
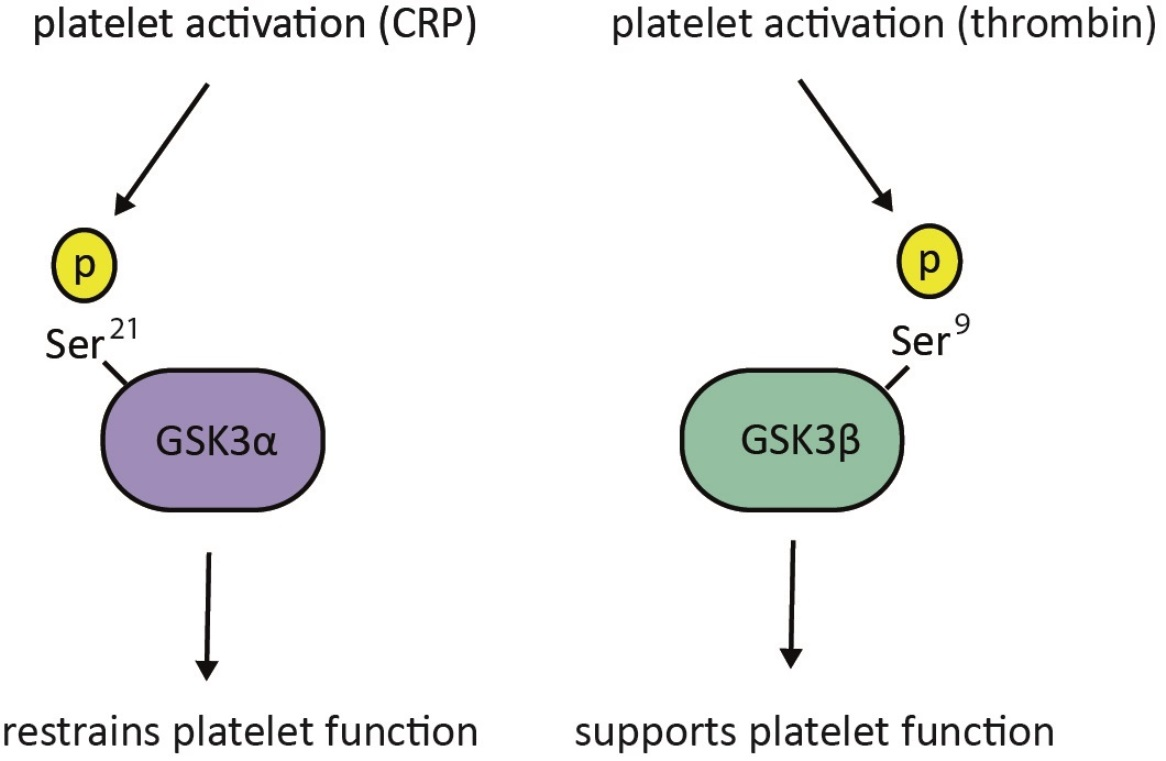
Opposing Roles of GSK3α and GSK3β Phosphorylation in Platelet Function and Thrombosis


Abstract
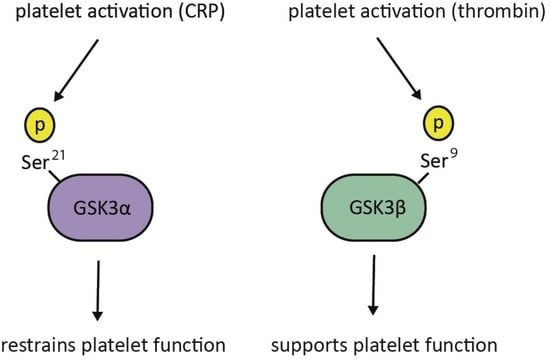
:
1. Introduction
2. Results
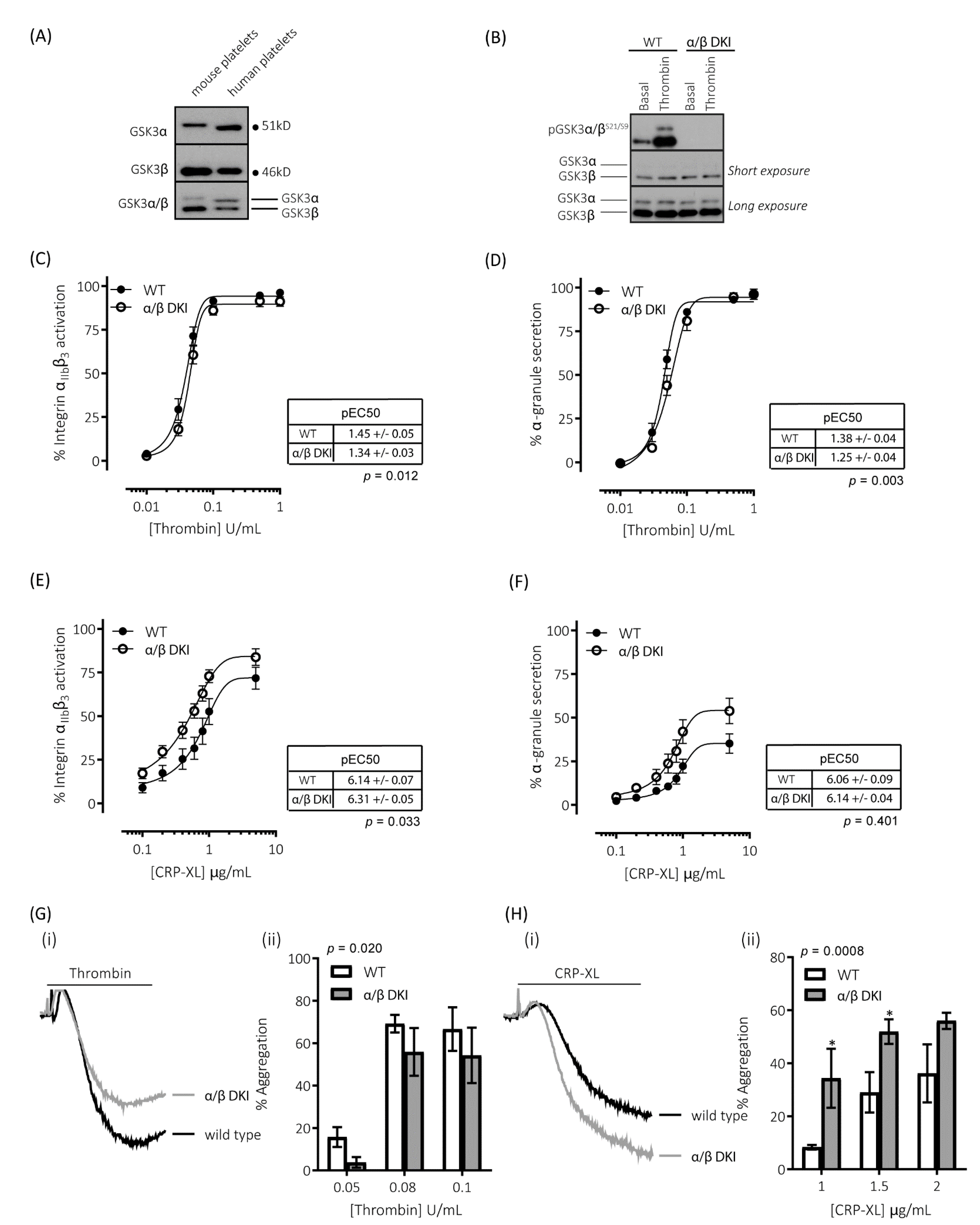
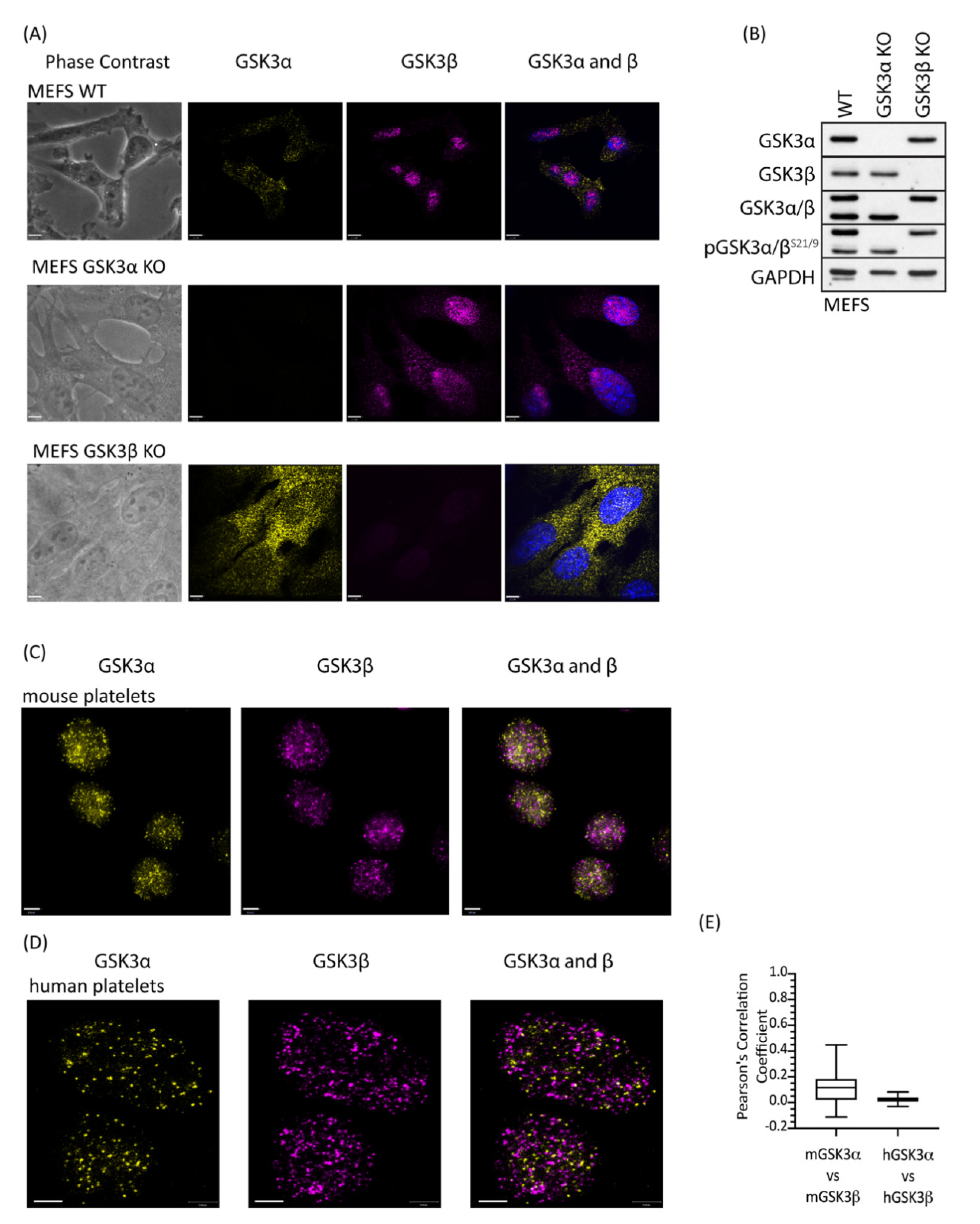
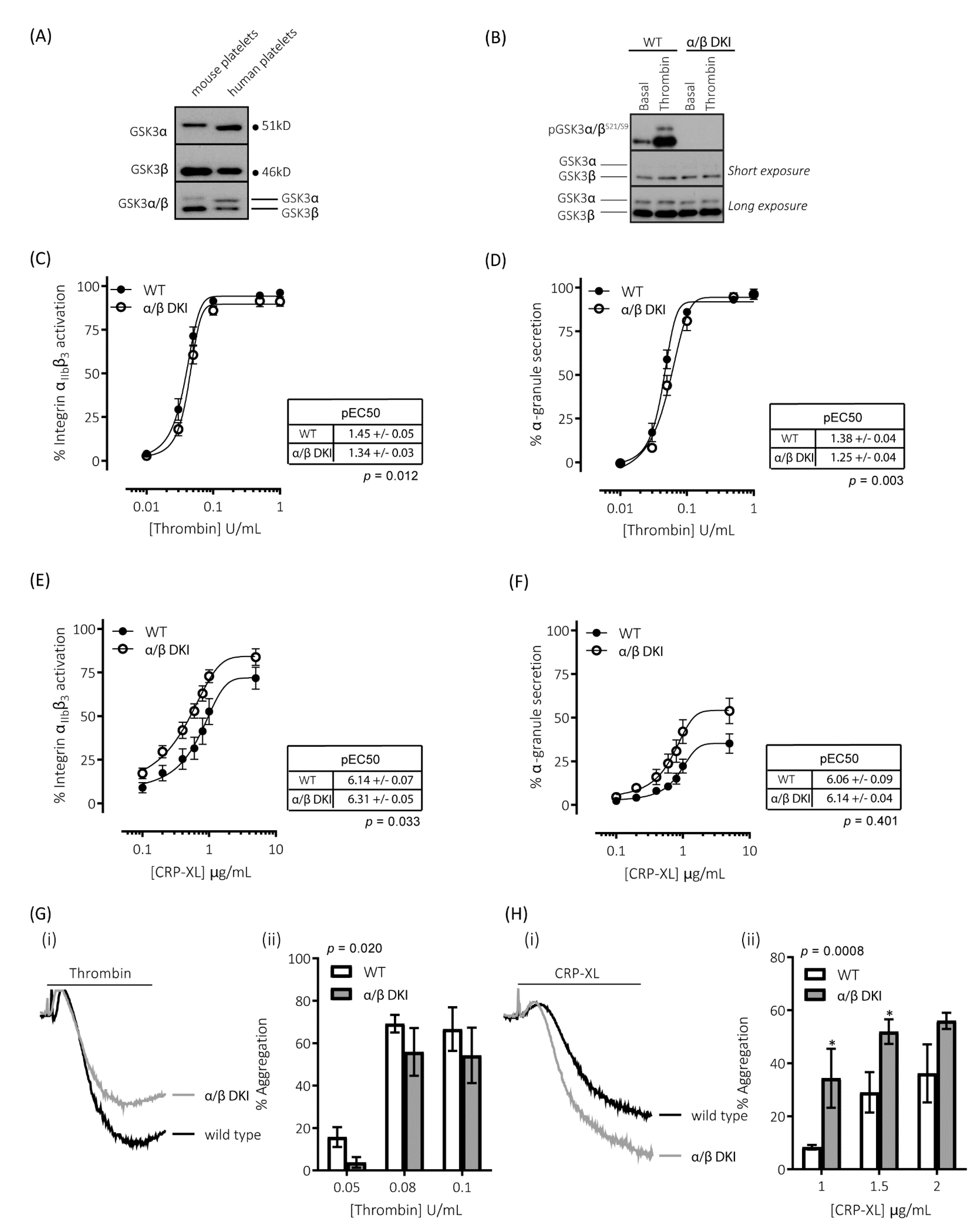
2.1. GSK3α and GSK3β Expression in Human and Mouse Platelets
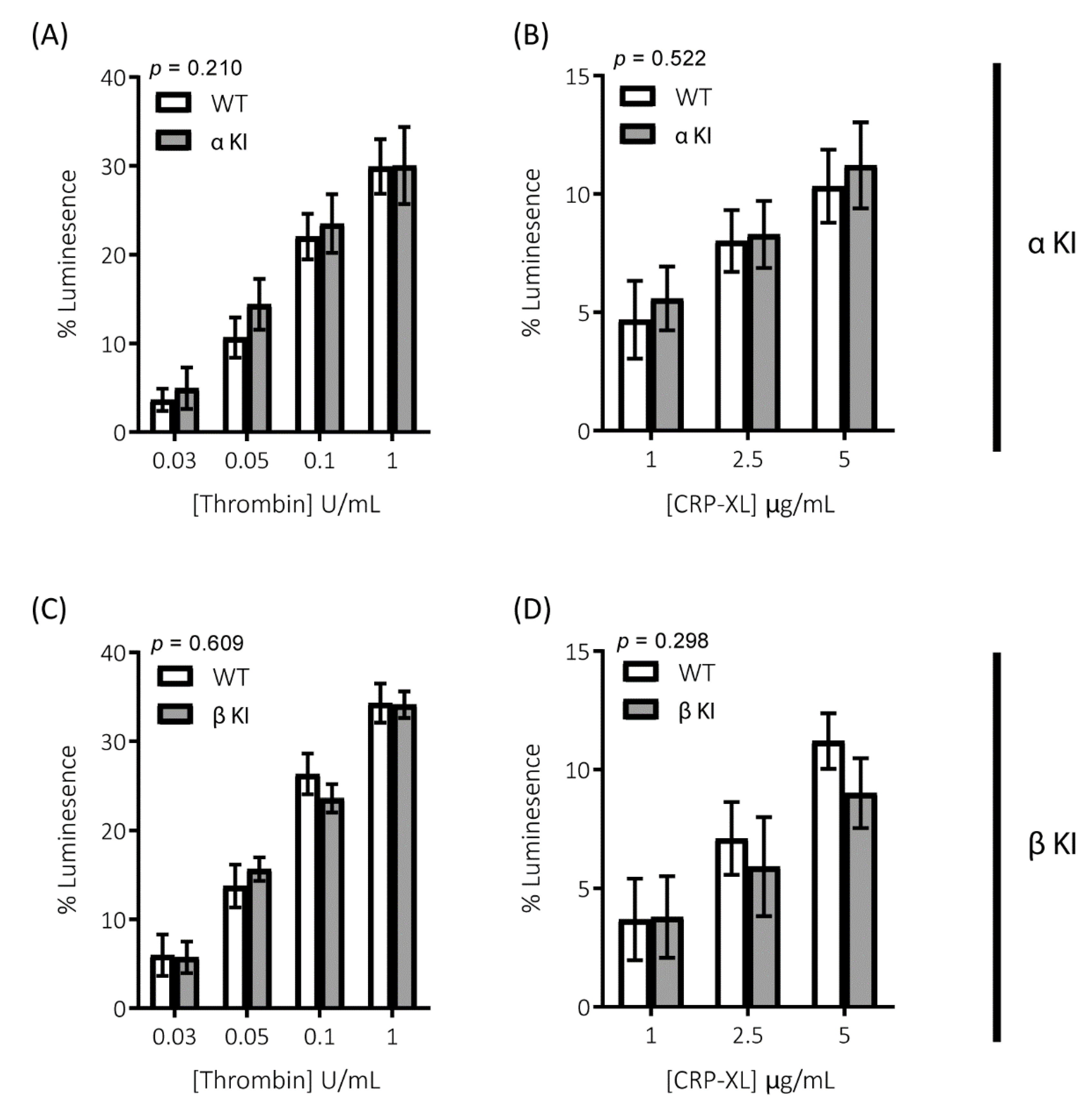
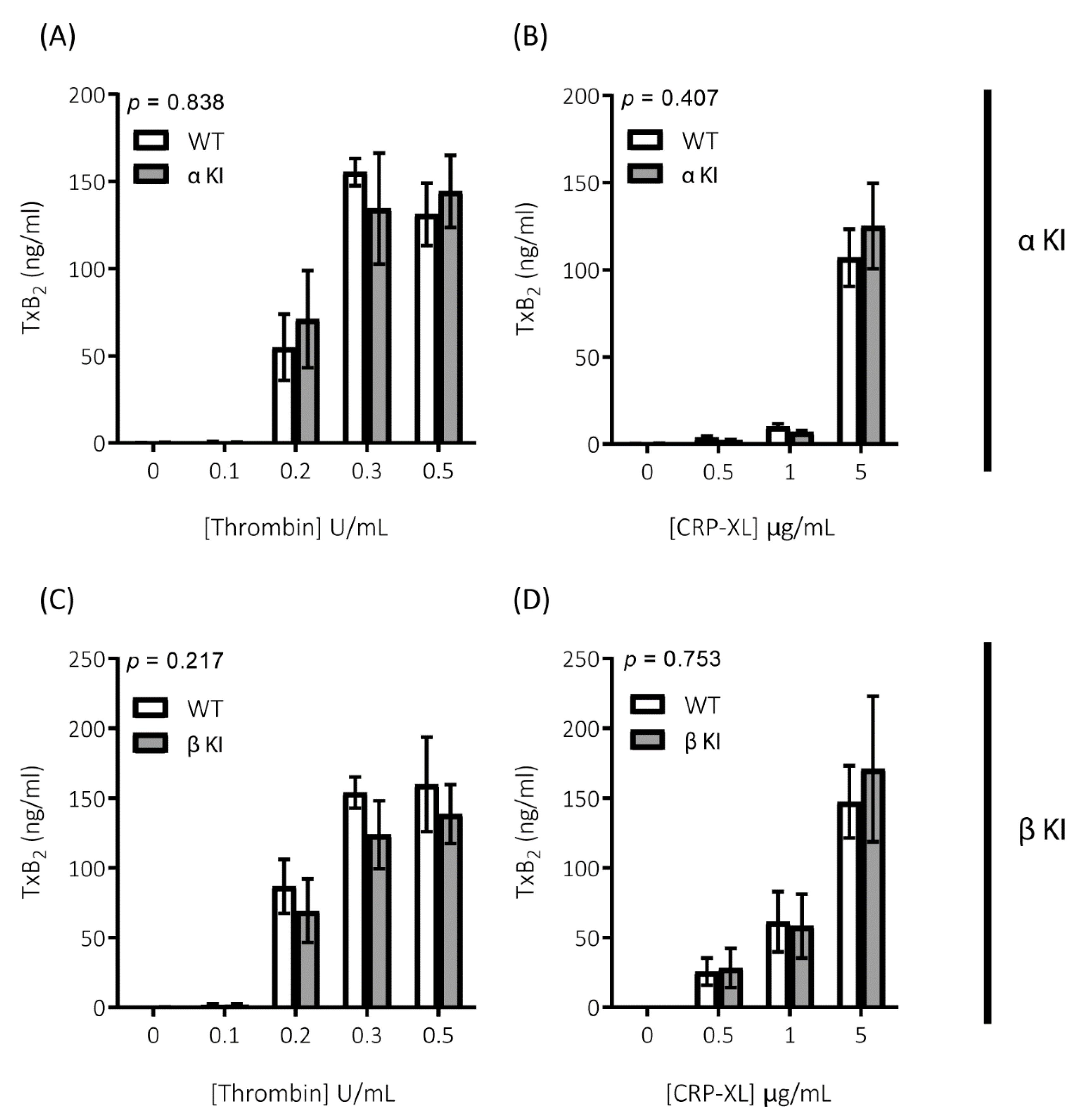
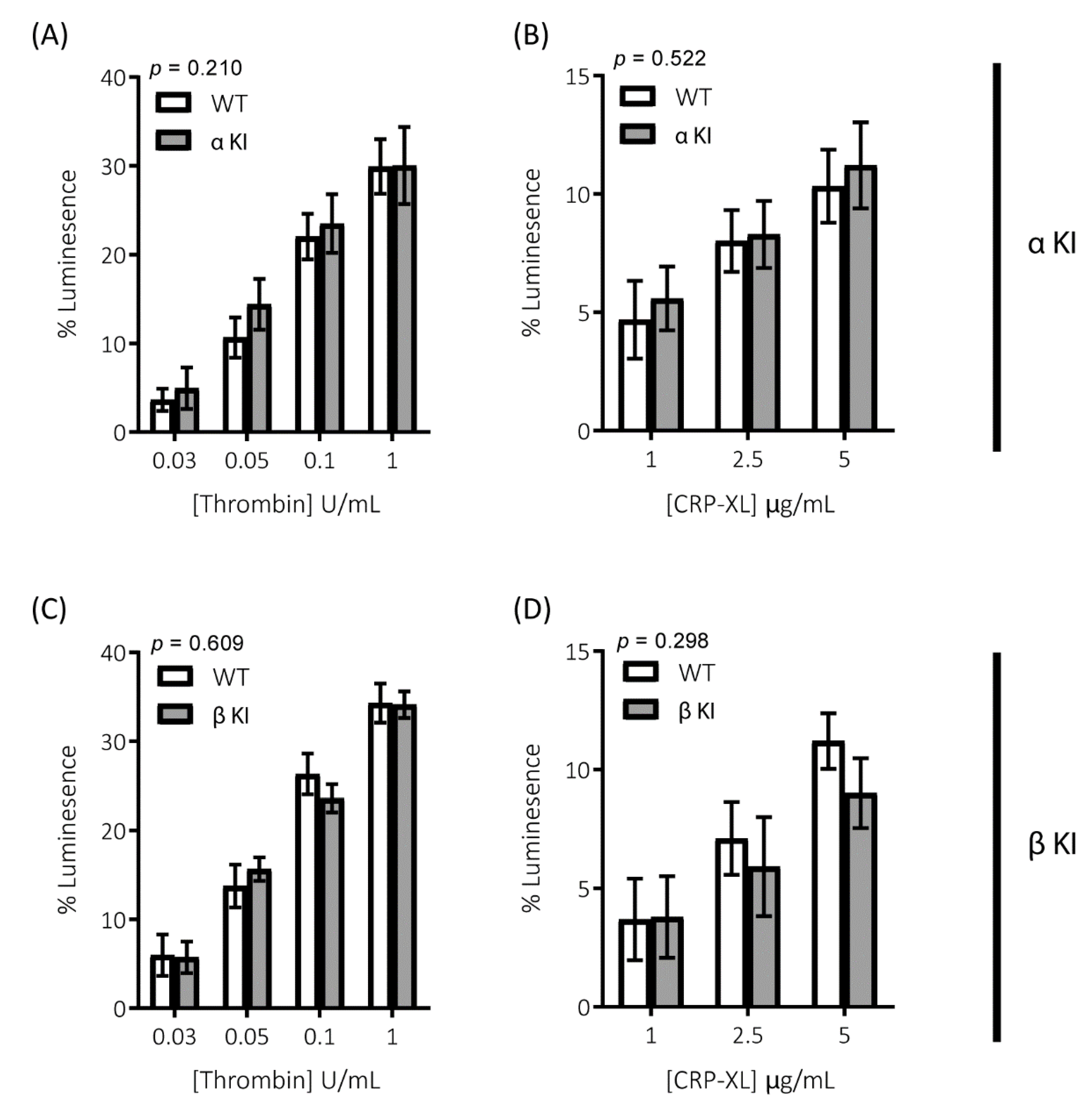
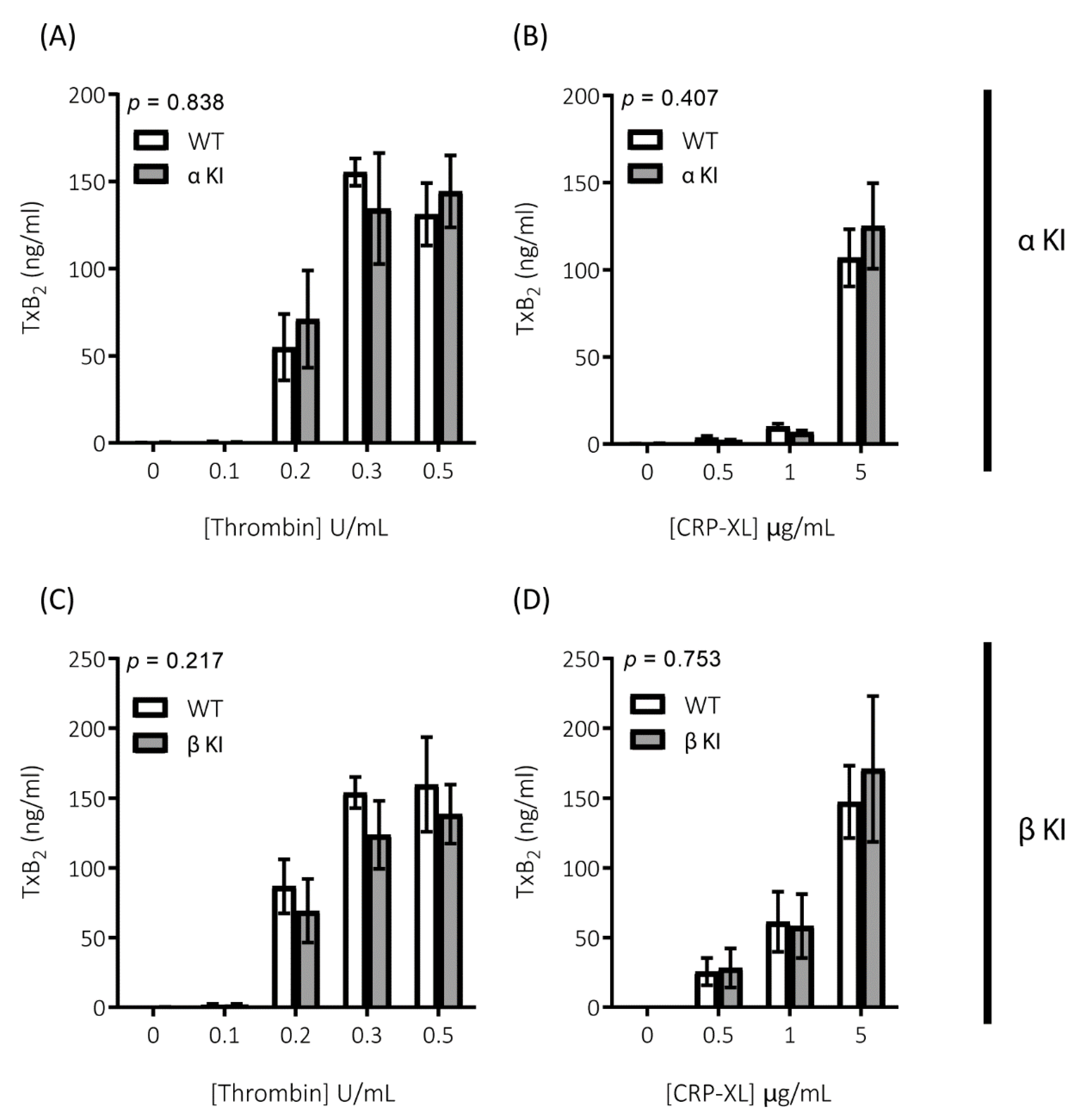
2.2. Thrombin-Mediated Responses Are Reduced in Phosphorylation-Resistant GSK3α/β Platelets
2.3. GPVI-Mediated Responses Are Enhanced in Phosphorylation-Resistant GSK3α/β Platelets
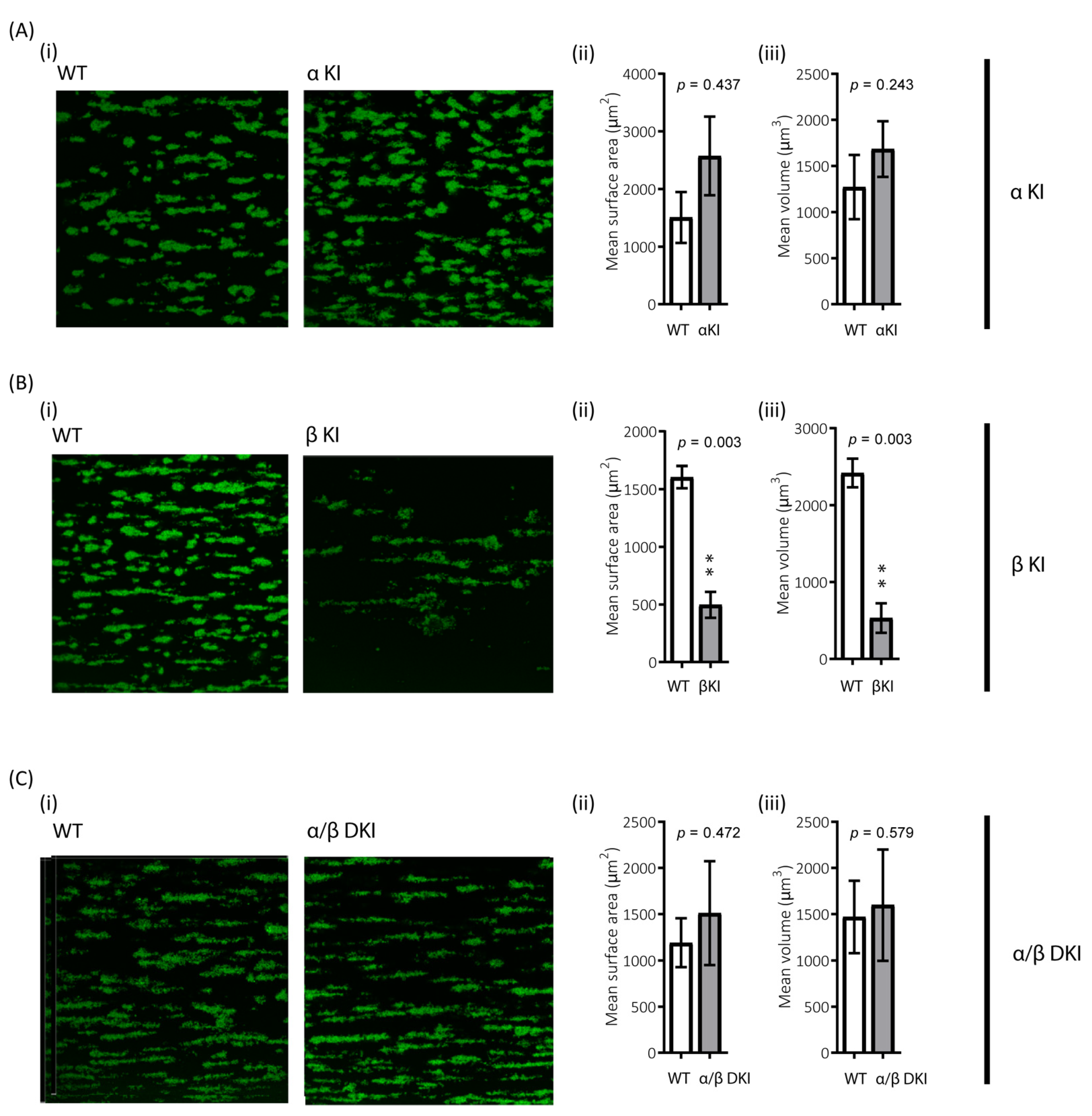
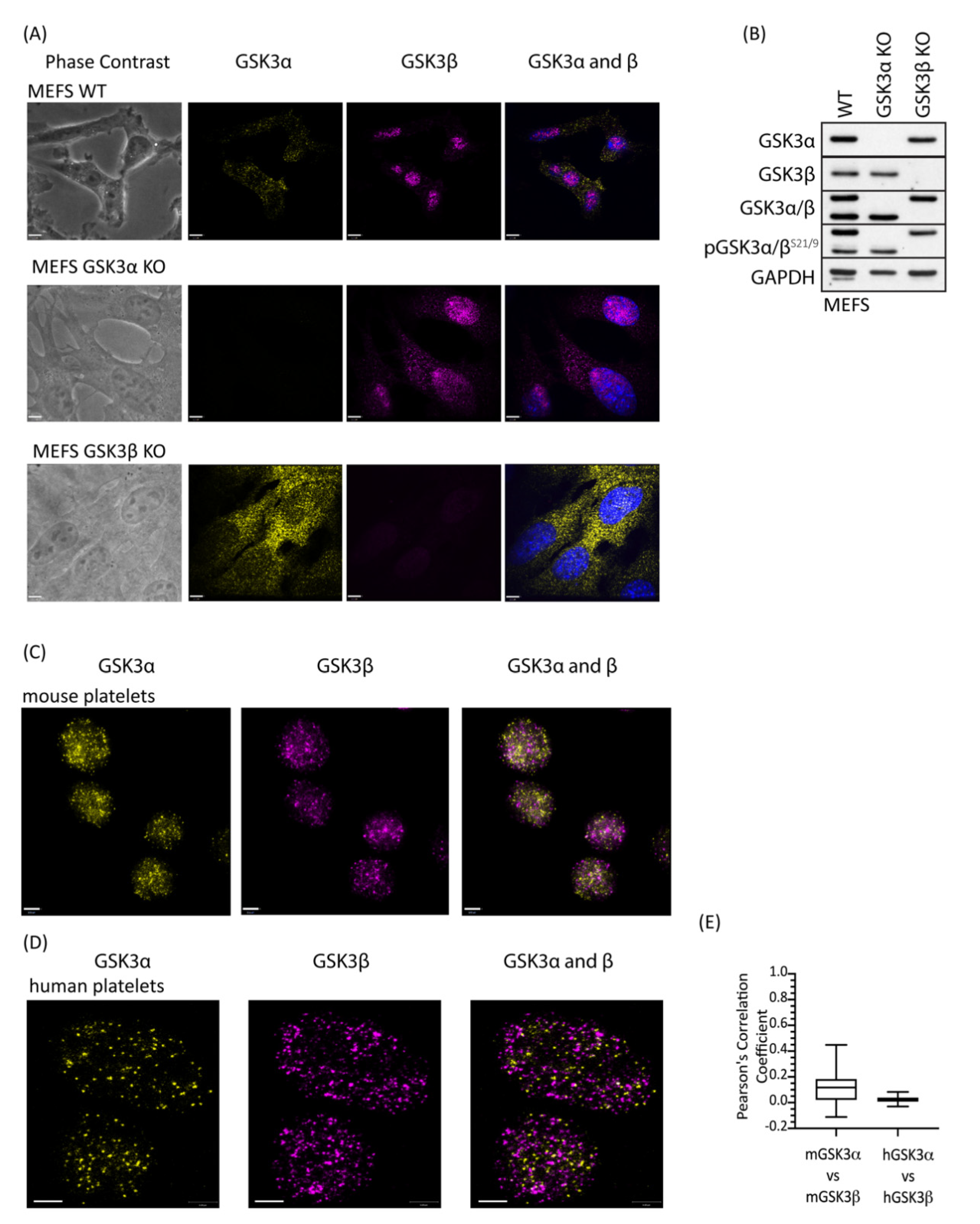
2.4. GSK3 Paralogs Localise to Distinct Locations in Mouse and Human Platelets
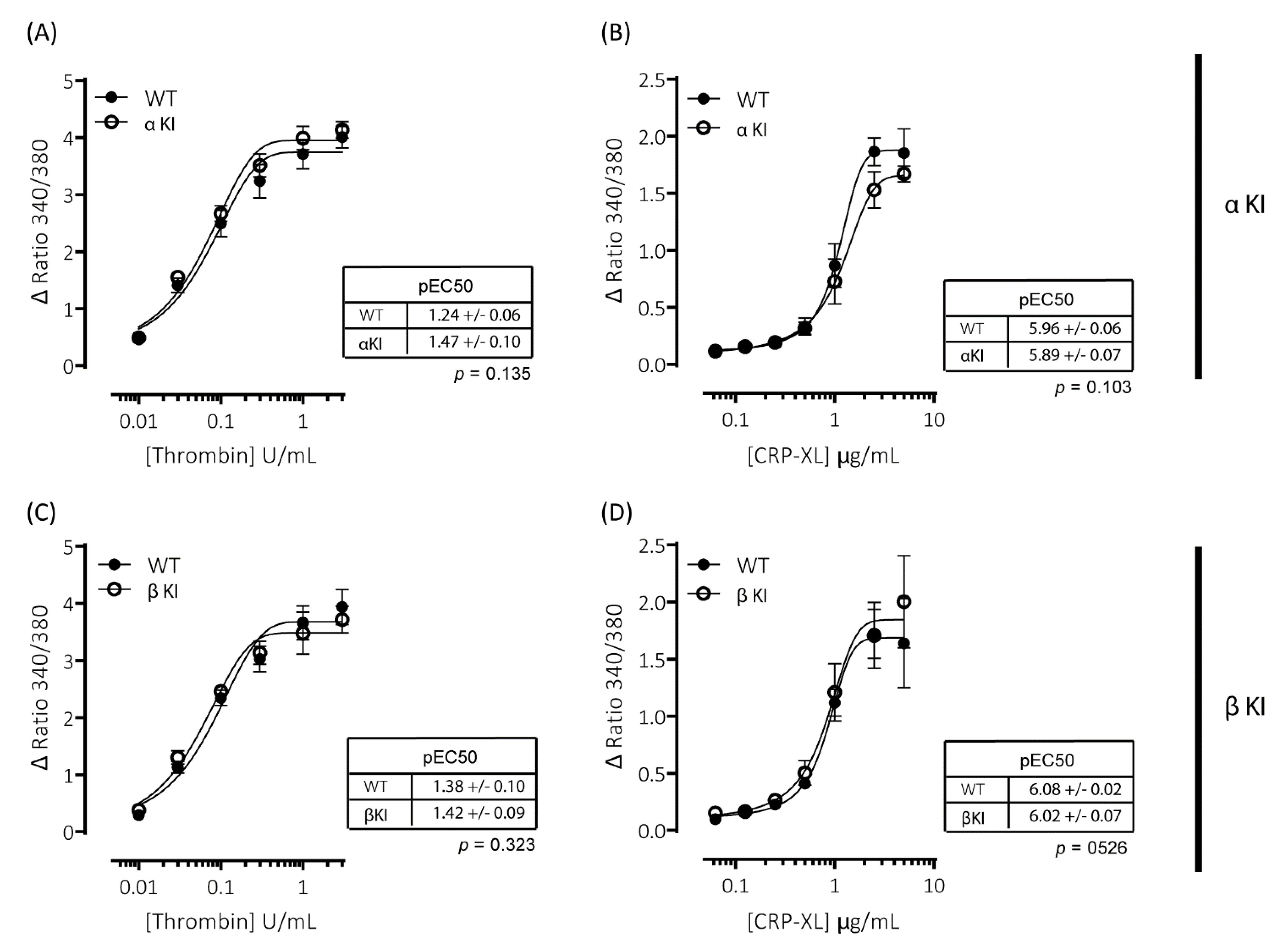
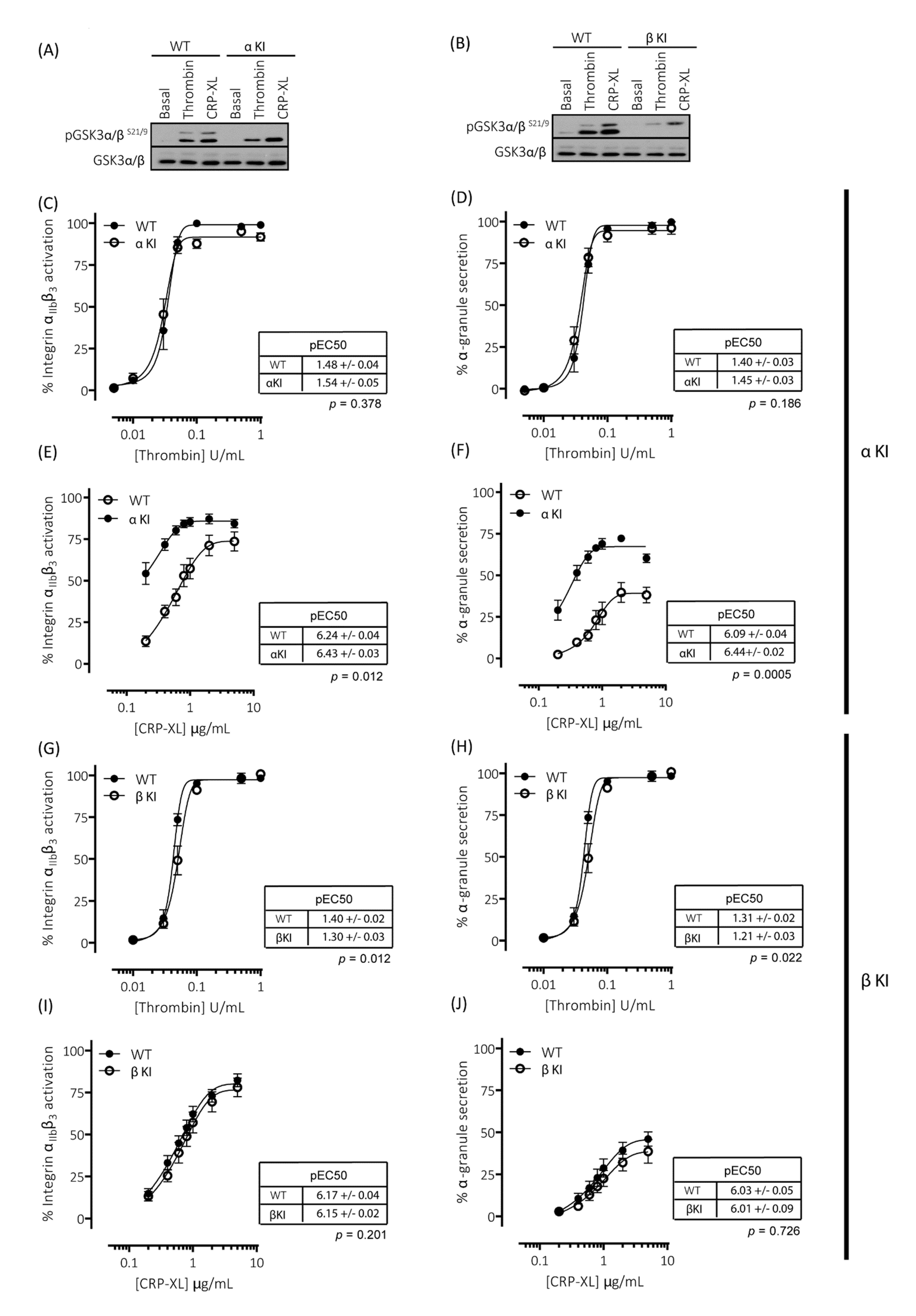
2.5. Expression of Phosphorylation-Resistant GSK3α Underlies Enhanced GPVI-Mediated Platelet Activation
2.6. Expression of Phosphorylation-Resistant GSK3β Underlies Reduced Thrombin-Mediated Platelet Activation
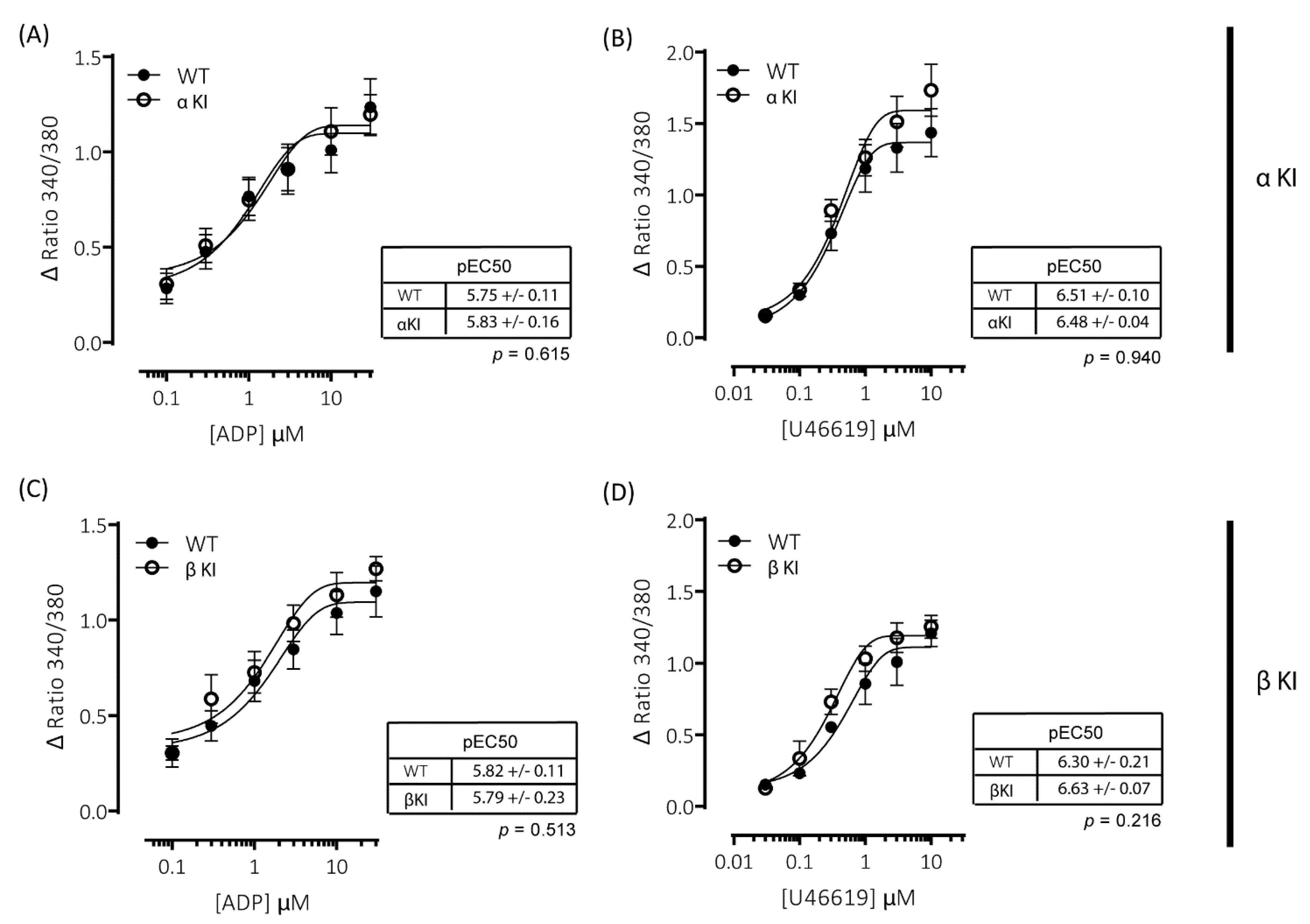
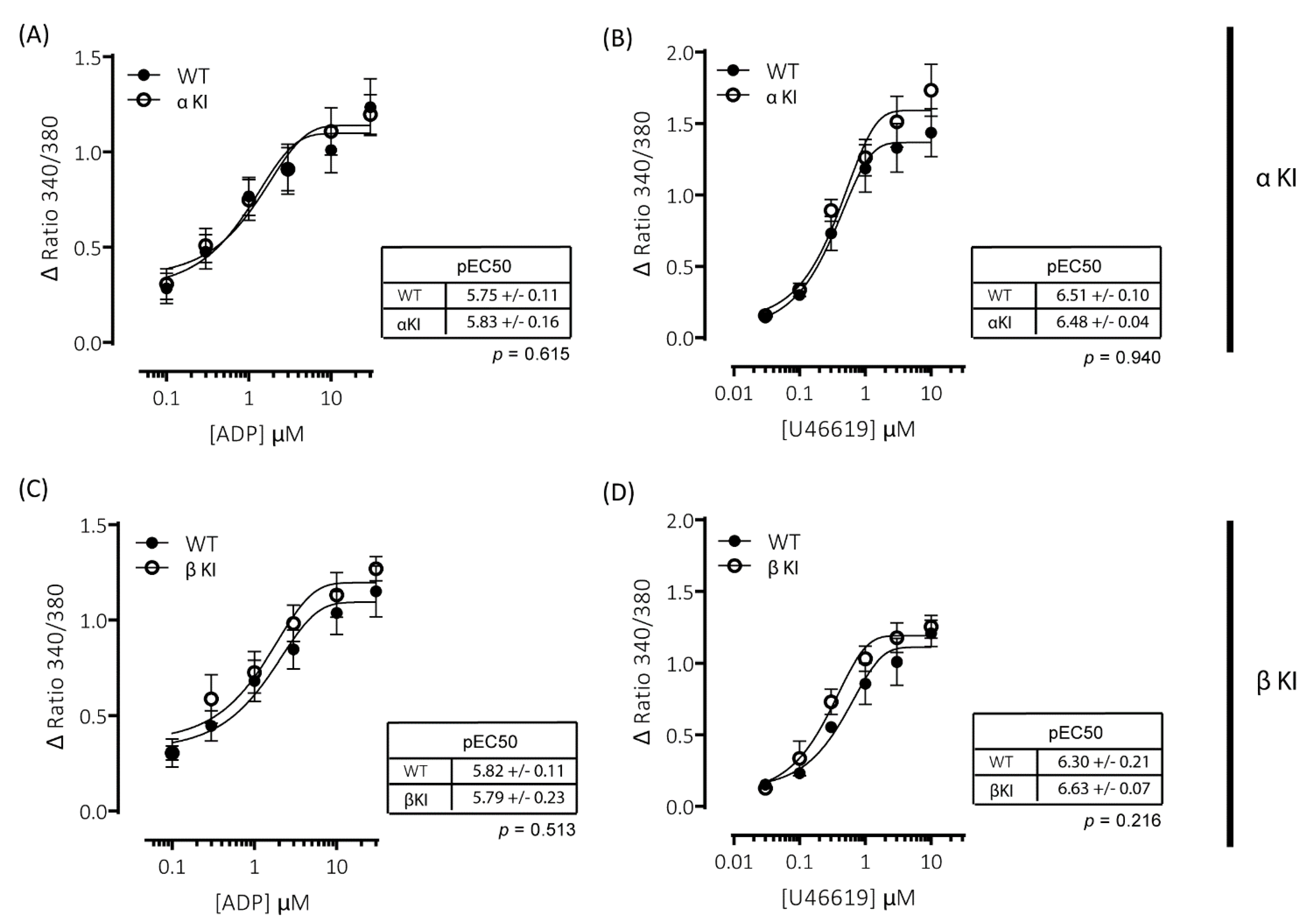
2.7. Intracellular Ca2+ Signalling Is Unaltered in Platelets Expressing Phosphorylation-Resistant GSKα or GSKβ
2.8. Pathways Involved in Amplification of Platelet Function Are Unaltered in Platelets Expressing Phosphorylation-Resistant GSKα or GSK3β
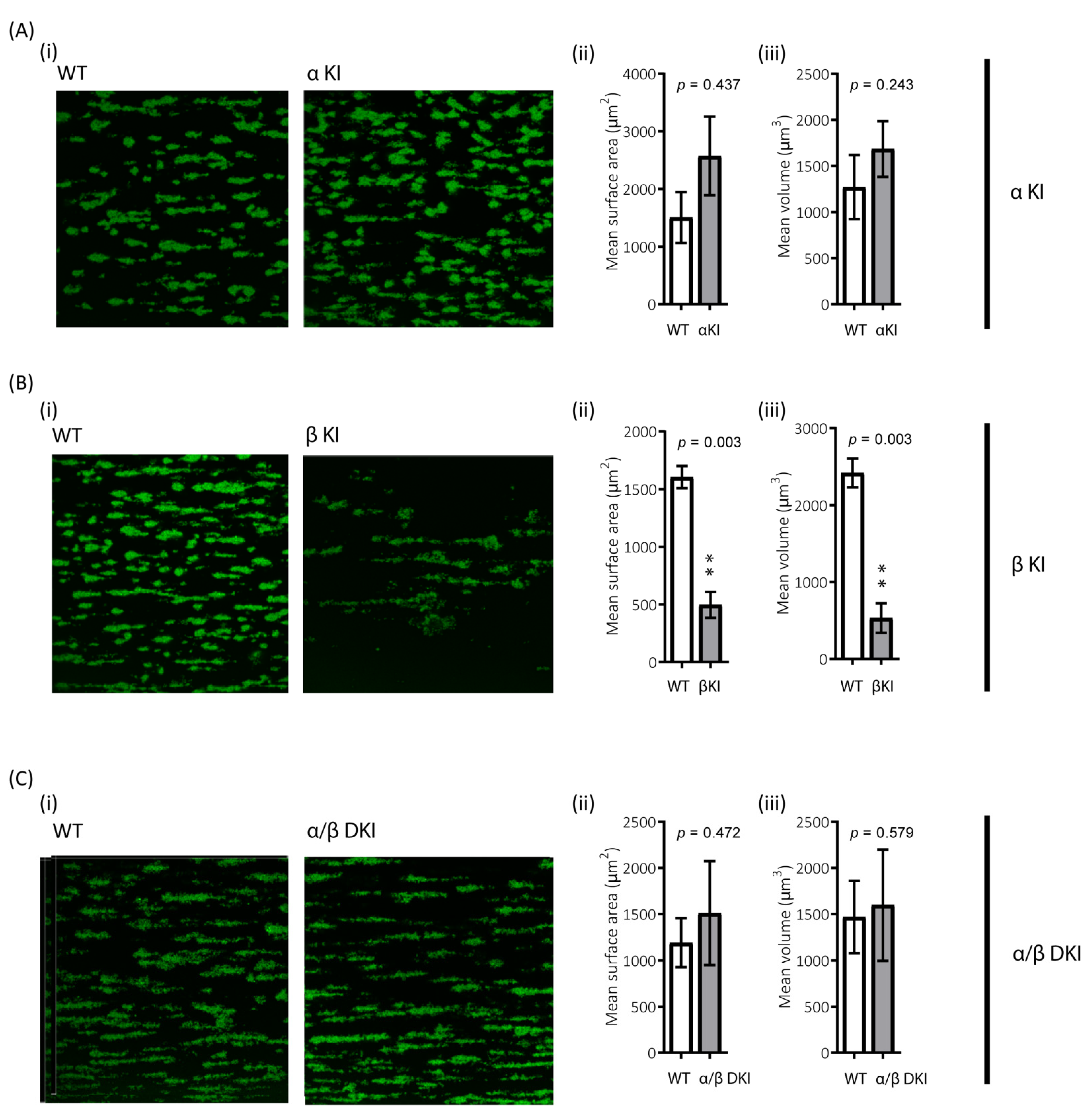
2.9. Role of GSK3α/β Phosphorylation in Thrombus Formation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Mice
4.3. Platelet Isolation
4.4. Protein Extraction and Immunoblotting
4.5. Flow Cytometry
4.6. Platelet Aggregation
4.7. Localisation of GSK3 Isoforms
4.8. Intracellular Ca2+ Measurements
4.9. δ-Granule Secretion
4.10. TxA2 Generation
4.11. In Vitro Thrombus Formation
4.12. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Guidetti, G.F.; Canobbio, I.; Torti, M. PI3K/Akt in platelet integrin signaling and implications in thrombosis. Adv. Biol. Regul. 2015, 59, 36–52. [Google Scholar] [CrossRef]
- Ribes, A.; Oprescu, A.; Viaud, J.; Hnia, K.; Chicanne, G.; Xuereb, J.M.; Severin, S.; Gratacap, M.P.; Payrastre, B. Phosphoinositide 3-kinases in platelets, thrombosis and therapeutics. Biochem. J. 2020, 477, 4327–4342. [Google Scholar] [CrossRef]
- Laurent, P.A.; Severin, S.; Gratacap, M.P.; Payrastre, B. Class I PI 3-kinases signaling in platelet activation and thrombosis: PDK1/Akt/GSK3 axis and impact of PTEN and SHIP1. Adv. Biol. Regul. 2014, 54, 162–174. [Google Scholar] [CrossRef]
- Durrant, T.N.; Hutchinson, J.L.; Heesom, K.J.; Anderson, K.E.; Stephens, L.R.; Hawkins, P.T.; Marshall, A.J.; Moore, S.F.; Hers, I. In-depth PtdIns(3,4,5)P3 signalosome analysis identifies DAPP1 as a negative regulator of GPVI-driven platelet function. Blood Adv. 2017, 1, 918–932. [Google Scholar] [CrossRef] [Green Version]
- Durrant, T.N.; Moore, S.F.; Bayliss, A.L.; Jiang, Y.; Aitken, E.W.; Wilson, M.C.; Heesom, K.J.; Hers, I. Identification of PtdIns(3,4)P2 effectors in human platelets using quantitative proteomics. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2020, 1865, 158575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battram, A.M.; Durrant, T.N.; Agbani, E.O.; Heesom, K.J.; Paul, D.S.; Piatt, R.; Poole, A.W.; Cullen, P.J.; Bergmeier, W.; Moore, S.F.; et al. The Phosphatidylinositol 3,4,5-trisphosphate (PI(3,4,5)P3) Binder Rasa3 Regulates Phosphoinositide 3-kinase (PI3K)-dependent Integrin alphaIIbbeta3 Outside-in Signaling. J. Biol. Chem. 2017, 292, 1691–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanini, L.; Paul, D.S.; Robledo, R.F.; Chan, E.R.; Getz, T.M.; Campbell, R.A.; Kechele, D.O.; Casari, C.; Piatt, R.; Caron, K.M.; et al. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J. Clin. Investig. 2015, 125, 1419–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, K.A.; Stojanovic-Terpo, A.; Hay, N.; Du, X. An important role for Akt3 in platelet activation and thrombosis. Blood 2011, 118, 4215–4223. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.F.; van den Bosch, M.T.; Hunter, R.W.; Sakamoto, K.; Poole, A.W.; Hers, I. Dual regulation of glycogen synthase kinase 3 (GSK3)alpha/beta by protein kinase C (PKC)alpha and Akt promotes thrombin-mediated integrin alphaIIbbeta3 activation and granule secretion in platelets. J. Biol. Chem. 2013, 288, 3918–3928. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; De, S.; Damron, D.S.; Chen, W.S.; Hay, N.; Byzova, T.V. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood 2004, 104, 1703–1710. [Google Scholar] [CrossRef] [Green Version]
- Woulfe, D.; Jiang, H.; Morgans, A.; Monks, R.; Birnbaum, M.; Brass, L.F. Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J. Clin. Investig. 2004, 113, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Durrant, T.N.; Hers, I. PI3K inhibitors in thrombosis and cardiovascular disease. Clin. Transl. Med. 2020, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990, 9, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Frame, S.; Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 2001, 359 Pt 1, 1–16. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296 Pt 1, 15–19. [Google Scholar] [CrossRef]
- Sutherland, C.; Cohen, P. The alpha-isoform of glycogen synthase kinase-3 from rabbit skeletal muscle is inactivated by p70 S6 kinase or MAP kinase-activated protein kinase-1 in vitro. FEBS Lett. 1994, 338, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Frame, S.; Cohen, P.; Biondi, R.M. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell. 2001, 7, 1321–1327. [Google Scholar] [CrossRef]
- Li, D.; August, S.; Woulfe, D. GSK3beta is a negative regulator of platelet function and thrombosis. Blood 2008, 111, 3522–3530. [Google Scholar] [CrossRef] [Green Version]
- Barry, F.A.; Graham, G.J.; Fry, M.J.; Gibbins, J.M. Regulation of glycogen synthase kinase 3 in human platelets: A possible role in platelet function? FEBS Lett. 2003, 553, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.F.; Hunter, R.W.; Harper, M.T.; Savage, J.S.; Siddiq, S.; Westbury, S.K.; Poole, A.W.; Mumford, A.D.; Hers, I. Dysfunction of the PI3 kinase/Rap1/integrin alpha(IIb)beta(3) pathway underlies ex vivo platelet hypoactivity in essential thrombocythemia. Blood 2013, 121, 1209–1219. [Google Scholar] [CrossRef] [Green Version]
- Laurent, P.A.; Severin, S.; Hechler, B.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.P. Platelet PI3Kbeta and GSK3 regulate thrombus stability at a high shear rate. Blood 2015, 125, 881–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, K.A.; Gartner, T.K.; Hay, N.; Du, X. ADP-stimulated activation of Akt during integrin outside-in signaling promotes platelet spreading by inhibiting glycogen synthase kinase-3beta. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2232–2240. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Zhang, W.; Zhu, C.; Liu, J.; Chen, Q. FUNDC2 regulates platelet activation through AKT/GSK-3beta/cGMP axis. Cardiovasc. Res. 2019, 115, 1672–1679. [Google Scholar] [CrossRef]
- Rowley, J.W.; Oler, A.J.; Tolley, N.D.; Hunter, B.N.; Low, E.N.; Nix, D.A.; Yost, C.C.; Zimmerman, G.A.; Weyrich, A.S. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood 2011, 118, e101–e111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiler, M.; Moser, M.; Mann, M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol. Cell. Proteom. 2014, 13, 3435–3445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005, 24, 1571–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moroi, A.J.; Watson, S.P. Impact of the PI3-kinase/Akt pathway on ITAM and hemITAM receptors: Haemostasis, platelet activation and antithrombotic therapy. Biochem. Pharmacol. 2015, 94, 186–194. [Google Scholar] [CrossRef] [Green Version]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, T.; Zhai, P.; Maejima, Y.; Hong, C.; Gao, S.; Tian, B.; Goto, K.; Takagi, H.; Tamamori-Adachi, M.; Kitajima, S.; et al. Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc. Natl. Acad. Sci. USA 2008, 105, 20900–20905. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.M.; Park, I.K.; Fiol, C.J.; Roach, P.J.; DePaoli-Roach, A.A. Isoform differences in substrate recognition by glycogen synthase kinases 3 alpha and 3 beta in the phosphorylation of phosphatase inhibitor 2. Biochemistry 1994, 33, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Caspi, M.; Zilberberg, A.; Eldar-Finkelman, H.; Rosin-Arbesfeld, R. Nuclear GSK-3beta inhibits the canonical Wnt signalling pathway in a beta-catenin phosphorylation-independent manner. Oncogene 2008, 27, 3546–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, A.; Lin, K.; Zhang, S.; Chen, Y.; Zhang, N.; Xue, J.; Wang, Z.; Aldape, K.D.; Xie, K.; Woodgett, J.R.; et al. Nuclear GSK3beta promotes tumorigenesis by phosphorylating KDM1A and inducing its deubiquitylation by USP22. Nat. Cell Biol. 2016, 18, 954–966. [Google Scholar] [CrossRef] [Green Version]
- Bechard, M.; Dalton, S. Subcellular localization of glycogen synthase kinase 3beta controls embryonic stem cell self-renewal. Mol. Cell. Biol 2009, 29, 2092–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moroi, A.J.; Watson, S.P. Akt and mitogen-activated protein kinase enhance C-type lectin-like receptor 2-mediated platelet activation by inhibition of glycogen synthase kinase 3alpha/beta. J. Thromb. Haemost. 2015, 13, 1139–1150. [Google Scholar] [CrossRef] [Green Version]
- Munnix, I.C.; Kuijpers, M.J.; Auger, J.; Thomassen, C.M.; Panizzi, P.; van Zandvoort, M.A.; Rosing, J.; Bock, P.E.; Watson, S.P.; Heemskerk, J.W. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: Regulation by transient integrin activation. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2484–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Gamez, G.; Myers, D.R.; Clemmons, W.; Lam, W.A.; Jobe, S.M. Mitochondrially mediated integrin alphaIIbbeta3 protein inactivation limits thrombus growth. J. Biol. Chem. 2013, 288, 30672–30681. [Google Scholar] [CrossRef] [Green Version]
- Loyau, S.; Dumont, B.; Ollivier, V.; Boulaftali, Y.; Feldman, L.; Ajzenberg, N.; Jandrot-Perrus, M. Platelet glycoprotein VI dimerization, an active process inducing receptor competence, is an indicator of platelet reactivity. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 778–785. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Westein, E.; Niego, B.; Hagemeyer, C.E. Shear-Dependent Platelet Aggregation: Mechanisms and Therapeutic Opportunities. Front. Cardiovasc. Med. 2019, 6, 141. [Google Scholar] [CrossRef]
- Steele, L.; Mannion, A.J.; Shaw, G.; Maclennan, K.A.; Cook, G.P.; Rudd, C.E.; Taylor, A. Non-redundant activity of GSK-3alpha and GSK-3beta in T cell-mediated tumor rejection. iScience 2021, 24, 102555. [Google Scholar] [CrossRef]
- Pardo, M.; Abrial, E.; Jope, R.S.; Beurel, E. GSK3beta isoform-selective regulation of depression, memory and hippocampal cell proliferation. Genes Brain Behav. 2016, 15, 348–355. [Google Scholar] [CrossRef]
- Li, J.; Ma, S.; Chen, J.; Hu, K.; Li, Y.; Zhang, Z.; Su, Z.; Woodgett, J.R.; Li, M.; Huang, Q. GSK-3beta Contributes to Parkinsonian Dopaminergic Neuron Death: Evidence From Conditional Knockout Mice and Tideglusib. Front. Mol. Neurosci. 2020, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, R.; Goswami, S.; Dey, S.; Gangoda, M.; Brothag, C.; Eisa, A.; Woodgett, J.; Phiel, C.; Kline, D.; Vijayaraghavan, S. Isoform-specific requirement for GSK3alpha in sperm for male fertility. Biol. Reprod. 2018, 99, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Werstuck, G. Characterizing the Role of Glycogen Synthase Kinase-3alpha/beta in Macrophage Polarization and the Regulation of Pro-Atherogenic Pathways in Cultured Ldlr(-/-) Macrophages. Front. Immunol. 2021, 12, 676752. [Google Scholar] [CrossRef]
- Ahmad, F.; Woodgett, J.R. Emerging roles of GSK-3alpha in pathophysiology: Emphasis on cardio-metabolic disorders. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118616. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.; Hers, I. Insulin/IGF-1 hybrid receptor expression on human platelets: Consequences for the effect of insulin on platelet function. J. Thromb. Haemost. 2009, 7, 2123–2130. [Google Scholar] [CrossRef]
- Agbani, E.O.; Williams, C.M.; Hers, I.; Poole, A.W. Membrane Ballooning in Aggregated Platelets is Synchronised and Mediates a Surge in Microvesiculation. Sci. Rep. 2017, 7, 2770. [Google Scholar] [CrossRef] [Green Version]
- Blair, T.A.; Moore, S.F.; Walsh, T.G.; Hutchinson, J.L.; Durrant, T.N.; Anderson, K.E.; Poole, A.W.; Hers, I. Phosphoinositide 3-kinase p110alpha negatively regulates thrombopoietin-mediated platelet activation and thrombus formation. Cell. Signal. 2018, 50, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Wersall, A.; Williams, C.M.; Brown, E.; Iannitti, T.; Williams, N.; Poole, A.W. Mouse Platelet Ral GTPases Control P-Selectin Surface Expression, Regulating Platelet-Leukocyte Interaction. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 787–800. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | α/β DKI | |||||
|---|---|---|---|---|---|---|
| Mean | s.e.m | Mean | s.e.m | n | p-Value | |
| Platelets 103/mm3 | 903 | 107 | 744 | 52 | 9 | 0.48 |
| MPV mm3 | 5.31 | 0.05 | 5.48 | 0.14 | 9 | 0.38 |
| WBC 103/mm3 | 10.51 | 1.10 | 8.87 | 1.04 | 9 | 0.42 |
| RBC 106/mm3 | 10.06 | 0.24 | 9.37 | 0.30 | 9 | 0.06 |
| Integrin αII mfi | 9672 | 882 | 8793 | 1000 | 7 | 0.46 |
| Integrin α2 mfi | 896 | 87 | 884 | 79 | 7 | >0.99 |
| GP1bα mfi | 4498 | 439 | 4346 | 705 | 7 | 0.84 |
| GPVI mfi | 1211 | 89 | 1397 | 135 | 7 | 0.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, S.F.; Agbani, E.O.; Wersäll, A.; Poole, A.W.; Williams, C.M.; Zhao, X.; Li, Y.; Hutchinson, J.L.; Hunter, R.W.; Hers, I. Opposing Roles of GSK3α and GSK3β Phosphorylation in Platelet Function and Thrombosis. Int. J. Mol. Sci. 2021, 22, 10656. https://doi.org/10.3390/ijms221910656
Moore SF, Agbani EO, Wersäll A, Poole AW, Williams CM, Zhao X, Li Y, Hutchinson JL, Hunter RW, Hers I. Opposing Roles of GSK3α and GSK3β Phosphorylation in Platelet Function and Thrombosis. International Journal of Molecular Sciences. 2021; 22(19):10656. https://doi.org/10.3390/ijms221910656
Chicago/Turabian StyleMoore, Samantha F., Ejaife O. Agbani, Andreas Wersäll, Alastair W. Poole, Chris M. Williams, Xiaojuan Zhao, Yong Li, James L. Hutchinson, Roger W. Hunter, and Ingeborg Hers. 2021. "Opposing Roles of GSK3α and GSK3β Phosphorylation in Platelet Function and Thrombosis" International Journal of Molecular Sciences 22, no. 19: 10656. https://doi.org/10.3390/ijms221910656
APA StyleMoore, S. F., Agbani, E. O., Wersäll, A., Poole, A. W., Williams, C. M., Zhao, X., Li, Y., Hutchinson, J. L., Hunter, R. W., & Hers, I. (2021). Opposing Roles of GSK3α and GSK3β Phosphorylation in Platelet Function and Thrombosis. International Journal of Molecular Sciences, 22(19), 10656. https://doi.org/10.3390/ijms221910656