, B. The Syk-kinase inhibitor R406 impairs platelet activation and monocyte tissue factor expression triggered by heparin-PF4 complex directed antibodies. J. Thromb. Haemost. 2011, 9, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.R.; Singh, M.V.; Dewhurst, S.; Schifitto, G.; Maggirwar, S.B. Platelets function as an acute viral reservoir during HIV-1 infection by harboring virus and T-cell complex formation. Blood Adv. 2020, 4, 4512–4521. [Google Scholar] [CrossRef]
- Heemskerk, J.W.M.; Mattheij, N.J.A.; Cosemans, J.M.E.M. Platelet-based coagulation: Different populations, different functions. J. Thromb. Haemost. 2013, 11, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I. Targeted therapy for patients with renal-cell carcinoma. Lancet Oncol. 2011, 12, 1085–1087. [Google Scholar] [CrossRef]
- Rini, B.I. Metastatic renal cell carcinoma: Many treatment options, one patient. J. Clin. Oncol. 2009, 27, 3225–3234. [Google Scholar] [CrossRef]
- Koksal, U.I.; Goffin, J.; Lewis, B.; Sartor, O.A.; Belyaeva, E.; Socola, F.; Barata, P.C. A case report with severe thrombocytopenia induced by axitinib. Case Rep. Hematol. 2020, 2020, 7520783. [Google Scholar] [CrossRef]
- Gunnarsson, O.; Pfanzelter, N.R.; Cohen, R.B.; Keefe, S.M. Evaluating the safety and efficacy of axitinib in the treatment of advanced renal cell carcinoma. Cancer Manag. Res. 2015, 7, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Sater, H.A.; Ghandi, A.S.; Dainer, P.; Pantin, J. Receptor Tyrosine Kinases in Human Platelets: A Review of Expression, Function and Inhibition in Relation to the Risk of Bleeding or Thrombocytopenia from Phase I through Phase III Trials. J. Cancer Prev. Curr. Res. 2017, 8, 00298. [Google Scholar]
- Li, R.; Grosser, T.; Diamond, S.L. Microfluidic whole blood testing of platelet response to pharmacological agents. Platelets 2017, 28, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.N.; Meinhardt, G.; Jacques, C.; Laurent, D.; Thomas, A.L. Vatalanib: The clinical development of a tyrosine kinase inhibitor of angiogenesis in solid tumours. Expert Opin. Investig. Drugs 2007, 16, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Gratacap, M.P.; Martin, V.; Valéra, M.C.; Allart, S.; Garcia, C.; Sié, P.; Recher, C.; Payrastre, B. The new tyrosine-kinase inhibitor and anticancer drug dasatinib reversibly affects platelet activation in vitro and in vivo. Blood 2009, 114, 1884–1892. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, Z.; Moghaddam, M.F. A systematic analysis of physicochemical and ADME properties of all small molecule kinase inhibitors approved by US FDA from january 2001 to october 2015. Curr. Med. Chem. 2017, 24, 3159–3184. [Google Scholar]
- Podoll, T.; Pearson, P.G.; Evarts, J.; Ingallinera, T.; Bibikova, E.; Sun, H.; Gohdes, M.; Cardinal, K.; Sanghvi, M.; Slatter, J.G. Bioavailability, biotransformation, and excretion of the covalent Bruton tyrosine kinase inhibitor acalabrutinib in rats, dogs, and humans. Drug Metab. Dispos. 2019, 47, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Owzar, K.; Gupta, P.; Larson, R.A.; Mulkey, F.; Miller, A.A.; Lewis, L.D.; Hurd, D.; Vij, R.; Ratain, M.J.; et al. Vatalanib population pharmacokinetics in patients with myelodysplastic syndrome: CALGB 10105 (Alliance). Br. J. Clin. Pharm. 2014, 78, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Brahmer, J.; Messersmith, W.; Hidalgo, M.; Baker, S.D. Binding of gefitinib, an inhibitor of epidermal growth factor receptor-tyrosine kinase, to plasma proteins and blood cells: In vitro and in cancer patients. Investig. New Drugs 2006, 24, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.W.; Raslan, Z.; Parfitt, L.; Khan, A.O.; Patel, P.; Senis, Y.A.; Mazharian, A. TREM-like transcript 1: A more sensitive marker of platelet activation than P-selectin in humans and mice. Blood Adv. 2018, 2, 2072–2078. [Google Scholar] [CrossRef]
- Washington, A.V.; Schubert, R.L.; Quigley, L.; Disipio, T.; Feltz, R.; Cho, E.H.; McVicar, D.W. A TREM family member, TLT-1, is found exclusively in the alpha-granules of megakaryocytes and platelets. Blood 2004, 104, 1042–1047. [Google Scholar] [CrossRef]
- Washington, A.V.; Gibot, S.; Acevedo, I.; Gattis, J.; Quigley, L.; Feltz, R.; De La Mota, A.; Schubert, R.L.; Gomez-Rodriguez, J.; Cheng, J.; et al. TREM-like transcript-1 protects against inflammation-associated hemorrhage by facilitating platelet aggregation in mice and humans. J. Clin. Investig. 2009, 119, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Morales-Ortiz, J.; Reyes, F.S.; Santiago, O.; Rivera, L.; Nahomy, L.; Manne, B.K.; Madera, B.; Rondicna, M.T.; Washington, A.V. TLT-1- controls early thrombus formation and stability by facilitating αIIbβ3 outside-in signaling in mice. Int. J. Adv. Res. 2018, 6, 1143–1149. [Google Scholar]
- Baaten, C.C.F.M.J.; Ten Cate, H.; van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet populations and priming in hematological diseases. Blood Rev. 2017, 31, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Bergmeier, W.; Bouvard, D.; Eble, J.A.; Mokhtari-Nejad, R.; Schulte, V.; Zirngibl, H.; Brakebusch, C.; Fassler, R.; Nieswandt, B. Rhodocytin (aggretin) activates platelets lacking alpha(2)beta(1) integrin, glycoprotein VI, and the ligand-binding domain of glycoprotein Ibalpha. J. Biol. Chem. 2001, 276, 25121–25126. [Google Scholar] [CrossRef]
- van der Meijden, P.E.J.; Feijge, M.A.; Giesen, P.L.; Huijberts, M.; van Raak, L.P.; Heemskerk, J.W.M. Platelet P2Y12 receptors enhance signalling towards procoagulant activity and thrombin generation. A study with healthy subjects and patients at thrombotic risk. Thromb. Haemost. 2005, 93, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Mattheij, N.J.A.; Gilio, K.; van Kruchten, R.; Jobe, S.M.; Wieschhaus, A.J.; Chishti, A.H.; Collins, P.; Heemskerk, J.W.M.; Cosemans, J.M.E.M. Dual mechanism of integrin αIIbβ3 closure in procoagulant platelets. J. Biol. Chem. 2013, 288, 13325–13336. [Google Scholar] [CrossRef] [PubMed]
- van Geffen, J.P.; Brouns, S.L.N.; Batista, J.; McKinney, H.; Kempster, C.; Nagy, M.; Sivapalaratnam, S.; Baaten, C.C.F.M.J.; Bourry, N.; Frontini, M.; et al. High-throughput elucidation of thrombus formation reveals sources of platelet function variability. Haematologica 2019, 104, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Feijge, M.A.; van Pampus, E.C.; Lacabaratz-Porret, C.; Hamulyàk, K.; Levy-Toledano, S.; Enouf, J.; Heemskerk, J.W. Inter-individual variability in Ca2+ signalling in platelets from healthy volunteers: Effects of aspirin and relationship with expression of endomembrane Ca2+-ATPases. Br. J. Haematol. 1998, 102, 850–859. [Google Scholar] [CrossRef]
- Monaco, G.; Chen, H.; Poidinger, M.; Chen, J.; de Magalhães, J.P.; Larbi, A. FlowAI: Automatic and interactive anomaly discerning tools for flow cytometry data. Bioinformatics 2016, 32, 2473–2480. [Google Scholar] [CrossRef]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T.; Saeys, Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry 2015, 87, 636–645. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TKI | Reported Targets | Targets in Platelets (MKs) | Types of Cancers Treated | Bleeding Reported | Ref. |
|---|---|---|---|---|---|
| Dasatinib | PDGFR, EFGR, BCR-ABL, EphA2, Kit, SFK | SFK: Src, Fyn, Lck, Lyn, Yes, Btk, (Kit) | CP CML, AP MB, LB CML | Yes | [32,33] |
| Fostamatinib | Syk | Syk, Src, Fgr, Fyn, lck, Lyn, Yes | B-cell lymphoma, CLL | No | [34] |
| Sunitinib | VEGFR, PDGFR, CSF-R, Ret, Kit, Flt3 | (Ret, Kit) | RCC, GIST, PNET | Yes | [29,31,32,35] |
| Pazopanib | VEGFR, PDGFR, FGFR, Kit, Fms, Itk, Lck | Lck (Kit) | NSCLC, OC, RCC, STS, TC | Yes | [31,32] |
| Axitinib | VEGFR, PDGFRβ | Lck, Yes, Axl, Tie | RCC | Yes | [31,32] |
| Cabozantinib | VEGFR, Met, Ret, Kit, Flt3, Axl, Tie | Axl, Tie (Ret, Kit) | TC, RCC | Yes | [31,36] |
| Lapatinib | EGFR, ErbB1-2 | ND | ErbB2+ HR− or HR+ BC | No | [32,37] |
| Vatalanib | VEGFR, PDGFRβ, Kit | (Kit) | MCRC | No | [38] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tullemans, B.M.E.; Veninga, A.; Fernandez, D.I.; Aarts, M.J.B.; Eble, J.A.; van der Meijden, P.E.J.; Heemskerk, J.W.M.; Kuijpers, M.J.E. Multiparameter Evaluation of the Platelet-Inhibitory Effects of Tyrosine Kinase Inhibitors Used for Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 11199. https://doi.org/10.3390/ijms222011199
Tullemans BME, Veninga A, Fernandez DI, Aarts MJB, Eble JA, van der Meijden PEJ, Heemskerk JWM, Kuijpers MJE. Multiparameter Evaluation of the Platelet-Inhibitory Effects of Tyrosine Kinase Inhibitors Used for Cancer Treatment. International Journal of Molecular Sciences. 2021; 22(20):11199. https://doi.org/10.3390/ijms222011199
Chicago/Turabian StyleTullemans, Bibian M. E., Alicia Veninga, Delia I. Fernandez, Maureen J. B. Aarts, Johannes A. Eble, Paola E. J. van der Meijden, Johan W. M. Heemskerk, and Marijke J. E. Kuijpers. 2021. "Multiparameter Evaluation of the Platelet-Inhibitory Effects of Tyrosine Kinase Inhibitors Used for Cancer Treatment" International Journal of Molecular Sciences 22, no. 20: 11199. https://doi.org/10.3390/ijms222011199
APA StyleTullemans, B. M. E., Veninga, A., Fernandez, D. I., Aarts, M. J. B., Eble, J. A., van der Meijden, P. E. J., Heemskerk, J. W. M., & Kuijpers, M. J. E. (2021). Multiparameter Evaluation of the Platelet-Inhibitory Effects of Tyrosine Kinase Inhibitors Used for Cancer Treatment. International Journal of Molecular Sciences, 22(20), 11199. https://doi.org/10.3390/ijms222011199