
ATP Synthase and Mitochondrial Bioenergetics Dysfunction in Alzheimer’s Disease


 ,
,  ,
, 
{kind=link}
{kind=link}
Abstract
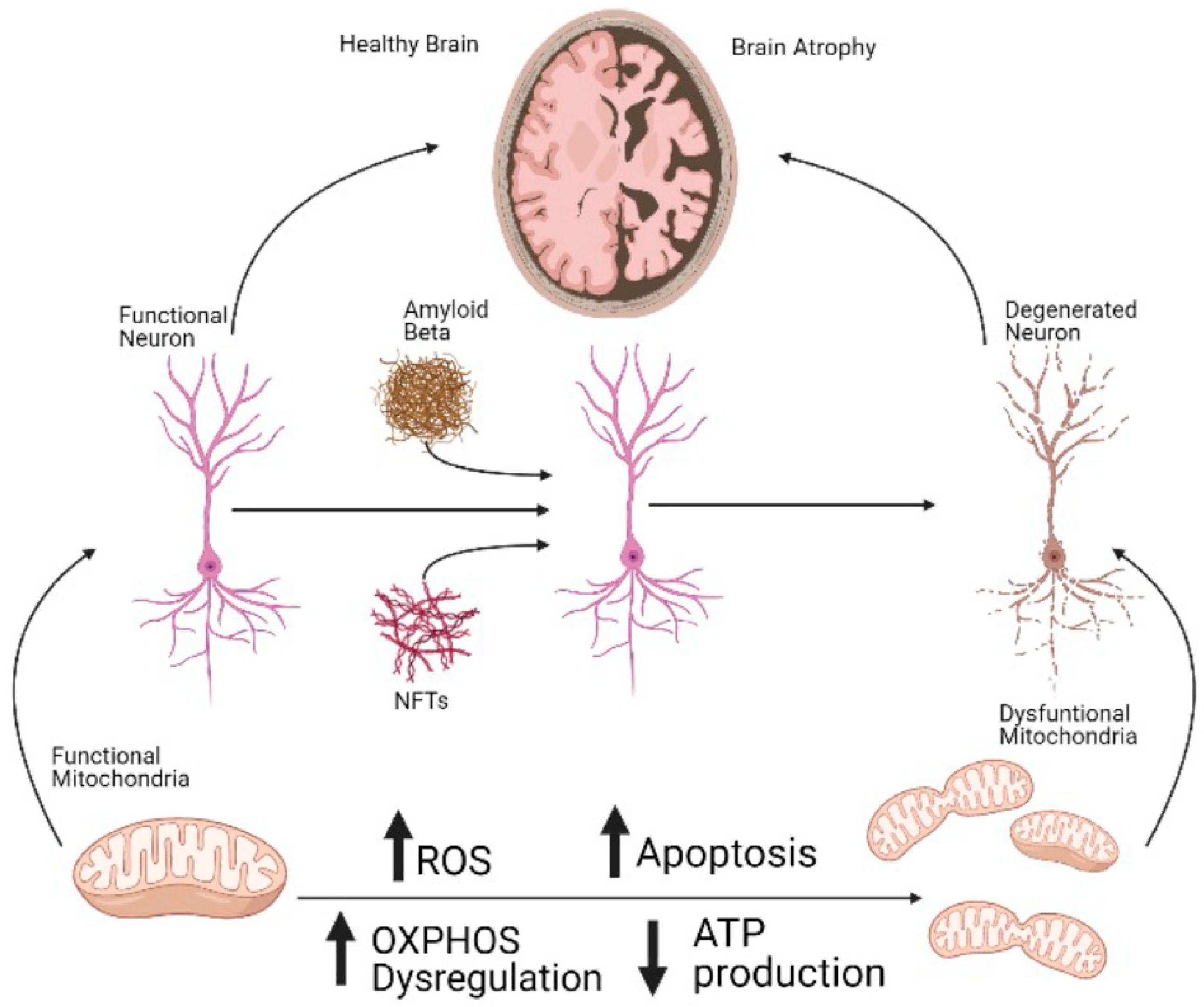
1. Introduction
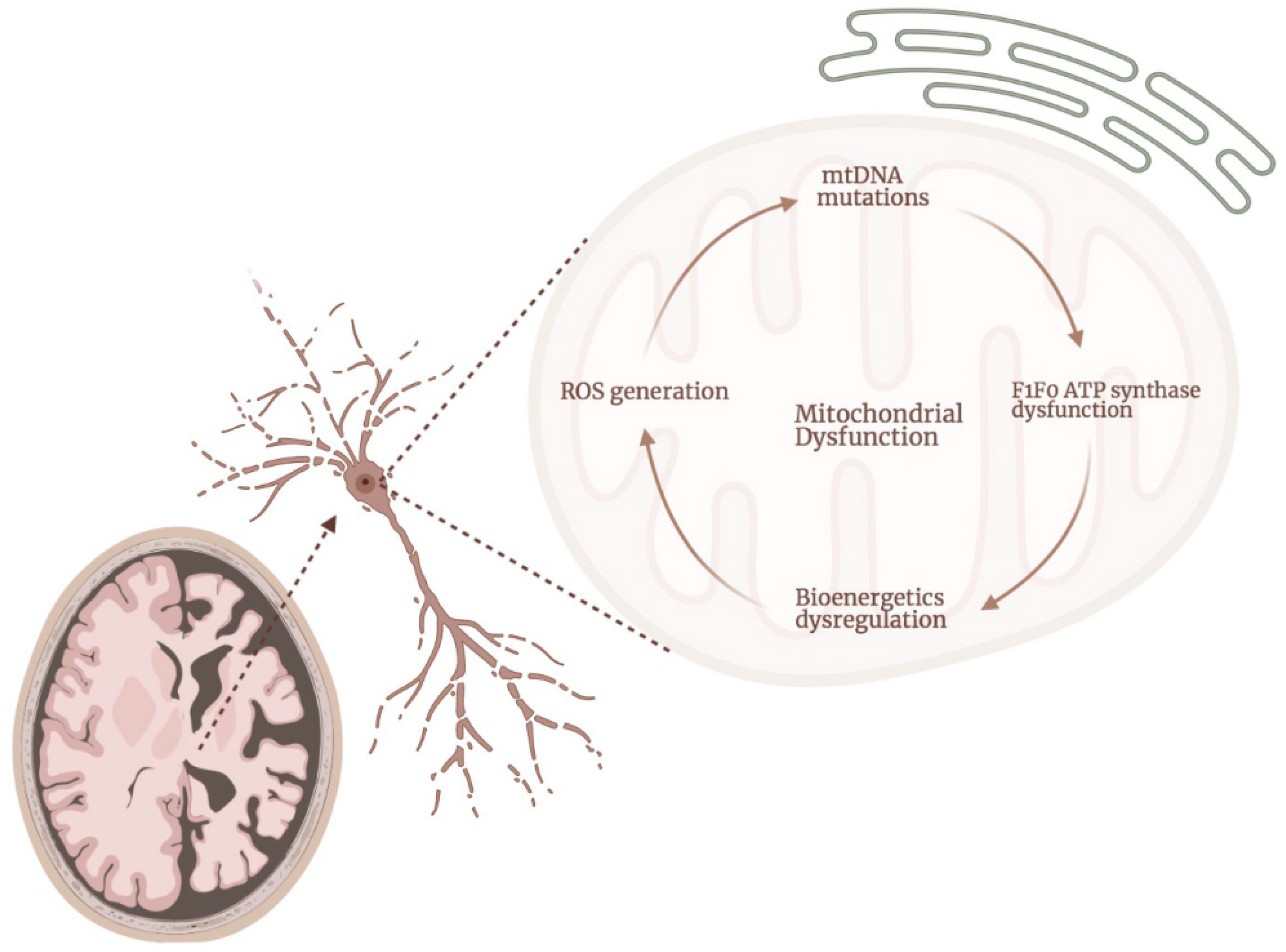
2. ATP Synthase Dysfunction in AD
3. Dysregulation of Other Complexes of the ETC in AD
4. Mitochondrial Calcium Homeostasis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Association, A.s. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Ten Kate, M.; Dicks, E.; Visser, P.J.; van der Flier, W.M.; Teunissen, C.E.; Barkhof, F.; Scheltens, P.; Tijms, B.M.; Alzheimer’s Disease Neuroimaging Initiative. Atrophy subtypes in prodromal Alzheimer’s disease are associated with cognitive decline. Brain 2018, 141, 3443–3456. [Google Scholar] [CrossRef]
- James, B.D.; Wilson, R.S.; Capuano, A.W.; Boyle, P.A.; Shah, R.C.; Lamar, M.; Ely, E.W.; Bennett, D.A.; Schneider, J.A. Hospitalization, Alzheimer’s Disease and Related Neuropathologies, and Cognitive Decline. Ann. Neurol. 2019, 86, 844–852. [Google Scholar] [CrossRef]
- Ahmed, S.; Haigh, A.M.; de Jager, C.A.; Garrard, P. Connected speech as a marker of disease progression in autopsy-proven Alzheimer’s disease. Brain 2013, 136, 3727–3737. [Google Scholar] [CrossRef]
- Duara, R.; Lopez-Alberola, R.F.; Barker, W.W.; Loewenstein, D.A.; Zatinsky, M.; Eisdorfer, C.E.; Weinberg, G.B. A comparison of familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Kvartsberg, H.; Lashley, T.; Murray, C.E.; Brinkmalm, G.; Cullen, N.C.; Hoglund, K.; Zetterberg, H.; Blennow, K.; Portelius, E. The intact postsynaptic protein neurogranin is reduced in brain tissue from patients with familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2019, 137, 89–102. [Google Scholar] [CrossRef]
- Hsu, S.; Pimenova, A.A.; Hayes, K.; Villa, J.A.; Rosene, M.J.; Jere, M.; Goate, A.M.; Karch, C.M. Systematic validation of variants of unknown significance in APP, PSEN1 and PSEN2. Neurobiol. Dis. 2020, 139, 104817. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Dowjat, W.K.; Permanne, B.; Palha, J.; Kumar, A.; Gallo, G.; Frangione, B. Presenilin-1 is associated with Alzheimer’s disease amyloid. Am. J. Pathol. 1997, 151, 601–610. [Google Scholar]
- Harman, D. Alzheimer’s disease pathogenesis: Role of aging. Ann. N. Y. Acad. Sci. 2006, 1067, 454–460. [Google Scholar] [CrossRef]
- Matthews, K.A.; Xu, W.; Gaglioti, A.H.; Holt, J.B.; Croft, J.B.; Mack, D.; McGuire, L.C. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015–2060) in adults aged >/=65 years. Alzheimers Dement. 2019, 15, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 17, 327–406. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Malpetti, M.; Kievit, R.A.; Passamonti, L.; Jones, P.S.; Tsvetanov, K.A.; Rittman, T.; Mak, E.; Nicastro, N.; Bevan-Jones, W.R.; Su, L.; et al. Microglial activation and tau burden predict cognitive decline in Alzheimer’s disease. Brain 2020, 143, 1588–1602. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Tsuji, T.; Shiozaki, A.; Kohno, R.; Yoshizato, K.; Shimohama, S. Proteomic profiling and neurodegeneration in Alzheimer’s disease. Neurochem. Res. 2002, 27, 1245–1253. [Google Scholar] [CrossRef]
- Nagele, R.G.; D’Andrea, M.R.; Lee, H.; Venkataraman, V.; Wang, H.Y. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003, 971, 197–209. [Google Scholar] [CrossRef]
- Wirths, O.; Multhaup, G.; Czech, C.; Blanchard, V.; Moussaoui, S.; Tremp, G.; Pradier, L.; Beyreuther, K.; Bayer, T.A. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci. Lett. 2001, 306, 116–120. [Google Scholar] [CrossRef]
- Welikovitch, L.A.; Do Carmo, S.; Magloczky, Z.; Malcolm, J.C.; Loke, J.; Klein, W.L.; Freund, T.; Cuello, A.C. Early intraneuronal amyloid triggers neuron-derived inflammatory signaling in APP transgenic rats and human brain. Proc. Natl. Acad. Sci. USA 2020, 117, 6844–6854. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Zotova, E.; Holmes, C.; Johnston, D.; Neal, J.W.; Nicoll, J.A.; Boche, D. Microglial alterations in human Alzheimer’s disease following Abeta42 immunization. Neuropathol. Appl. Neurobiol. 2011, 37, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.C.; Muratore, C.R.; Gierahn, T.M.; Sullivan, S.E.; Srikanth, P.; De Jager, P.L.; Love, J.C.; Young-Pearse, T.L. Single-Cell Detection of Secreted Abeta and sAPPalpha from Human IPSC-Derived Neurons and Astrocytes. J. Neurosci. 2016, 36, 1730–1746. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Choi, H.; Kim, C.; Song, H.; Cha, M.Y.; Cho, H.J.; Son, S.M.; Kim, H.J.; Mook-Jung, I. Amyloid beta-induced elevation of O-GlcNAcylated c-Fos promotes neuronal cell death. Aging Cell 2019, 18, e12872. [Google Scholar] [CrossRef]
- Shimohama, S. Apoptosis in Alzheimer’s disease—An update. Apoptosis 2000, 5, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Vanmechelen, E.; Hampel, H. CSF total tau, Abeta42 and phosphorylated tau protein as biomarkers for Alzheimer’s disease. Mol. Neurobiol. 2001, 24, 87–97. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhang, S.; Ma, X.; Jia, C.; Liu, Z.; Huang, C.; Liu, C.; Li, D. Structural basis of the interplay between alpha-synuclein and Tau in regulating pathological amyloid aggregation. J. Biol. Chem. 2020, 295, 7470–7480. [Google Scholar] [CrossRef]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef]
- Drummond, E.; Pires, G.; MacMurray, C.; Askenazi, M.; Nayak, S.; Bourdon, M.; Safar, J.; Ueberheide, B.; Wisniewski, T. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain 2020, 143, 2803–2817. [Google Scholar] [CrossRef]
- Estus, S.; Tucker, H.M.; van Rooyen, C.; Wright, S.; Brigham, E.F.; Wogulis, M.; Rydel, R.E. Aggregated amyloid-beta protein induces cortical neuronal apoptosis and concomitant “apoptotic” pattern of gene induction. J. Neurosci. 1997, 17, 7736–7745. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef]
- Onyango, I.G.; Dennis, J.; Khan, S.M. Mitochondrial Dysfunction in Alzheimer’s Disease and the Rationale for Bioenergetics Based Therapies. Aging Dis. 2016, 7, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Baltanas, A.; Solesio, M.E.; Zalba, G.; Galindo, M.F.; Fortuno, A.; Jordan, J. The senescence-accelerated mouse prone-8 (SAM-P8) oxidative stress is associated with upregulation of renal NADPH oxidase system. J. Physiol. Biochem. 2013, 69, 927–935. [Google Scholar] [CrossRef]
- Galindo, M.F.; Solesio, M.E.; Atienzar-Aroca, S.; Zamora, M.J.; Jordan Bueso, J. Mitochondrial dynamics and mitophagy in the 6-hydroxydopamine preclinical model of Parkinson’s disease. Parkinsons Dis. 2012, 2012, 131058. [Google Scholar] [CrossRef] [PubMed]
- Solesio, M.E.; Prime, T.A.; Logan, A.; Murphy, M.P.; Del Mar Arroyo-Jimenez, M.; Jordan, J.; Galindo, M.F. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim. Biophys. Acta 2013, 1832, 174–182. [Google Scholar] [CrossRef]
- Solesio, M.E.; Saez-Atienzar, S.; Jordan, J.; Galindo, M.F. Characterization of mitophagy in the 6-hydoxydopamine Parkinson’s disease model. Toxicol. Sci. 2012, 129, 411–420. [Google Scholar] [CrossRef]
- Solesio, M.E.; Saez-Atienzar, S.; Jordan, J.; Galindo, M.F. 3-Nitropropionic acid induces autophagy by forming mitochondrial permeability transition pores rather than activating the mitochondrial fission pathway. Br. J. Pharmacol. 2013, 168, 63–75. [Google Scholar] [CrossRef]
- Lunnon, K.; Keohane, A.; Pidsley, R.; Newhouse, S.; Riddoch-Contreras, J.; Thubron, E.B.; Devall, M.; Soininen, H.; Kloszewska, I.; Mecocci, P.; et al. Mitochondrial genes are altered in blood early in Alzheimer’s disease. Neurobiol. Aging 2017, 53, 36–47. [Google Scholar] [CrossRef]
- Stefanova, N.A.; Ershov, N.I.; Kolosova, N.G. Suppression of Alzheimer’s Disease-Like Pathology Progression by Mitochondria-Targeted Antioxidant SkQ1: A Transcriptome Profiling Study. Oxid. Med. Cell. Longev. 2019, 2019, 3984906. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 Ameliorates Synaptic Depression, Abeta Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef]
- Solesio, M.E.; Peixoto, P.M.; Debure, L.; Madamba, S.M.; de Leon, M.J.; Wisniewski, T.; Pavlov, E.V.; Fossati, S. Carbonic anhydrase inhibition selectively prevents amyloid beta neurovascular mitochondrial toxicity. Aging Cell 2018, 17, e12787. [Google Scholar] [CrossRef]
- Fossati, S.; Giannoni, P.; Solesio, M.E.; Cocklin, S.L.; Cabrera, E.; Ghiso, J.; Rostagno, A. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol. Dis. 2016, 86, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Solesio, M.E.; Xie, L.; McIntyre, B.; Ellenberger, M.; Mitaishvili, E.; Bhadra-Lobo, S.; Bettcher, L.F.; Bazil, J.N.; Raftery, D.; Jakob, U.; et al. Depletion of mitochondrial inorganic polyphosphate (polyP) in mammalian cells causes metabolic shift from oxidative phosphorylation to glycolysis. Biochem. J. 2021, 478, 1631–1646. [Google Scholar] [CrossRef]
- McIntyre, B.; Solesio, M.E. Mitochondrial inorganic polyphosphate (polyP): The missing link of mammalian bioenergetics. Neural. Regen. Res. 2021, 16, 2227–2228. [Google Scholar] [CrossRef]
- Borden, E.A.; Furey, M.; Gattone, N.J.; Hambardikar, V.D.; Liang, X.H.; Scoma, E.R.; Abou Samra, A.; LR, D.G.; Dennis, D.J.; Fricker, D.; et al. Is there a link between inorganic polyphosphate (polyP), mitochondria, and neurodegeneration? Pharmacol. Res. 2021, 163, 105211. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A.; Ruffo, A.; Johnson, M.; Eggleston, L.V.; Hems, R. Oxidative phosphorylation. Biochem. J. 1953, 54, 107–116. [Google Scholar] [CrossRef]
- Zhu, X.H.; Qiao, H.; Du, F.; Xiong, Q.; Liu, X.; Zhang, X.; Ugurbil, K.; Chen, W. Quantitative imaging of energy expenditure in human brain. Neuroimage 2012, 60, 2107–2117. [Google Scholar] [CrossRef]
- Markesbery, W.R. The role of oxidative stress in Alzheimer disease. Arch. Neurol. 1999, 56, 1449–1452. [Google Scholar] [CrossRef]
- Tzagoloff, A.; Myers, A.M. Genetics of mitochondrial biogenesis. Annu. Rev. Biochem. 1986, 55, 249–285. [Google Scholar] [CrossRef]
- Lazarou, M.; Smith, S.M.; Thorburn, D.R.; Ryan, M.T.; McKenzie, M. Assembly of nuclear DNA-encoded subunits into mitochondrial complex IV, and their preferential integration into supercomplex forms in patient mitochondria. FEBS J. 2009, 276, 6701–6713. [Google Scholar] [CrossRef] [PubMed]
- Lemarie, A.; Grimm, S. Mitochondrial respiratory chain complexes: Apoptosis sensors mutated in cancer? Oncogene 2011, 30, 3985–4003. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Menz, R.I.; Walker, J.E.; Leslie, A.G. Structure of bovine mitochondrial F(1)-ATPase with nucleotide bound to all three catalytic sites: Implications for the mechanism of rotary catalysis. Cell 2001, 106, 331–341. [Google Scholar] [CrossRef]
- Pinke, G.; Zhou, L.; Sazanov, L.A. Cryo-EM structure of the entire mammalian F-type ATP synthase. Nat. Struct. Mol. Biol. 2020, 27, 1077–1085. [Google Scholar] [CrossRef]
- Suzuki, T.; Ueno, H.; Mitome, N.; Suzuki, J.; Yoshida, M. F(0) of ATP synthase is a rotary proton channel. Obligatory coupling of proton translocation with rotation of c-subunit ring. J. Biol. Chem. 2002, 277, 13281–13285. [Google Scholar] [CrossRef]
- Dittrich, M.; Hayashi, S.; Schulten, K. On the mechanism of ATP hydrolysis in F1-ATPase. Biophys. J. 2003, 85, 2253–2266. [Google Scholar] [CrossRef]
- Xu, T.; Pagadala, V.; Mueller, D.M. Understanding structure, function, and mutations in the mitochondrial ATP synthase. Microb. Cell 2015, 2, 105–125. [Google Scholar] [CrossRef]
- Antoniel, M.; Giorgio, V.; Fogolari, F.; Glick, G.D.; Bernardi, P.; Lippe, G. The oligomycin-sensitivity conferring protein of mitochondrial ATP synthase: Emerging new roles in mitochondrial pathophysiology. Int. J. Mol. Sci. 2014, 15, 7513–7536. [Google Scholar] [CrossRef] [PubMed]
- Devenish, R.J.; Prescott, M.; Boyle, G.M.; Nagley, P. The oligomycin axis of mitochondrial ATP synthase: OSCP and the proton channel. J. Bioenerg. Biomembr. 2000, 32, 507–515. [Google Scholar] [CrossRef]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef]
- Amodeo, G.F.; Solesio, M.E.; Pavlov, E.V. From ATP synthase dimers to C-ring conformational changes: Unified model of the mitochondrial permeability transition pore. Cell Death Dis. 2017, 8, 1. [Google Scholar] [CrossRef]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17.e12. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Sergeant, N.; Wattez, A.; Galvan-valencia, M.; Ghestem, A.; David, J.P.; Lemoine, J.; Sautiere, P.E.; Dachary, J.; Mazat, J.P.; Michalski, J.C.; et al. Association of ATP synthase alpha-chain with neurofibrillary degeneration in Alzheimer’s disease. Neuroscience 2003, 117, 293–303. [Google Scholar] [CrossRef]
- Manczak, M.; Park, B.S.; Jung, Y.; Reddy, P.H. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: Implications for early mitochondrial dysfunction and oxidative damage. Neuromol. Med. 2004, 5, 147–162. [Google Scholar] [CrossRef]
- Kawamoto, E.M.; Munhoz, C.D.; Glezer, I.; Bahia, V.S.; Caramelli, P.; Nitrini, R.; Gorjao, R.; Curi, R.; Scavone, C.; Marcourakis, T. Oxidative state in platelets and erythrocytes in aging and Alzheimer’s disease. Neurobiol. Aging 2005, 26, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Terni, B.; Boada, J.; Portero-Otin, M.; Pamplona, R.; Ferrer, I. Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Pathol. 2010, 20, 222–233. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH(2)-tau fragment and Abeta in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef]
- Xing, S.L.; Chen, B.; Shen, D.Z.; Zhu, C.Q. beta-amyloid peptide binds and regulates ectopic ATP synthase alpha-chain on neural surface. Int. J. Neurosci. 2012, 122, 290–297. [Google Scholar] [CrossRef]
- Cha, M.Y.; Cho, H.J.; Kim, C.; Jung, Y.O.; Kang, M.J.; Murray, M.E.; Hong, H.S.; Choi, Y.J.; Choi, H.; Kim, D.K.; et al. Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer’s disease. Hum. Mol. Genet. 2015, 24, 6492–6504. [Google Scholar] [CrossRef]
- Veas-Perez de Tudela, M.; Delgado-Esteban, M.; Maestre, C.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Vecino, R.; Bolanos, J.P.; Almeida, A. Regulation of Bcl-xL-ATP Synthase Interaction by Mitochondrial Cyclin B1-Cyclin-Dependent Kinase-1 Determines Neuronal Survival. J. Neurosci. 2015, 35, 9287–9301. [Google Scholar] [CrossRef]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S.; et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 2016, 7, 11483. [Google Scholar] [CrossRef] [PubMed]
- Gauba, E.; Guo, L.; Du, H. Cyclophilin D Promotes Brain Mitochondrial F1FO ATP Synthase Dysfunction in Aging Mice. J. Alzheimers Dis. 2017, 55, 1351–1362. [Google Scholar] [CrossRef]
- Gauba, E.; Chen, H.; Guo, L.; Du, H. Cyclophilin D deficiency attenuates mitochondrial F1Fo ATP synthase dysfunction via OSCP in Alzheimer’s disease. Neurobiol. Dis. 2019, 121, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Galber, C.; Carissimi, S.; Baracca, A.; Giorgio, V. The ATP Synthase Deficiency in Human Diseases. Life 2021, 11, 325. [Google Scholar] [CrossRef]
- Ebanks, B.; Ingram, T.L.; Chakrabarti, L. ATP synthase and Alzheimer’s disease: Putting a spin on the mitochondrial hypothesis. Aging 2020, 12, 16647–16662. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Chou, J.L.; Shenoy, D.V.; Thomas, N.; Choudhary, P.K.; Laferla, F.M.; Goodman, S.R.; Breen, G.A. Early dysregulation of the mitochondrial proteome in a mouse model of Alzheimer’s disease. J. Proteomics 2011, 74, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, K.; Hatanpaa, K.; Rapoport, S.I.; Brady, D.R. Decreased expression of nuclear and mitochondrial DNA-encoded genes of oxidative phosphorylation in association neocortex in Alzheimer disease. Brain Res. Mol. Brain Res. 1997, 44, 99–104. [Google Scholar] [CrossRef]
- Reddy, P.H.; McWeeney, S.; Park, B.S.; Manczak, M.; Gutala, R.V.; Partovi, D.; Jung, Y.; Yau, V.; Searles, R.; Mori, M.; et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum. Mol. Genet. 2004, 13, 1225–1240. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef]
- Hundal, T.; Norling, B.; Ernster, L. The oligomycin sensitivity conferring protein (OSCP) of beef heart mitochondria: Studies of its binding to F1 and its function. J. Bioenerg. Biomembr. 1984, 16, 535–550. [Google Scholar] [CrossRef]
- Liu, F.; Shi, J.; Tanimukai, H.; Gu, J.; Gu, J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain 2009, 132, 1820–1832. [Google Scholar] [CrossRef]
- Hanover, J.A.; Krause, M.W.; Love, D.C. Bittersweet memories: Linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol. 2012, 13, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W.; Akimoto, Y. The O-GlcNAc Modification. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor: New York, NY, USA, 2009. [Google Scholar]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Drose, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef]
- Parks, J.K.; Smith, T.S.; Trimmer, P.A.; Bennett, J.P., Jr.; Parker, W.D., Jr. Neurotoxic Abeta peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J. Neurochem. 2001, 76, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Therriault, J.; Kang, M.S.P.; Savard, M.; Pascoal, T.A.; Lussier, F.; Tissot, C.; Wang, Y.T.; Benedet, A.; Matsudaira, T.; et al. Mitochondrial complex I abnormalities is associated with tau and clinical symptoms in mild Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 28. [Google Scholar] [CrossRef] [PubMed]
- David, D.C.; Hauptmann, S.; Scherping, I.; Schuessel, K.; Keil, U.; Rizzu, P.; Ravid, R.; Drose, S.; Brandt, U.; Muller, W.E.; et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005, 280, 23802–23814. [Google Scholar] [CrossRef] [PubMed]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Parks, J.; Filley, C.M.; Kleinschmidt-DeMasters, B.K. Electron transport chain defects in Alzheimer’s disease brain. Neurology 1994, 44, 1090–1096. [Google Scholar] [CrossRef]
- Aksenov, M.Y.; Tucker, H.M.; Nair, P.; Aksenova, M.V.; Butterfield, D.A.; Estus, S.; Markesbery, W.R. The expression of several mitochondrial and nuclear genes encoding the subunits of electron transport chain enzyme complexes, cytochrome c oxidase, and NADH dehydrogenase, in different brain regions in Alzheimer’s disease. Neurochem. Res. 1999, 24, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.W.; Farivar, N.; Butcher, L.L. Alzheimer dementia and reduced nicotinamide adenine dinucleotide (NADH)-diaphorase activity in senile plaques and the basal forebrain. Neurosci. Lett. 1985, 53, 39–44. [Google Scholar] [CrossRef]
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain 2019, 12, 8. [Google Scholar] [CrossRef]
- Li, Y.; Park, J.S.; Deng, J.H.; Bai, Y. Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J. Bioenerg. Biomembr. 2006, 38, 283–291. [Google Scholar] [CrossRef]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef]
- Cottrell, D.A.; Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Turnbull, D.M. The role of cytochrome c oxidase deficient hippocampal neurones in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2002, 28, 390–396. [Google Scholar] [CrossRef]
- Nagy, Z.; Esiri, M.M.; LeGris, M.; Matthews, P.M. Mitochondrial enzyme expression in the hippocampus in relation to Alzheimer-type pathology. Acta Neuropathol. 1999, 97, 346–354. [Google Scholar] [CrossRef]
- Canevari, L.; Clark, J.B.; Bates, T.E. beta-Amyloid fragment 25-35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 1999, 457, 131–134. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Hatanpaa, K.; Brady, D.R.; Stoll, J.; Rapoport, S.I. Downregulation of oxidative phosphorylation in Alzheimer disease: Loss of cytochrome oxidase subunit mRNA in the hippocampus and entorhinal cortex. Brain Res. 1998, 796, 13–19. [Google Scholar] [CrossRef]
- Behl, C.; Davis, J.B.; Lesley, R.; Schubert, D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 1994, 77, 817–827. [Google Scholar] [CrossRef]
- Wei, W.; Wang, X.; Kusiak, J.W. Signaling events in amyloid beta-peptide-induced neuronal death and insulin-like growth factor I protection. J. Biol. Chem. 2002, 277, 17649–17656. [Google Scholar] [CrossRef]
- Pratico, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Surh, Y.J. beta-Amyloid induces oxidative DNA damage and cell death through activation of c-Jun N terminal kinase. Ann. N. Y. Acad. Sci. 2002, 973, 228–236. [Google Scholar] [CrossRef]
- Tamagno, E.; Guglielmotto, M.; Aragno, M.; Borghi, R.; Autelli, R.; Giliberto, L.; Muraca, G.; Danni, O.; Zhu, X.; Smith, M.A.; et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 2008, 104, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef]
- Antonyova, V.; Kejik, Z.; Brogyanyi, T.; Kaplanek, R.; Pajkova, M.; Talianova, V.; Hromadka, R.; Masarik, M.; Sykora, D.; Miksatkova, L.; et al. Role of mtDNA disturbances in the pathogenesis of Alzheimer’s and Parkinson’s disease. DNA Repair 2020, 91–92, 102871. [Google Scholar] [CrossRef] [PubMed]
- Gredilla, R.; Bohr, V.A.; Stevnsner, T. Mitochondrial DNA repair and association with aging—An update. Exp. Gerontol. 2010, 45, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, A.; Fantin, D.J.; Tustanoff, E.R. Role of phospholipid fatty acids on the kinetics of high and low affinity sites of cytochrome c oxidase. Biochem. Cell Biol. 1986, 64, 1195–1210. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Peroxidative damage to cardiac mitochondria: Cytochrome oxidase and cardiolipin alterations. FEBS Lett. 1998, 424, 155–158. [Google Scholar] [CrossRef]
- Richardson, J.S. Free radicals in the genesis of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1993, 695, 73–76. [Google Scholar] [CrossRef]
- Chagnon, P.; Betard, C.; Robitaille, Y.; Cholette, A.; Gauvreau, D. Distribution of brain cytochrome oxidase activity in various neurodegenerative diseases. Neuroreport 1995, 6, 711–715. [Google Scholar] [CrossRef]
- Kish, S.J.; Bergeron, C.; Rajput, A.; Dozic, S.; Mastrogiacomo, F.; Chang, L.J.; Wilson, J.M.; DiStefano, L.M.; Nobrega, J.N. Brain cytochrome oxidase in Alzheimer’s disease. J. Neurochem. 1992, 59, 776–779. [Google Scholar] [CrossRef]
- Cadenas, E.; Boveris, A.; Ragan, C.I.; Stoppani, A.O. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 1977, 180, 248–257. [Google Scholar] [CrossRef]
- Ahn, B.W.; Song, D.U.; Jung, Y.D.; Chay, K.O.; Chung, M.A.; Yang, S.Y.; Shin, B.A. Detection of beta-amyloid peptide aggregation using DNA electrophoresis. Anal. Biochem. 2000, 284, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Nunomura, A.; Moreira, P.I.; Lee, H.G.; Perry, G.; Smith, M.A.; Zhu, X. Oxidative stress signaling in Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 525–532. [Google Scholar] [CrossRef]
- Korolainen, M.A.; Goldsteins, G.; Nyman, T.A.; Alafuzoff, I.; Koistinaho, J.; Pirttila, T. Oxidative modification of proteins in the frontal cortex of Alzheimer’s disease brain. Neurobiol. Aging 2006, 27, 42–53. [Google Scholar] [CrossRef]
- Suthammarak, W.; Yang, Y.Y.; Morgan, P.G.; Sedensky, M.M. Complex I function is defective in complex IV-deficient Caenorhabditis elegans. J. Biol. Chem. 2009, 284, 6425–6435. [Google Scholar] [CrossRef] [PubMed]
- Demarin, V.; Podobnik, S.S.; Storga-Tomic, D.; Kay, G. Treatment of Alzheimer’s disease with stabilized oral nicotinamide adenine dinucleotide: A randomized, double-blind study. Drugs Exp. Clin. Res. 2004, 30, 27–33. [Google Scholar]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Maezawa, I.; Trushin, S.; Minhas, P.; Pinto, M.; Jin, L.W.; Prasain, K.; Nguyen, T.D.; Yamazaki, Y.; et al. Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s Disease. EBioMedicine 2015, 2, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, M.; Priebe, S.; Groth, M.; Hartmann, N.; Menzel, U.; Pandolfini, L.; Koch, P.; Felder, M.; Ristow, M.; Englert, C.; et al. Longitudinal RNA-Seq Analysis of Vertebrate Aging Identifies Mitochondrial Complex I as a Small-Molecule-Sensitive Modifier of Lifespan. Cell Syst. 2016, 2, 122–132. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T.; Mamun, A.A.; Barreto, G.E.; Rashid, M.; Perveen, A.; Ashraf, G.M. Pharmacological approaches to mitigate neuroinflammation in Alzheimer’s disease. Int. Immunopharmacol. 2020, 84, 106479. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Akter, S.; Akundi, R.S. The two-hit hypothesis for neuroinflammation: Role of exogenous ATP in modulating inflammation in the brain. Front. Cell. Neurosci. 2014, 8, 260. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Marmisolle, I.; Tarallo, D.; Quijano, C. Mitochondrial Bioenergetics and Dynamics in Secretion Processes. Front. Endocrinol. 2020, 11, 319. [Google Scholar] [CrossRef]
- Dixit, S.; Fessel, J.P.; Harrison, F.E. Mitochondrial dysfunction in the APP/PSEN1 mouse model of Alzheimer’s disease and a novel protective role for ascorbate. Free Radic. Biol. Med. 2017, 112, 515–523. [Google Scholar] [CrossRef]
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.; Gerasimenko, O.V. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 2009, 284, 20796–20803. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Rizzuto, R.; Simpson, A.W.; Brini, M.; Pozzan, T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 1992, 358, 325–327. [Google Scholar] [CrossRef]
- Montero, M.; Alonso, M.T.; Carnicero, E.; Cuchillo-Ibanez, I.; Albillos, A.; Garcia, A.G.; Garcia-Sancho, J.; Alvarez, J. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat. Cell Biol. 2000, 2, 57–61. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [CrossRef] [PubMed]
- Solesio, M.E.; Garcia Del Molino, L.C.; Elustondo, P.A.; Diao, C.; Chang, J.C.; Pavlov, E.V. Inorganic polyphosphate is required for sustained free mitochondrial calcium elevation, following calcium uptake. Cell Calcium 2020, 86, 102127. [Google Scholar] [CrossRef]
- Solesio, M.E.; Demirkhanyan, L.; Zakharian, E.; Pavlov, E.V. Contribution of inorganic polyphosphate towards regulation of mitochondrial free calcium. Biochim. Biophys. Acta 2016, 1860, 1317–1325. [Google Scholar] [CrossRef]
- Solesio, M.E.; Elustondo, P.A.; Zakharian, E.; Pavlov, E.V. Inorganic polyphosphate (polyP) as an activator and structural component of the mitochondrial permeability transition pore. Biochem. Soc. Trans. 2016, 44, 7–12. [Google Scholar] [CrossRef]
- Khong, M.L.; Li, L.; Solesio, M.E.; Pavlov, E.V.; Tanner, J.A. Inorganic polyphosphate controls cyclophilin B-mediated collagen folding in osteoblast-like cells. FEBS J. 2020, 287, 4500–4524. [Google Scholar] [CrossRef]
- Sheehan, J.P.; Swerdlow, R.H.; Miller, S.W.; Davis, R.E.; Parks, J.K.; Parker, W.D.; Tuttle, J.B. Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer’s disease. J. Neurosci. 1997, 17, 4612–4622. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Jin, H.; Huang, Y. Mitochondria-associated membranes (MAMs): A potential therapeutic target for treating Alzheimer’s disease. Clin. Sci. 2021, 135, 109–126. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Jaworska, A.; Dzbek, J.; Styczynska, M.; Kuznicki, J. Analysis of calcium homeostasis in fresh lymphocytes from patients with sporadic Alzheimer’s disease or mild cognitive impairment. Biochim. Biophys. Acta 2013, 1833, 1692–1699. [Google Scholar] [CrossRef][Green Version]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Ronnback, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [PubMed]
- Boyman, L.; Greiser, M.; Lederer, W.J. Calcium influx through the mitochondrial calcium uniporter holocomplex, MCUcx. J. Mol. Cell Cardiol. 2021, 151, 145–154. [Google Scholar] [CrossRef]
- Perez, M.J.; Ponce, D.P.; Aranguiz, A.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial permeability transition pore contributes to mitochondrial dysfunction in fibroblasts of patients with sporadic Alzheimer’s disease. Redox. Biol. 2018, 19, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef]
- Supnet, C.; Bezprozvanny, I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 2010, 47, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.T.; Sharpe, G.R. Thapsigargin raises intracellular free calcium levels in human keratinocytes and inhibits the coordinated expression of differentiation markers. Exp. Cell Res. 1994, 210, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Deniaud, A.; Sharaf el dein, O.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Zhang, H.; Toral-Barza, L.; Szolosi, S.; Tofel-Grehl, B. Calcium stores in cultured fibroblasts and their changes with Alzheimer’s disease. Biochim. Biophys. Acta 1996, 1316, 71–77. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patro, S.; Ratna, S.; Yamamoto, H.A.; Ebenezer, A.T.; Ferguson, D.S.; Kaur, A.; McIntyre, B.C.; Snow, R.; Solesio, M.E. ATP Synthase and Mitochondrial Bioenergetics Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11185. https://doi.org/10.3390/ijms222011185
Patro S, Ratna S, Yamamoto HA, Ebenezer AT, Ferguson DS, Kaur A, McIntyre BC, Snow R, Solesio ME. ATP Synthase and Mitochondrial Bioenergetics Dysfunction in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(20):11185. https://doi.org/10.3390/ijms222011185
Chicago/Turabian StylePatro, Somya, Sujay Ratna, Hianny A. Yamamoto, Andrew T. Ebenezer, Dillon S. Ferguson, Amanpreet Kaur, Brendan C. McIntyre, Ryan Snow, and Maria E. Solesio. 2021. "ATP Synthase and Mitochondrial Bioenergetics Dysfunction in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 20: 11185. https://doi.org/10.3390/ijms222011185
APA StylePatro, S., Ratna, S., Yamamoto, H. A., Ebenezer, A. T., Ferguson, D. S., Kaur, A., McIntyre, B. C., Snow, R., & Solesio, M. E. (2021). ATP Synthase and Mitochondrial Bioenergetics Dysfunction in Alzheimer’s Disease. International Journal of Molecular Sciences, 22(20), 11185. https://doi.org/10.3390/ijms222011185