1. Introduction
Platelets are small anucleate cells of the hematopoietic system and play a major role in hemostasis. At sites of vascular injury, platelets adhere to the damaged vessel wall to form a hemostatic plug important to avoid blood loss [
1]. However, uncontrolled platelet activation can trigger vessel occlusion leading to myocardial infarction or stroke [
2]. Injury of the endothelium induces the exposure of the extracellular matrix (ECM) that serves as substrate to initiate the activation, adhesion, and aggregation of circulating platelets [
3,
4].
The primary physical support for endothelial cells is provided by the vascular endothelial basement membrane and is exposed to circulating platelets following denudation of the endothelium. ECM proteins of the basement membrane are laminin, fibronectin, entactin, several proteoglycans, and collagen type IV [
4]. The layers below the basement membrane vary according to the vessel type [
3]. The smooth muscle layer between the basement membrane and the internal elastic lamina has been termed vascular ECM and is composed of elastin, microfibrils, collagens, fibronectin, proteoglycans, and vitronectin. The ECM proteins that mediate platelet adhesion and activation vary depending on the type of vessel and the severity of the injury. The major components of the vascular matrix are fibrillar collagens (type I and III) known to capture circulating von Willebrand factor (VWF) to mediate adhesion and activation of platelets through GPVI and integrin α
2β
1 [
4]. It is well known that major components of the vascular extracellular matrix responsible for the interaction with platelets are laminin, fibronectin, vitronectin, and collagen. Moreover, different structural proteins such as VWF, fibrinogen, and thrombospondin are known to be recruited to the ECM upon injury [
3].
The subendothelial ECM regulates platelet adhesion and thrombus formation upon vessel injury. After denudation of endothelial cells, ECM proteins are exposed to the flowing blood, followed by capturing (tethering) of platelets to counter the high shear rates in arteries. Tethering of platelets is mediated by VWF binding to subendothelial collagens via glycoprotein (GP)Ib–IX–V and enables other ECM proteins with slower on-rate to interact with platelet membrane receptors [
5]. The interaction of fibrillar collagens with GPVI is a major pathway that induces the activation of platelets, including inside-out activation of integrins to allow stable platelet adhesion at sites of vessel injury via integrin α
IIbβ
3, α
2β
1, α
5β
1, and α
6β
1 [
4,
6].
Integrin α
2β
1 binds to collagen to induce stable adhesion especially under conditions of reduced VWF plasma concentrations [
4]. Moreover, integrin α
2β
1 is known to bind to the small proteoglycan decorin to induce platelet adhesion [
7]. Biglycan is another small proteoglycan of the ECM and is involved in cell adhesion and migration of fibroblasts and smooth muscle cells in the lung [
8,
9]. De novo synthesis of biglycan can be triggered in different cell types such as endothelial cells, smooth muscle cells, fibroblasts, and macrophages. Transforming growth factor-beta and platelet-derived growth factor regulate the production of biglycan [
10,
11]. In macrophages, IL-6 and IL-1β stimulate the synthesis of biglycan [
12]. Biglycan is found in the ECM of arterioles and capillaries and is important for thrombin generation upon inflammation and atherosclerosis [
13]. Beside its role as a component of the ECM, biglycan may act as a signaling molecule to stimulate proinflammatory signaling such as inflammatory responses to microbial invasion induced by biglycan-mediated clustering of several types of receptors on the cell surface [
14]. To this end, biglycan is implicated in kidney disease [
15], the regulation of inflammation and autophagy in cancer [
16], cardiac hypertrophy [
17], erythropoietin production [
18], autoimmune perimyocarditis [
19], and adaptive remodeling after myocardial infarction [
20]. However, the impact of small proteoglycans as ECM proteins of the vessel wall—especially of biglycan—in platelet adhesion and activation in hemostasis and arterial thrombosis is poorly understood. Thus, the present study explored the role and importance of biglycan for platelet adhesion and thrombus formation in vitro and in vivo.
3. Discussion
In this study, we have shown that genetic deletion of the small proteoglycan biglycan interferes with hemostasis and protects against arterial thrombosis induced by reduced platelet binding to the injured vessel. Mechanistically, platelets are capable to bind to immobilized and soluble biglycan to increase GPVI signaling via tyrosine phosphorylation, leading to platelet degranulation and β3-integrin activation important for platelet adhesion and thrombus formation. Therefore, the current findings add important new information about the role of small proteoglycans, especially of biglycan, in arterial thrombosis and hemostasis.
The impact of biglycan in cardiovascular diseases was already shown earlier. Loss of biglycan led to enhanced thrombin generation and platelet activation in ApoE-deficient mice, suggesting a role for biglycan in inflammation and atherosclerosis [
13]. In contrast, no major alterations were observed in thrombin plasma levels in biglycan-deficient mice on a C57BL/6 genetic background (
Supplementary Figure S3A–E), suggesting that the role of biglycan as regulator of thrombin activity by heparin cofactor II is more pronounced in proinflammatory, atherosclerotic mice compared to healthy mice. Accordingly, platelet activation was unaltered in biglycan-deficient mice compared to control mice (
Supplementary Figure S2B,C). In contrast to enhanced platelet adhesion at the uninjured carotid artery in ApoE/biglycan double-deficient mice, reduced platelet adhesion was measured at the ligated carotid artery in
Bgn-/0 mice in the present study. Therefore, it is apparent that biglycan has different roles in regulating platelet function in hypercholesterinemic ApoE-deficient mice compared to wildtype controls. Possible mechanisms may include the presence of an intact glycocalyx in the uninjured carotid artery of ApoE-deficient mice versus the destruction of the glycocalyx and exposure of the subendothelial matrix in the present model. In summary, both studies revealed that the function of biglycan in the glycocalyx varies compared to its functions in the arterial wall.
After myocardial infarction, biglycan plays a major role for stable collagen matrix formation and for the preservation of cardiac left ventricle function [
20]. In addition, an involvement of biglycan in aortic dissection of mice has been described [
22]. Different studies provided evidence that biglycan exerts proinflammatory effects via toll-like receptor (TLR)2 and TLR4, suggesting a role in pathogen-mediated and sterile inflammation [
12,
14,
23,
24,
25]. Here we provided evidence for a role of biglycan in platelet adhesion. So far, only one study exists that has analyzed the role of proteoglycans in platelet adhesion and activation. In specific, the small proteoglycan decorin was shown to support platelet adhesion and activation. Unfortunately, the authors did not analyze the impact of decorin in platelet adhesion and thrombus formation in arterial thrombosis and hemostasis in vivo [
7]. Interestingly, decorin was shown to bind to the collagen receptor α
2β
1 integrin to induce tyrosine phosphorylation. Here, we provided evidence for biglycan-mediated platelet adhesion and activation via the collagen receptor GPVI, because blocking of the receptor by antibody treatment reduced platelet adhesion to biglycan, collagen, and a mixed biglycan-collagen matrix. As biglycan is described to bind and stabilize the collagen fibrils in the ECM of the vessels [
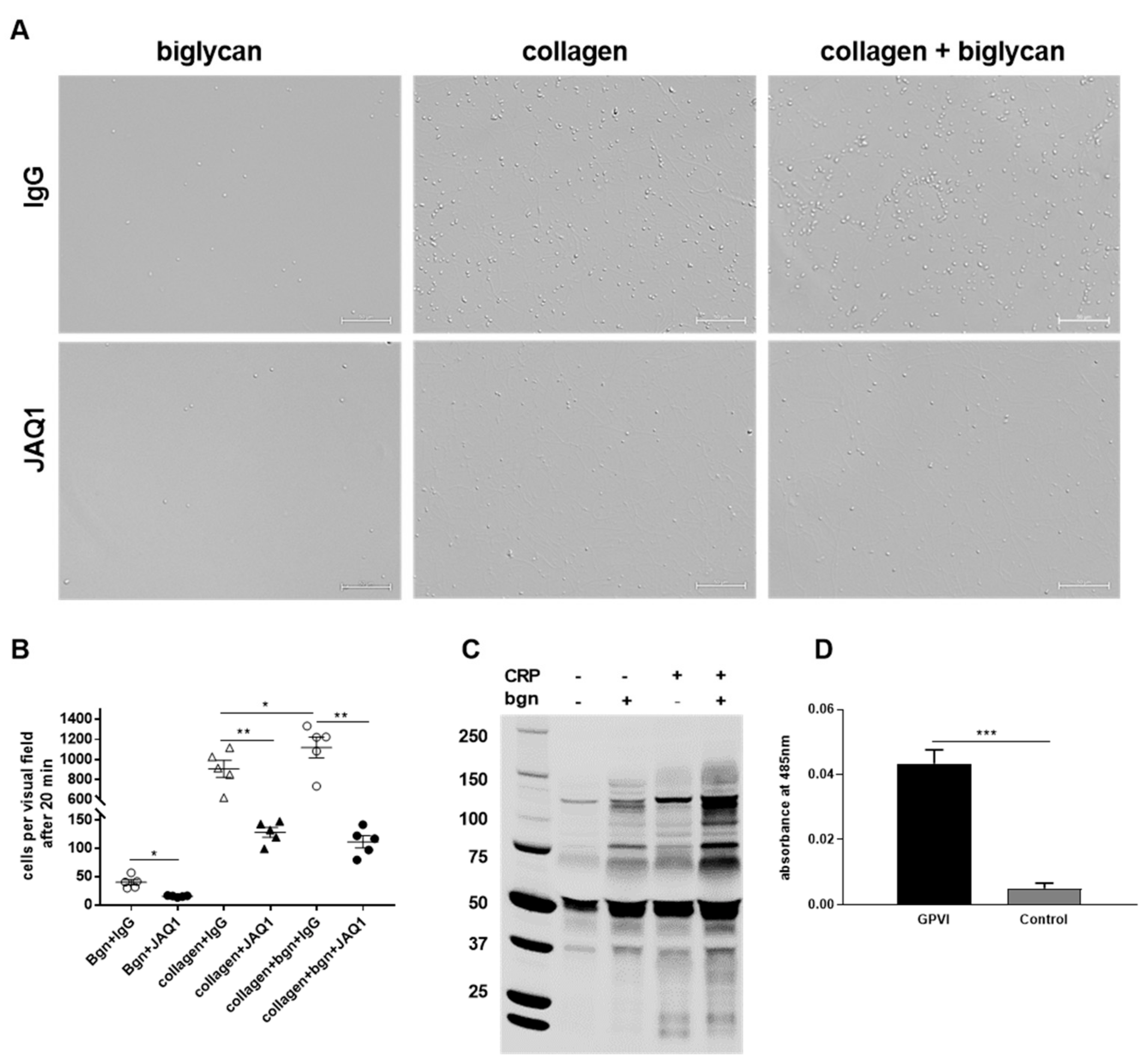
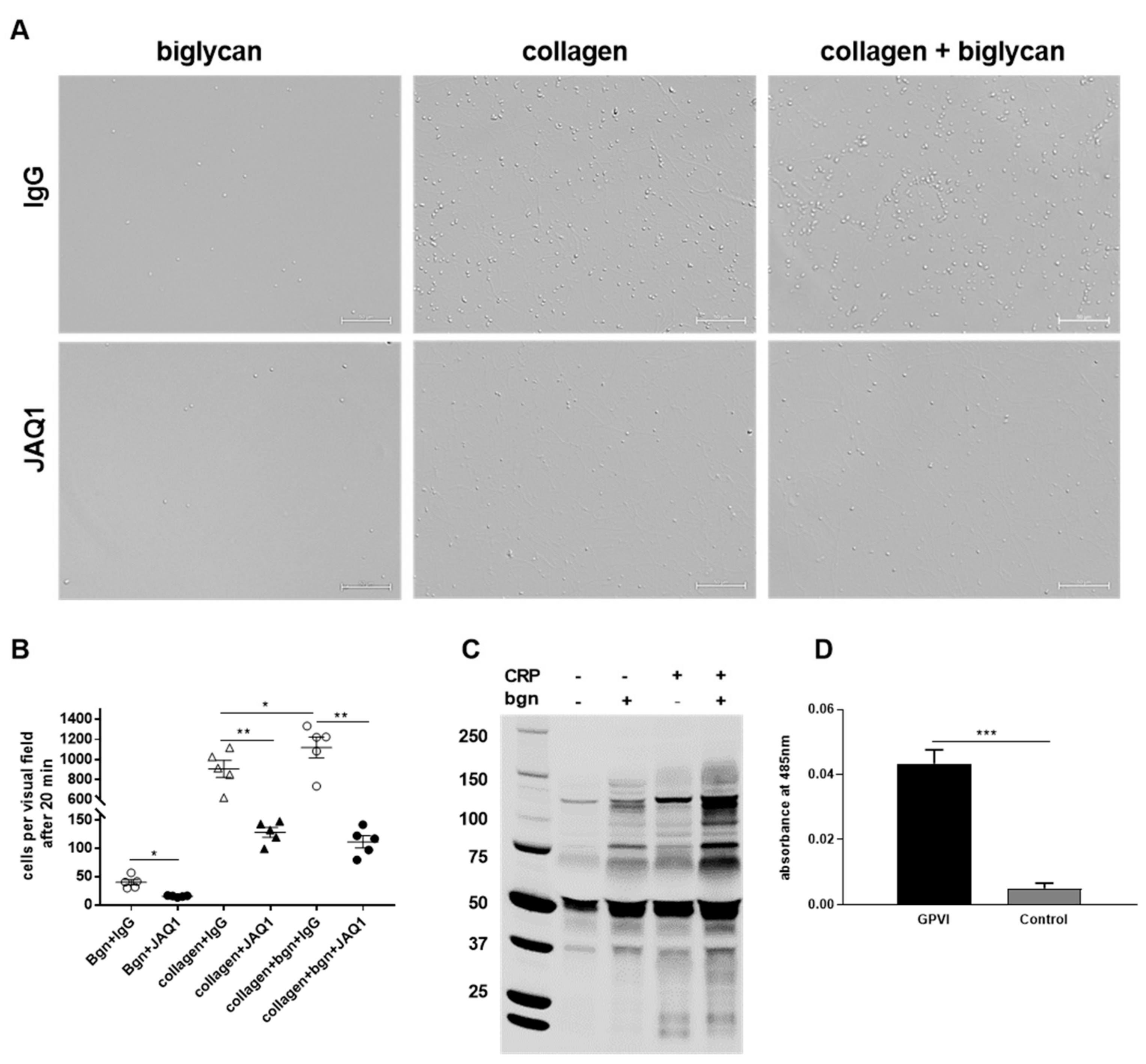
21] it can be feasible that biglycan functions as a co-factor for collagen binding to GPVI, thus supporting an enhancement in platelet adhesion, integrin activation, and thrombus formation. However, we observed significant differences in platelet adhesion to biglycan alone when we blocked GPVI with an antibody (JAQ1,
Figure 5B). This supports the hypothesis that biglycan is able to induce signaling via GPVI in the absence of collagen.
Furthermore, we detected tyrosine phosphorylation of biglycan in a GPVI-dependent manner. In contrast to Guidetti and colleagues, we found a role for biglycan in arterial thrombosis that is causative for myocardial infarction and stroke. Decreased platelet adhesion of biglycan-deficient platelets to the injured carotid artery was observed at early and intermediate time points (5 and 10 min), while we were not able to detect any differences compared to control mice after 30 min (data not shown), suggesting that the defects induced by the loss of biglycan can be compensated over time. Strikingly, defects in hemostasis were observed in individual biglycan-deficient mice, while others were able to compensate the loss of biglycan and showed normal bleeding times compared to control mice. Importantly, measurements of bleeding time had to be terminated in biglycan-deficient mice with defective hemostasis after 570 s because of elevated blood loss, suggesting that mice, which cannot compensate for the loss of biglycan, exert strong platelet activation defects.
In the last years, new GPVI ligands have been identified, including diesel exhaust particles (DEP) and large polysaccharides such as fucoidan and dextran sulfate [
26]. Recently, fibrin has been identified as ligand for monomeric GPVI [
27]. For the first time, we provide evidence for biglycan as novel ligand for GPVI in the presented study. First, tyrosine phosphorylation in a GPVI-dependent manner was detected when we stimulated platelets with soluble biglycan. Second, antibody-mediated inhibition of GPVI reduced the adhesion of platelets to immobilized biglycan, suggesting that biglycan can exert its effects via GPVI in an immobilized as well as in a soluble conformation. Direct binding of biglycan and GPVI was initially assumed when we measured reduced platelet adhesion to a pure biglycan matrix in the presence of GPVI-blocking antibodies but in the absence of collagen, which suggests a direct and collagen-independent interaction of biglycan and GPVI. Indeed, Microarray AVEXIS screening confirmed direct binding of biglycan and GPVI.
However, it is not clear if biglycan-induced effects on GPVI activation are mediated via GPVI clustering as shown for platelet adhesion to collagen that induced GPVI dimer clustering. In principle, biglycan is able to cluster several types of receptors and orchestrate their signaling. In a multiple receptor crosstalk, biglycan induces clustering of TRL2/4 and P2X7, which leads to proinflammatory signaling via TNF-α and IL-1β [
25]. Thus, it is tempting to speculate that biglycan exerts GPVI signaling via GPVI clustering. Interestingly, fibrin binds selectively to monomeric GPVI, in contrast to collagen, which binds primarily to dimeric GPVI [
28,
29]. Thus, further analysis of the mechanism of how biglycan affects GPVI signaling is important to validate if specific blocking of biglycan binding to GPVI might be a new antithrombotic target in future.
Beside its role in cardiovascular diseases, biglycan is important for the mineralization of bones [
30] and is highly expressed in cancer with metastatic activity and lower survival [
31]. Upon tissue stress or injury, the sequestered form of biglycan—that cannot act as a signaling molecule [
12]—becomes proteolytically released from the matrix to signal to the immune system [
32]. Binding of soluble biglycan to TLR2/4 mediates the activation of the NLRP3 inflammasome [
25], suggesting a prominent role for biglycan in different inflammatory diseases. Since platelets express TLR4 and bind to neutrophils in the capillaries of lung and liver upon sepsis [
33], it is tempting to speculate that soluble biglycan is able to bind to platelet TLR4 to mediate platelet-induced inflammatory responses. Furthermore, biglycan-mediated activation of the NLRP3 inflammasome includes the formation of reactive oxygen species (ROS) [
34,
35]. Interestingly, GPVI activation is also involved in ROS generation [
36], suggesting that biglycan might be able to induce ROS production of platelets via GPVI. Whether biglycan is also involved in platelet-mediated inflammation and/or ROS generation must be investigated in the future.
4. Materials and Methods
4.1. Animals
Gene-targeted mice lacking biglycan (Bgn-/0) and the corresponding wildtype (WT) littermates were bred from breeder pairs and genotyped by PCR. Experiments were performed with male mice aged 2–4 months. C57BL/6J mice (Janvier Labs, Le Genest-Saint-Isle, France) were used for blood withdrawal and organ removal. All animal experiments were conducted according to the Declaration of Helsinki and German law for the welfare of animals. The protocol was approved by the Heinrich-Heine-University Animal Care Committee and by the district government of North Rhine-Westphalia. Permit numbers: 84-02.04.2014.A303; 84-02.05.20.12.284; 84-02.04.2013.A210; 84-02.05.40.16.073.
4.2. Murine Platelet Preparation
Platelets were prepared as previously described [
37,
38]. Murine blood was withdrawn from the retro-orbital plexus under anesthesia and collected in 300 μL heparin (20 U/mL) (Ratiopharm GmbH, Ulm, Germany, # 002304). The blood was centrifuged at 250×
g for 5 min at 22 °C. The gained supernatant was centrifuged at 50×
g for 6 min to obtain platelet-rich plasma (PRP). PRP was washed twice at 650 g for 5 min. The remaining pellet was resuspended in murine Tyrode’s buffer (136 mM NaCl, 0.4 mM Na
2HPO
4, 2.7 mM KCl, 12 mM NaHCO
3, 0.1% glucose, 0.35% bovine serum albumin (BSA), pH 7.4), apyrase (0.02 U/mL) (Grade III, from potatoes, Sigma-Aldrich, Burlington, MA, USA, A7646), and prostacyclin (0.5 µM) (Calbiochem, San Diego, CA, USA) and centrifuged at 650×
g for 5 min. Depending on the following experiment, platelets were either resuspended in Tyrode’s buffer or Tyrode’s buffer plus added CaCl
2 (0.1 M).
4.3. Human Platelet Preparation
Blood samples from healthy volunteers were collected with the approval of the ethics committee of the Heinrich-Heine-University. Participants provided informed consent to their participation in the study (patient’s consent): Permit/Study Number 4669, ID No. 2014042327.
Platelets were prepared as previously described [
39]. Fresh citrate-anticoagulated blood was collected from healthy blood donors. The blood samples were centrifuged at 200×
g for 10 min. The PRP was separated, and phosphate-buffered saline (PBS) (pH 6.5, D8537), 2.5 U/mL apyrase Grade III from potatoes (Sigma-Aldrich, Burlington, MA, USA), and 1 μM prostacyclin (Calbiochem, San Diego, CA, USA, # 538925) were added in a 1:1 volumetric ratio and centrifuged at 1000×
g for 6 min. The platelet pellet was resuspended in human Tyrode’s buffer (137 mM NaCl, 2.8 mM KCl, 12 mM NaHCO
3, 0.4 mM Na
2HPO
4, 5.5 mM Glucose, 0.1% HIBSA, pH 7.4).
4.4. Microarray AVEXIS Screening
Printed slides were rinsed three times with MilliQ H2O and blocked with PBS (pH 6.5, # D8537) containing 1% BSA and 10 mM D-biotin for 45 min before interaction screening. Slides were incubated with normalized prey proteins for 1 h, washed three times in PBS/0.5% Tween, and incubated with 1:1000 anti-Flag horseradish peroxidase antibody (Sigma-Aldrich, Burlington, MA, USA) for 1 h. Detection was performed with TSA Alexa 555 substrate (Thermo Fisher Scientific Inc., Waltham, MA, USA) for 1 h. Slides were washed three times in PBS containing 0.1% Tween 20 with gentle rocking between all incubation steps. All steps were performed under condition of 22 °C. Identification of positive interactions was done by scanning slides with a ScanArray Express Microarray Scanner (PerkinElmer, Waltham, MA, USA) at a 550 nm wavelength.
4.5. Flow Cytometry
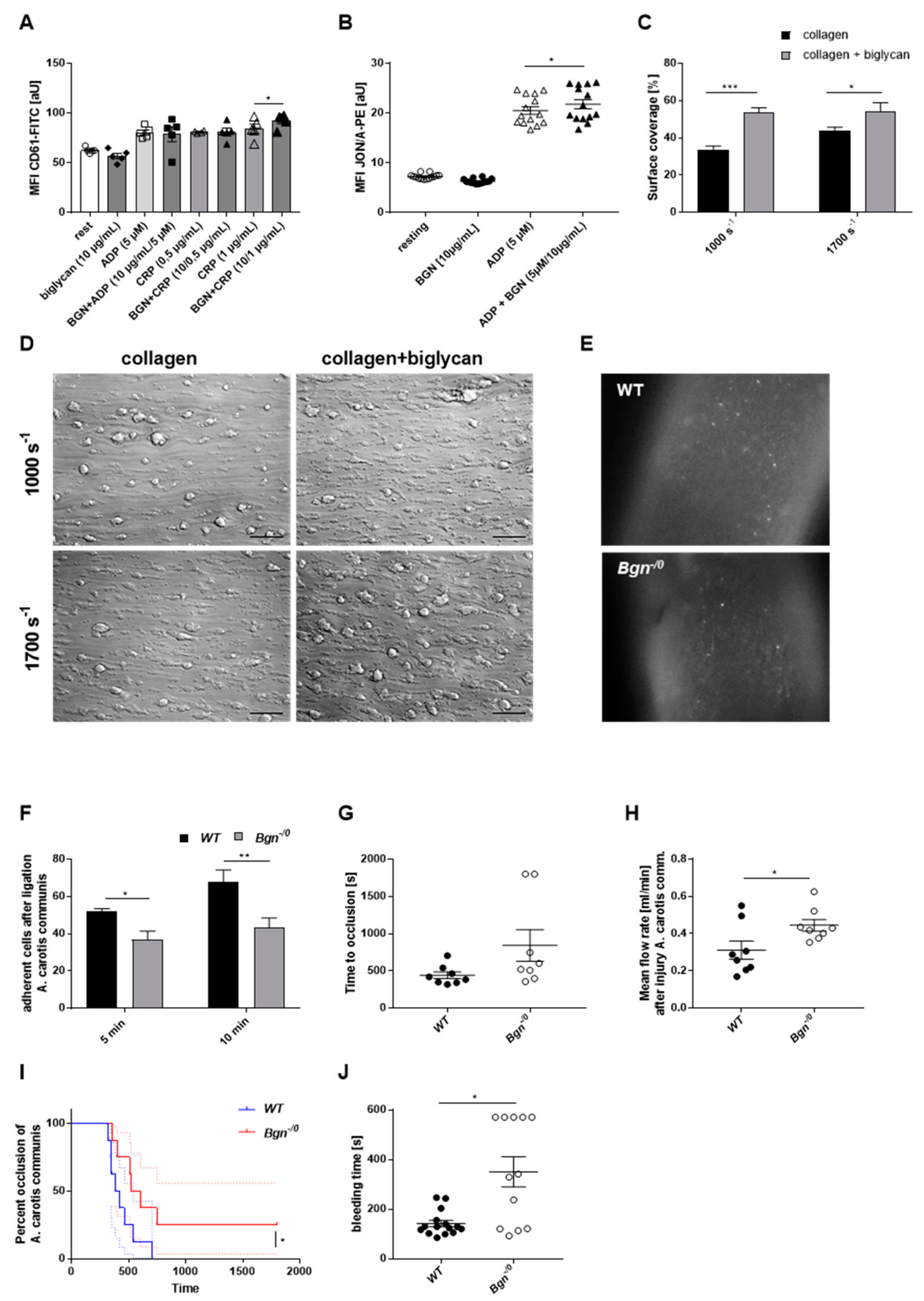
An amount of 100 μL murine blood was collected in 300 μL heparin (20 U/mL), then 500 μL murine Tyrode’s buffer was added and samples were centrifuged at 650× g for 5 min. The remaining pellet was resuspended in 500 μL Tyrode’s buffer and washed again two times as described above. Afterwards, the remaining pellet was resuspended in 500 μL Tyrode’s buffer supplemented with CaCl2 (0.1 M). Platelets were activated with one of the following agonists: collagen-related peptide (CRP)-XL (University of Cambridge, United Kingdom), Adenosine 5’-diphosphate (ADP) (Sigma-Aldrich, Burlington, MA, USA, # 01905), U46619 (Tocris Bioscience, Bristol, United Kingdom, # 1932), recombinant human biglycan (R&D Systems, Minneapolis, MI, USA, # 2667-CM-050), protease-activated receptor 4 (PAR-4) peptide (JPT Peptide Technologies GmbH, Berlin, Germany # 16035374). FITC-labeled rat anti-mouse P-selectin (JON/A-PE) monoclonal antibody (M130-1), PE-labeled rat anti-mouse integrin αIIbβ3, monoclonal antibody (M023-2), and FITC-labeled CD61 antibody (M031-1) (all purchased from Emfret Analytics, Würzburg, Germany) were used for labeling. The mean fluorescence intensity (MFI) was measured by flow cytometry.
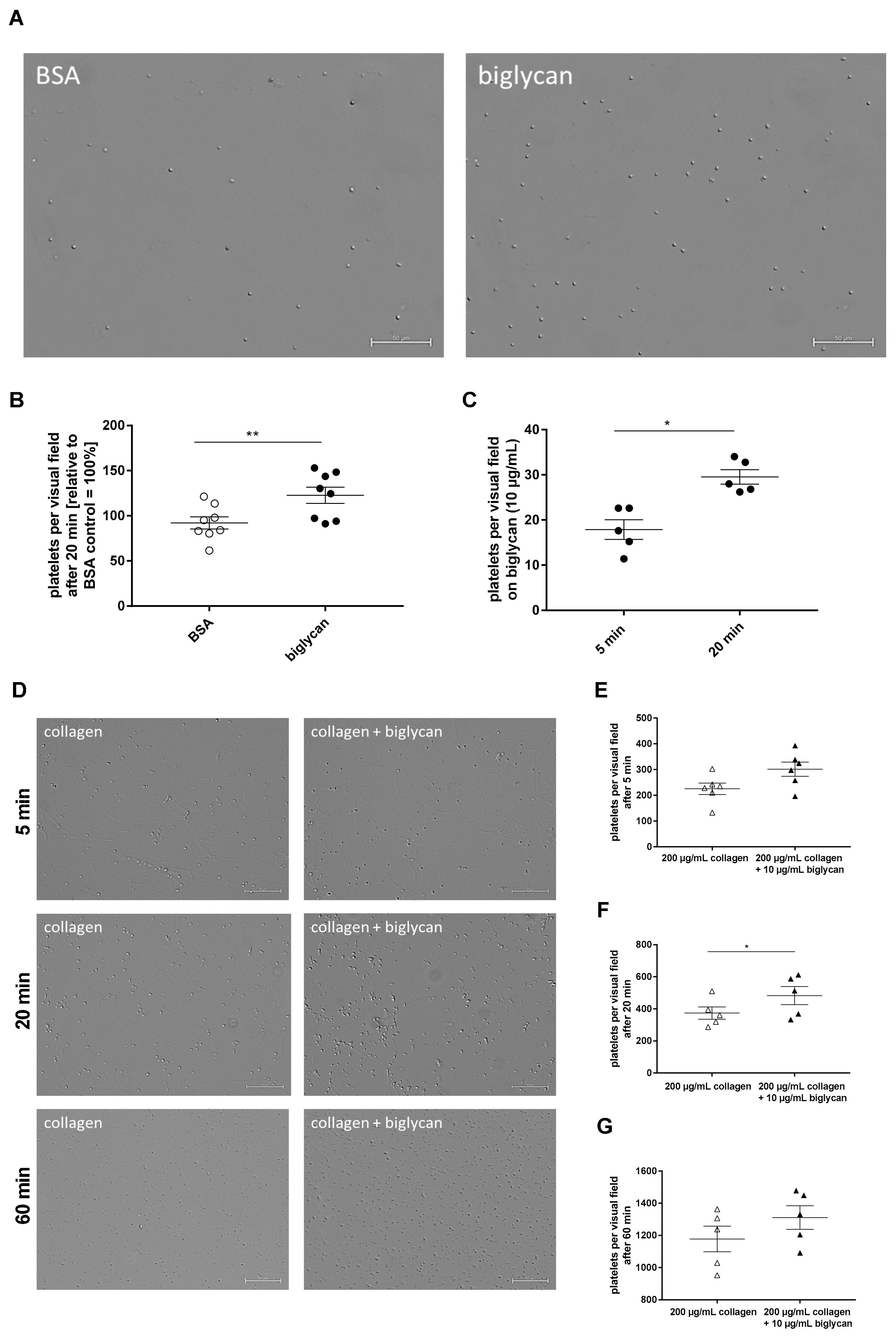
4.6. Platelet Adhesion and Spreading on Immobilized Recombinant Biglycan and Collagen
Experiments were performed as previously described [
37,
40]. Cover slips were coated with 200 μg/mL type I collagen (Takeda GmbH, Konstanz, Germany), 10 μg/mL recombinant human biglycan (R&D Systems, Minneapolis, MI, USA, # 2667-CM-050), 200 μg/mL type I collagen plus 10 μg/mL recombinant biglycan, or BSA (Sigma-Aldrich, Burlington, MA, USA, # A7906) at a defined area (10 × 10 mm) and incubated at 4 °C overnight. Afterwards, they were blocked with 1% BSA for 60 min. Isolated platelets (8 × 10
4) (see
Section 4.2) were resuspended in 70 μL Tyrode’s buffer supplemented with CaCl
2 (0.1 M), applicated on the prepared cover slips, and incubated at room temperature for indicated time points. Non-adherent platelets were carefully removed by rinsing with PBS (pH 6.5, D8537). The adherent platelets were fixed with phosphate-buffered formaldehyde (Roti
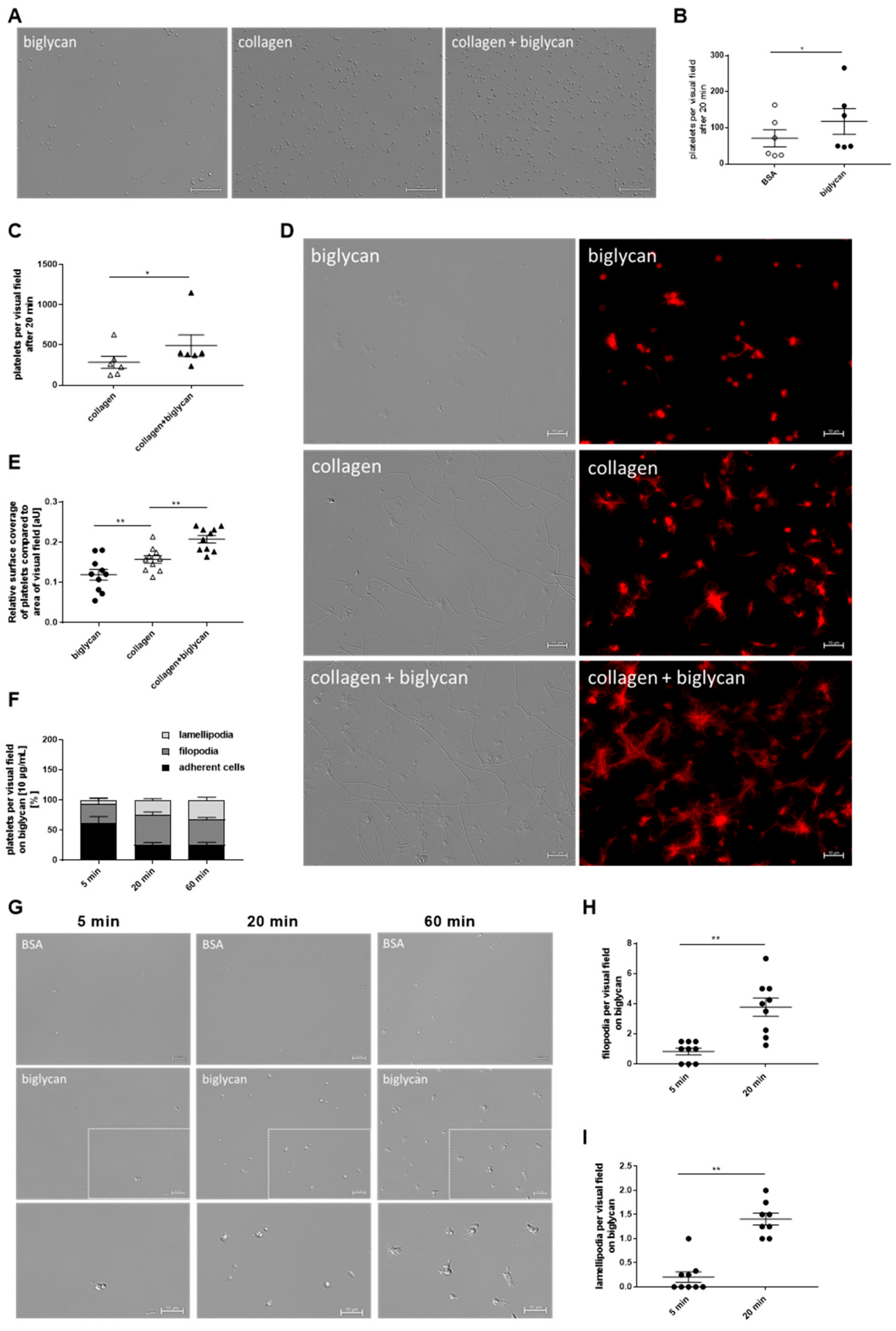
®-Histofix 4%, Carl Roth GmbH + Co. KG, Karlsruhe Germany), covered using Fluoromount™ (Sigma-Aldrich, Burlington, MA, USA, # F4680), and imaged using Microscope Axio Observer.D1 (Carl Zeiss Microscopy GmbH, Oberkochem, Germany, AxioCam MRm, objective LD Plan-Neofluar 40× 0.6 Korr. Ph2 M27) for 40 × magnification. The adherent cells were counted with ImageJ software. In case of the static adhesion assays with the GPVI inhibitor JAQ1, platelets were preincubated with 2 μg/mL JAQ1 (Emfret Analytics, Würzburg, Germany, # M011-0) and IgG control for 5 min prior to the application on the cover slips.
For spreading analysis of isolated human platelets, the adherent platelets were rinsed, fixed, and covered as described above. The samples were imaged using Microscope Axio Observer.D1 (Carl Zeiss Microscopy GmbH, Oberkochem, Germany, AxioCam MRm, objective Plan Apochromat 100×/1.40 Oil DIC M27) for differential interference contrast imaging. Platelets that show only filopodia were counted as filopodia-forming cells, platelets that start to form lamellipodia and fully spread platelets were counted as lamellipodia-forming platelets, and landing and attached platelets were counted as adherent cells according to Aslan and McCarty, 2012 [
41] using Carl Zeiss Software “ZEN 2012“ (blue edition).
For F-actin staining, 4 × 104 platelets were allowed to adhere on the above-described matrices for 20 min, rinsed with PBS, and fixed with PHEM buffer (100 mM PIPES, 5.25 mM HEPES, 10 mM EGTA, 20 mM MgCl2, pH 6.8), blocked with BSA (5%), washed with PBS (pH 6.5, D8537), and incubated with Alexa Phalloidin 568 (Thermo Fisher Scientific Inc., Waltham, MA, USA, # R415) overnight at 4 °C. The samples were rinsed and mounted using Fluoromount™ (Sigma-Aldrich, Burlington, MA, USA, # F4680), and visualized with the Microscope Axio Observer.D1 (Carl Zeiss Microscopy GmbH, Oberkochem, Germany, AxioCam MRm, objective Plan Apochromat 100×/1.40 Oil DIC M27) in differential interference contrast and Cy3 fluorescence microscopy (excitation wavelength 550/emission wavelength 570) channels. In this experiment, the surface coverage of the adherent platelets in relation to the total amount of platelets per visual field was analyzed using ImageJ software (ImageJ-win64).
4.7. Thrombus Formation in a Parallel-Plate Flow Chamber System
Thrombus formation under flow was analyzed using a parallel-plate flow chamber system as previously described [
39]. Cover slips were coated with 200 μg/mL type I collagen (Collagen Reagens HORM
®, Takeda GmbH, Konstanz, Germany) or 10 μg/mL recombinant human biglycan (R&D Systems, Minneapolis, MI, USA, # 2667-CM-050) and 200 μg/mL type I collagen at a defined area at 4 °C overnight. The coated surfaces were blocked with 1% BSA (Sigma-Aldrich, Burlington, MA, USA) for 60 min, and the cover slips were installed in the parallel-plate flow chamber system. Whole heparinized (20 U/mL) murine blood was pooled from mice with the same genotype to reach the requested amount of 0.9 mL. The blood was perfused through the flow chamber with continuous flow and different shear rates (1000 s
−1 and 1700 s
−1). After 3 min of flow, 5 pictures were taken from different representative areas (400× total magnification, Microscope Axio Observer.D1 (Carl Zeiss Microscopy GmbH, Oberkochem, Germany). The total surface coverage of the thrombi was analyzed by calculating the area covered by three-dimensional thrombi using the Carl Zeiss Software “ZEN 2012“ (blue edition).
4.8. Total Thrombus-Formation Analysis System (T-TAS®)
The T-TAS
® (Fujimori Kogyo Co. Ltd., Tokio, Japan) is a microchip-based whole blood flow chamber system to analyze in vitro thrombus formation [
42,
43,
44]. Whole blood collected from mice supplemented with Ca-Corn Trypsin Inhibitor (Fujimori Kogyo Co. Ltd., Tokio, Japan) was perfused over a microchip (AR-Chip) (Fujimori Kogyo Co. Ltd., Tokio, Japan) coated with collagen and thromboplastin at a flow rate of 240 s
−1 and 600 s
−1. An integrated pressure sensor continuously measured the pressure in the artificial vessel. In case of thrombus formation, the artificial vessel was occluded. At 100 kPa the experiment was ended owing to near complete occlusion of the artificial vessel due to thrombi, and the occlusion time (start of flow until 100 kPa was reached) was measured.
4.9. Histology and Fluorescence Microscopy
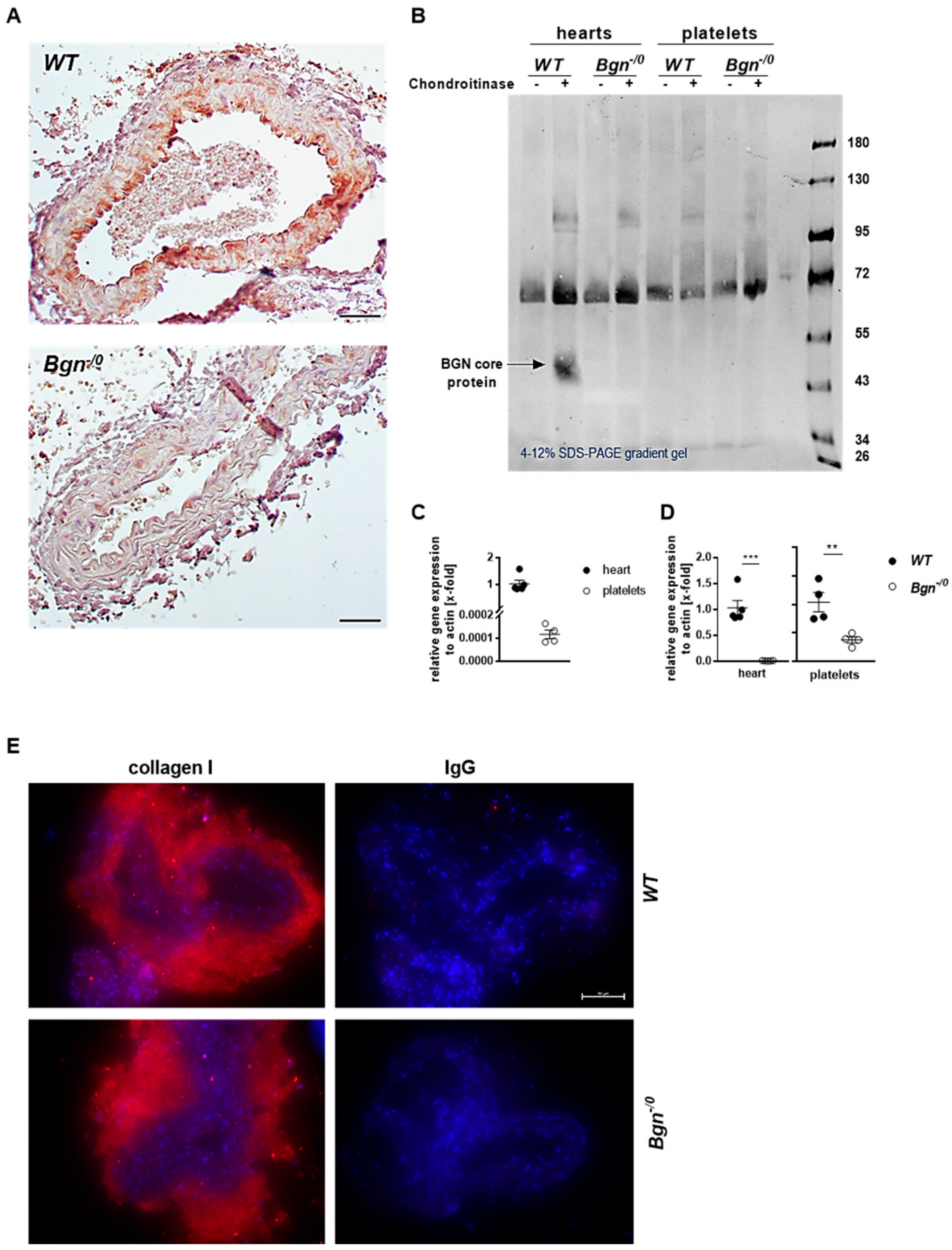
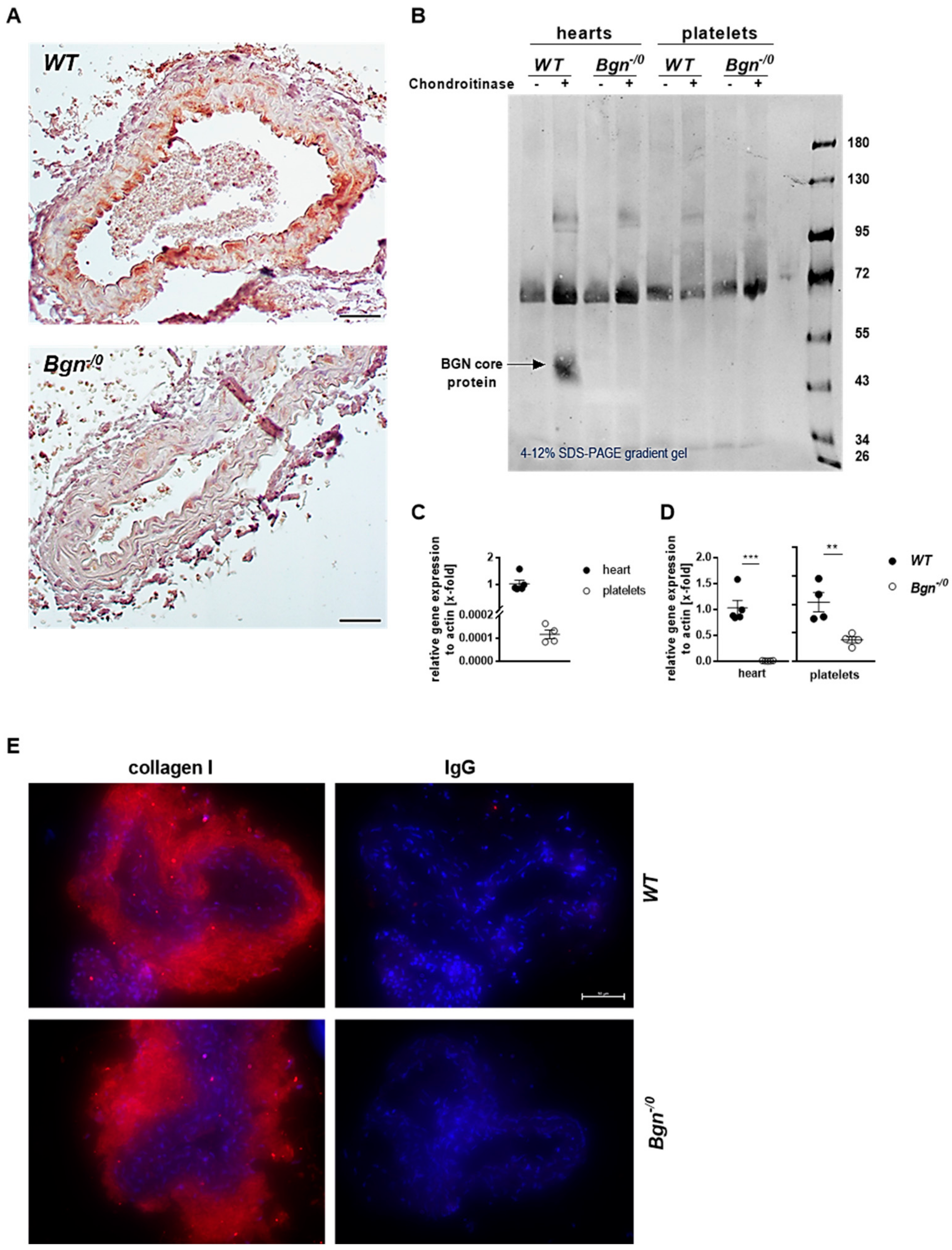
After FeCl3-induced injury, the occluded carotid artery was removed and embedded in paraffin. Paraffin sections were stained with biglycan-specific antibody (biglycan polyclonal rabbit antibody, abcam, Cambridge, United Kingdom, # 49701, 1:100) following the streptavidin–biotin immuno-peroxidase method (Dako, Glostrup, Denmark). Sample slices of healthy carotid arteries were prepared from paraffin-embedded samples, dewaxed and dehydrated, blocked (5% goat serum, 0.1% BSA and 0.3% triton (Sigma-Aldrich, Burlington, MA, USA, # T8787-100)), and incubated with antibodies against collagen I (rabbit anti-mouse collagen I antibody, Thermo Fisher Scientific Inc., Waltham, MA, USA, # PA1-85319) and collagen IV (rabbit polyclonal collagen IV antibody, abcam, Cambridge, United Kingdom, # ab6586). Then, secondary antibody incubation was performed for 1 h (Alexa Fluor™ 555 goat anti-rabbit IgG (Thermo Fisher Scientific Inc., Waltham, MA, USA # A21428)), and cell nuclei were stained with DAPI (Roche Diagnostics, Mannheim, Germany, final concentration 1.6 µg/mL) and inundated with FluoromountTM (Sigma-Aldrich, Burlington, MA, USA). The documentation was done after drying at 4 °C overnight with microscope Axio Observer.D1 (Carl Zeiss Microscopy GmbH, Oberkochem, Germany, AxioCam MRm, objective LD Plan-Neofluar 40× 0.6 Korr. Ph2 M27).
4.10. Biglycan ELISA
Biglycan levels in the supernatant of stimulated platelets were determined using an ELISA (Cusabio Technology LLC, Houstomn, TX, USA # CSB-EL002683MO) according to the manufacturer’s protocol. Blood from C57BL/6J mice was collected in 200 μL acid-citrate-dextrose buffer (85 mM Na3-Citrat, 71 mM citric acid, 2% glucose (all purchased from Carl Roth GmbH + Co.), pH 4.69), and platelets were isolated as described above. Platelets (7 × 105) were stimulated with CRP-XL (University of Cambridge, United Kingdom) or thrombin (Roche, Basel, Switzerland, # 10602400001) for 10 min at 37 °C, followed by centrifugation at 845× g for 10 min at 4 °C. Murine Tyrode’s buffer supplemented with CaCl2 (0.1 M) was added to the control group. The obtained supernatant was used for the ELISA.
4.11. RT-PCR
Isolation of RNA from murine platelets (C57BL/6J) was performed as described in 4.2. A minimum of 1 billion platelets were pooled from at least 3 mice for each biological replicate, while the purity of the platelet isolation was checked using a Sysmex cell analyzer. Subsequently, platelets were lysed using 500 µL TRI Reagent® (Sigma-Aldrich, Burlington, MA, USA, #T9424) incubated for 5 min at RT. Chloroform (100 µL) was added, samples were vortexed and incubated for 3 min at RT following centrifugation at 12,000× g at 4 °C. The upper phase was collected and mixed with 250 µL isopropanol and incubated for 10 min at RT following a centrifugation at 12,000× g at 4 °C. Afterwards, supernatant was discarded and the pellet was washed twice with 75% ethanol following centrifugation at 7500× g for 5 min. Finally, the pellet was dried for 5–10 min at RT and dissolved in 40 µL RNAse-free water. RNA concentration was measured with an Eppendorf BioPhotometer® D30 using 2 µL sample in an Eppendorf µCuvette® G1.0. Purity of the RNA was checked with the 260/230 nm and 260/280 nm ratio. Afterwards, samples were stored at −80 °C until use.
RNA isolation from murine hearts of wildtype and knockout mice was performed with the ReliaPrepTM RNA Tissue Miniprep System (Promega, Walldorf, Germany, # Z6111) according to the manufacturer’s protocol. Complementary DNA was synthesized by using the ImProm- II™ Reverse Transcription System Kit (Promega, Walldorf, Germany, # A3800) following the manufacturer’s protocol. The qRT-PCR was performed by using the 7500 Fast Real-Time PCR System (Applied Biosystems by Thermo Fisher Scientific, Waltham, MA, USA). SYBR Green (Fast SYBRTM Green Master Mix, Applied Biosystems by Thermo Fisher Scientific Inc., Waltham, MA, USA, # 4385162) was used as a fluorescence dye. Actin was used as housekeeping gene. Quantitative PCR amplification was performed using the following oligonucleotide primers: biglycan for “ctgagggaacttcacttgga” rev “cagatagacaacctggaggag”; actin for “ctaaggccaaccgtgaaaag” rev “accagaggcatacagggaca”.
4.12. Calibrated Automated Thrombography
The endogenous thrombin generation was determined using calibrated automated thrombography in murine platelet-poor plasma (PPP) as described previously by Tchaikovski et al. with slight modifications [
45]. Briefly, murine blood was anticoagulated with sodium citrate (final concentration 0.32%), followed by two times centrifugation at 21,000×
g for 10 min at 22 °C to obtain PPP. PPP-reagent LOW, thrombin calibrator, and FluCa-Kit were purchased from Stago Deutschland GmbH (Düsseldorf, Germany). PBS was used to dilute PPP-reagent LOW and thrombin calibrator each at a ratio of 10/15 with PBS. In the experiment, 25 μL of diluted PPP-reagent LOW or thrombin calibrator was mixed with 15 μL PPP and incubated for 10 min at 33 °C. Subsequently the reaction was started by adding 20 μL FluCa buffer. The measurement of the fluorescent signal in triplicates at 33 °C for 30 min was made with a Fluoroscan Ascent plate reader (excitation: 390 nm, emission: 460 nm; Thermo Labsystems), and automatic data analysis was performed with Thrombinoscope™ Software (Stago Deutschland GmbH, Düsseldorf, Germany).
4.13. Western Blot Analysis
Western blot analysis was performed as described earlier [
40] to detect GPVI-dependent phosphorylation in platelets upon incubation with recombinant human biglycan (R&D Systems, 2667-CM-050), and biglycan plus CRP. Platelets were separated by centrifugation, and lysates were made, separated on a gradient SDS polyacrylamide gel, and transferred onto nitrocellulose membrane. The membrane was blocked using 5% powdered skim milk in PBST (PBS with 0.1% Tween 20) and probed with anti-phosphotyrosine antibody, clone 4G10
® (Merck KgaA, Darmstadt, Germany, 05-321). The Novex Wedge Well kit (Thermo Fisher Scientific Inc., Waltham, MA, USA) was used as described in the manufacturers’ protocol.
4.14. Generation of Bone Marrow Chimera
The bone marrow donors (Bgn-/0 and wildtype mice) were sacrificed by cervical dislocation, and the bone marrow cells were isolated from the tibias and femurs. After centrifugation at 500× g for 10 min, the remaining pellet was resuspended in red blood cell lysis buffer (155 mM NH4Cl (Merck KgaA, Darmstadt, Germany), 10 mM KHCO3 (Sigma-Aldrich, Burlington, MA, USA), 0.1 mM EDTA (Sigma-Aldrich, Burlington, MA, USA), pH 7.2–7.4, adjusted with HCl (Merck KgaA, Darmstadt, Germany)), followed by centrifugation at 500× g for 10 min. The pellet was resuspended in 500 μL PBS (Sigma-Aldrich Burlington, MA, USA). The recipients of bone marrow cells (12-week-old Bgn-/0 mice and wildtype littermates) were preconditioned by irradiation with 10 gray (Gy) for 3.25 min using the Biobeam GM 2000 (Gamma-Service Medical GmbH, Leipzig, Germany). Irradiated Bgn-/0 mice received isolated bone marrow cells from wildtype littermates, and irradiated wildtype littermates received isolated bone marrow cells from Bgn-/0 mice (each 5 × 106 cells). The animals were treated with neomycin (65.2 mg/50 mL drinking water) for 2 weeks to reduce the risk of infections. Six weeks after transplantation, the blood cells of the chimera were genotyped to confirm the successful generation of bone marrow chimeric mice. Moreover, an automated hematology cell counter (KX-21N) (Sysmex Deutschland GmbH, Norderstedt, Germany) was used to confirm the reconstitution of blood cells.
4.15. Genotyping of Blood Cells
The preparation of the eluates was performed with the ReliaPrepTM Blood gDNA Miniprep System (Promega, Walldorf, Germany, A5081). The KAPA Mouse Genotyping Kit (KAPA Biosystems Hoffmann-La Roche, Basel, Switzerland) was used according to the manufacturer’s protocol for the PCR. The generated pro-ducts (Bgn-/0, 319 bp; wildtype littermates, 212 bp) were detected with gel electrophoresis using a 1% agarose gel (Sigma-Aldrich, Burlington, MA, USA, A9539-250G)/TRIS-Acetate-EDTA (TAE)-buffer (50 × (121 g Trisbase (Sigma-Aldrich, Burlington, MA, USA), 28.5 mL acetic acid (100%, Merck KgaA, Darmstadt, Germany), 50 mL 0.5 M EDTA (Sigma-Aldrich, Burlington, MA, USA) pH = 8.3)) and Midori Green Advance (Nippon Genetics Europe GmbH, Düren, Germany, 617004).
4.16. Determination of Bleeding Time
Bleeding times in mice were determined as described in Gowert et al. (2017) [
46]. Mice were anesthetized with Ketavet (90 mg/kg) (Zoetis Deutschland GmbH, Berlin, Germany) and Xylazin 2% (10 mg/kg) (Serumwerk Bernburg AG, Bernburg a. d. Saale, Germany) via intraperitoneal injection. The tip of the tail was amputated at a defined diameter (1.5 mm) using a gauge. The tail was immediately placed in a Falcon tube containing pre-warmed 0.9% NaCl, and bleeding was continuously monitored. The time was measured until bleeding had entirely stopped. In case of no cessation of bleeding and too much blood loss, the experiment was ended after 570 s and the bleeding stopped via cauterization.
4.17. Platelet Adhesion at the Injured Carotid Artery
This experiment was performed slightly modified as described in Grandoch et al. (2016) [
13]. Platelets from Bgn
-/0 mice and wildtype littermates were isolated. Platelets (8 × 10
6) were stained with 5-Carboxyfluorescein diacetate N-succinimidyl ester (4.48 mM) (Sigma-Aldrich, Burlington, MA, USA, 87444-5MG-F). Bgn
-/0 mice and wildtype littermates were anesthetized as described above. The right common carotid artery was dissected, the stained platelets injected into the retro-orbital plexus, and the wall of the carotid artery injured by performing a ligation with a surgical thread (PROLENE* Polypropylene Suture 6-0 (0.7 Ph. Eur., Ethicon, Inc., Raritan, NJ, USA, V396H) for 5 min. The ligation was re-opened, and the adhesion of the fluorescently-labeled platelets to the injured vessel wall was visualized at different time points by intravital microscopy (Leica Microsystems, Wetzlar, Germany).
4.18. Vessel Occlusion after FeCl3-Injury of the Carotid Artery
Wildtype and Bgn-/0 mice and chimeras underwent FeCl3 injury of the carotid artery while anesthetized with ketamine (Zoetis) and xylacine (Serumwerk Bernburg AG) and put on a heating pad. Arteria carotis communis dextra was prepared, placed into an ultrasonic flow probe (Transonic Systems, 0.5 PSB, AD Instruments, Sydney, Australia), and moisturized with 0.9% physiological saline while baseline blood flow was measured. A 0.5 × 1 mm filter paper (Whatman No.1) saturated with 20% FeCl3 (Sigma-Aldrich, Burlington, MA, USA) was placed at the dried area of the carotid artery below the measuring head for 3 min, followed by recording of thrombus formation by the software (LabChart8, AD Instruments, Sydney, Australia) until 5 min after firm occlusion or until 30 min passed (in case of no occlusion). LabChart8Reader software (AD Instruments, Sydney, Australia) was used for analysis.
4.19. Expression of Surface Proteins
To analyze the expression of surface proteins, 100 μL murine blood was collected in 300 μL heparin (20 U/mL) (Ratiopharm GmbH, Ulm, Germany # 002304). An amount of 300 μL murine Tyrode’s buffer was added. For each approach, 30 μL of the diluted blood with 5 μL of FITC-labeled rat anti-mouse GPVI monoclonal antibody (M011-1), Phycoerythrin (PE)-labeled rat anti-mouse GPIbα (M040-2), FITC-labeled rat anti-mouse/human integrin β3 monoclonal antibody (M031-1) or FITC-labeled rat anti-mouse/human integrin α5 chain monoclonal antibody (M080-1) (all Emfret Analytics, Würzburg, Germany) were incubated for 15 min. Mean fluorescence intensity (MFI) was measured.
4.20. Clot Retraction Assay of Murine Platelet Rich Plasma
Clot retraction was performed with citrated murine PRP (300,000 platelets/μL) as described earlier [
37]. Adding of thrombin (5 U/mL final; Roche, Basel, Switzerland, # 10602400001) and CaCl
2 (20 mM final) to 250 μL of PRP started the clot formation. Reaction tubes were carefully mixed and incubated at 37 °C. Pictures were taken at indicated time points. Retraction was quantified by measuring the remaining fluid in relation to the starting volume.
4.21. Statistical Analysis
All statistical tests were performed using Graph Pad Prism software 7.0 (Graphpad Holdings LLC, San Diego, CA, USA) following its recommendations based on experimental design. Data are demonstrated as mean ± standard error of the mean (SEM). Statistical significance was analyzed by Student´s two-tailed, unpaired and paired t-tests, one-way or two-way ANOVA followed by the recommended post hoc test, in case of normality and otherwise by two-tailed Wilcoxon matched-pairs signed-rank test or for unpaired samples Mann–Whitney test. For the survival curve comparison in carotid artery, occlusion log-rank (Mantel–Cox) test was applied. p-values < 0.05 were considered to be statistically significant.



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}