
Olaparib Is a Mitochondrial Complex I Inhibitor That Kills Temozolomide-Resistant Human Glioblastoma Cells

 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
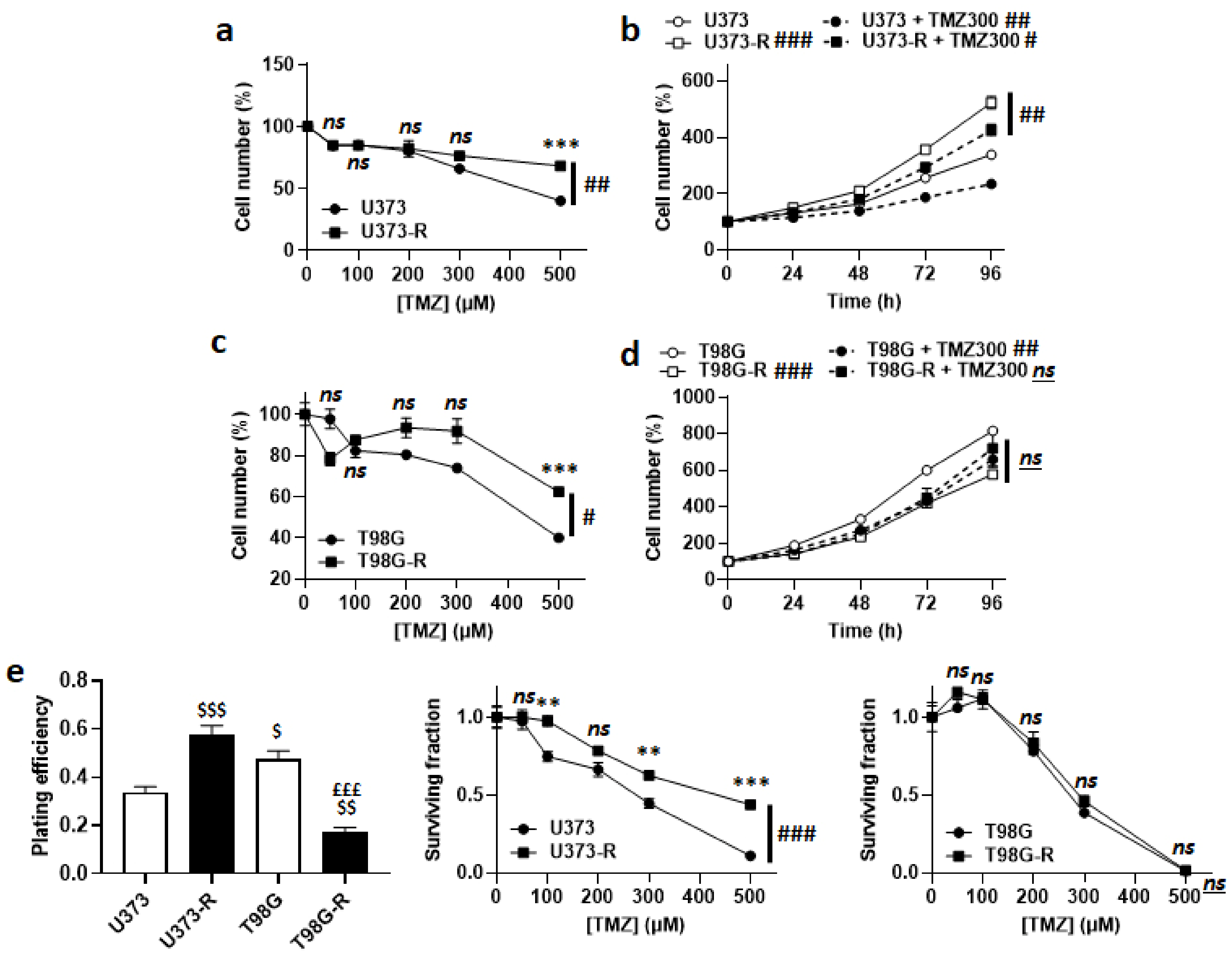
2.1. Selection of Temozolomide-Resistant Human Glioblastoma Cells
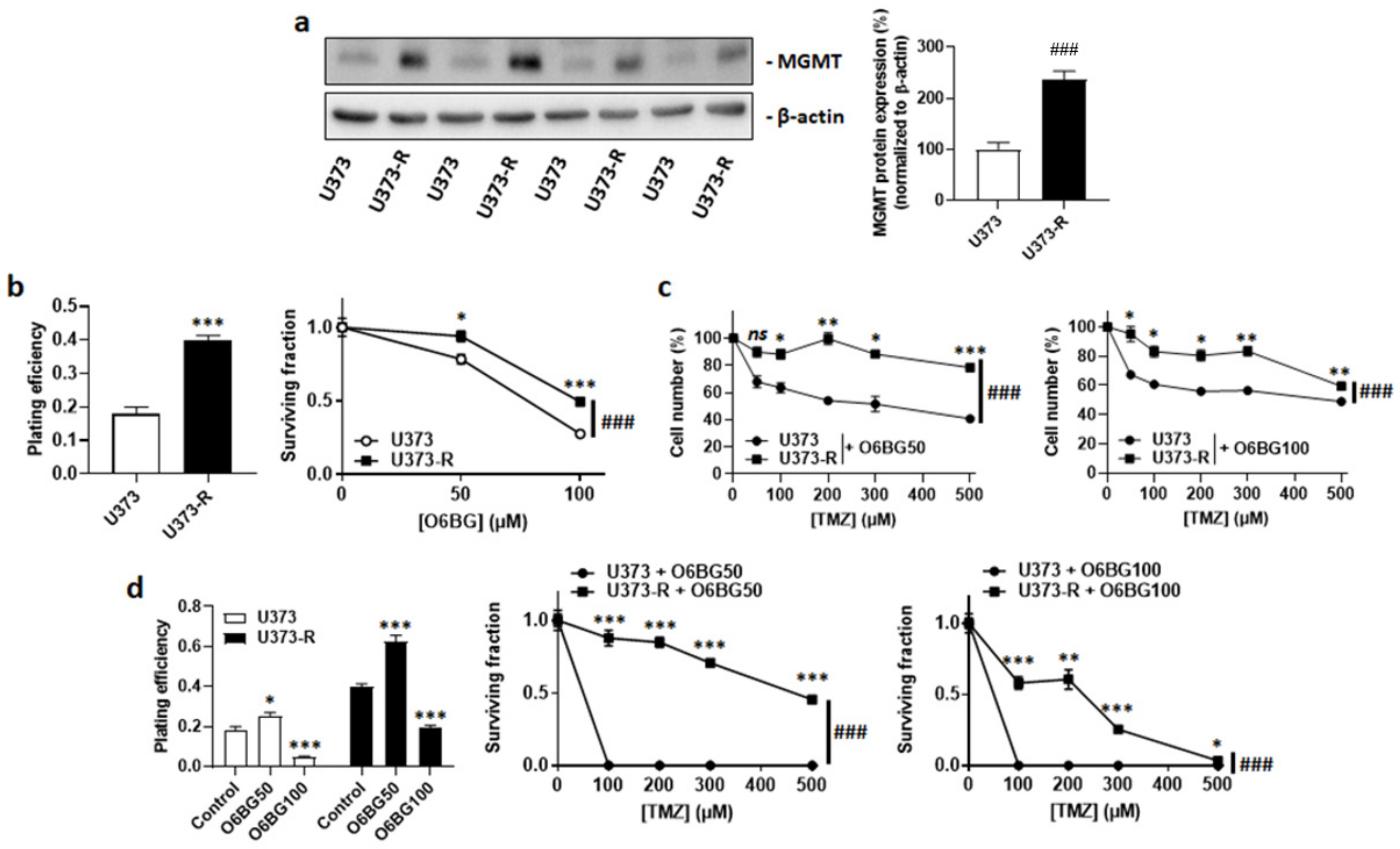
2.2. High MGMT Expression Does Not Account for the Acquired Resistance of Human U373-R Glioblastoma Cells to TMZ
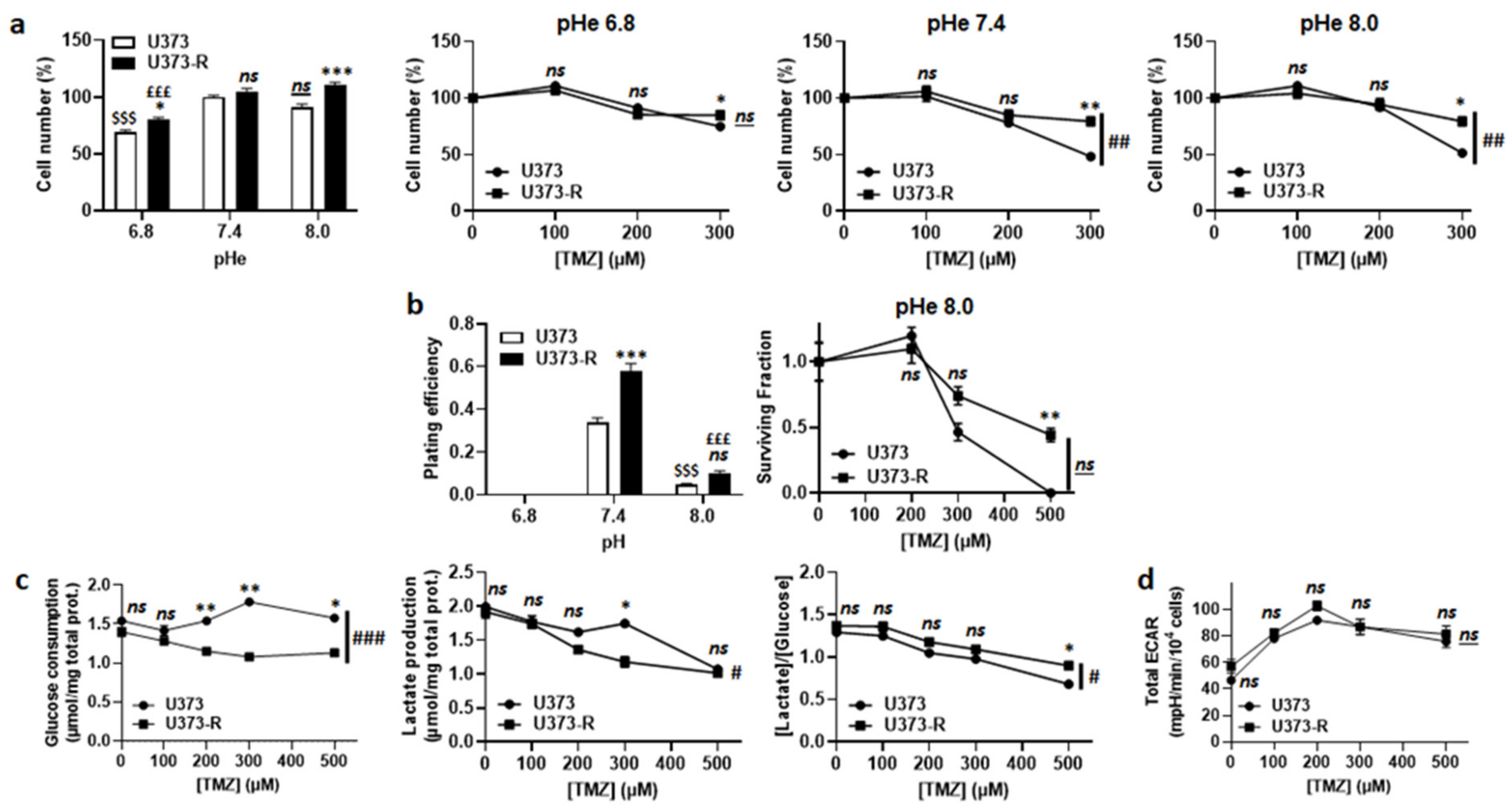
2.3. Extracellular Acidification Inactivates TMZ, but This Metabolic Strategy Is Not Used by Human U373-R Glioblastoma Cells to Resist to TMZ
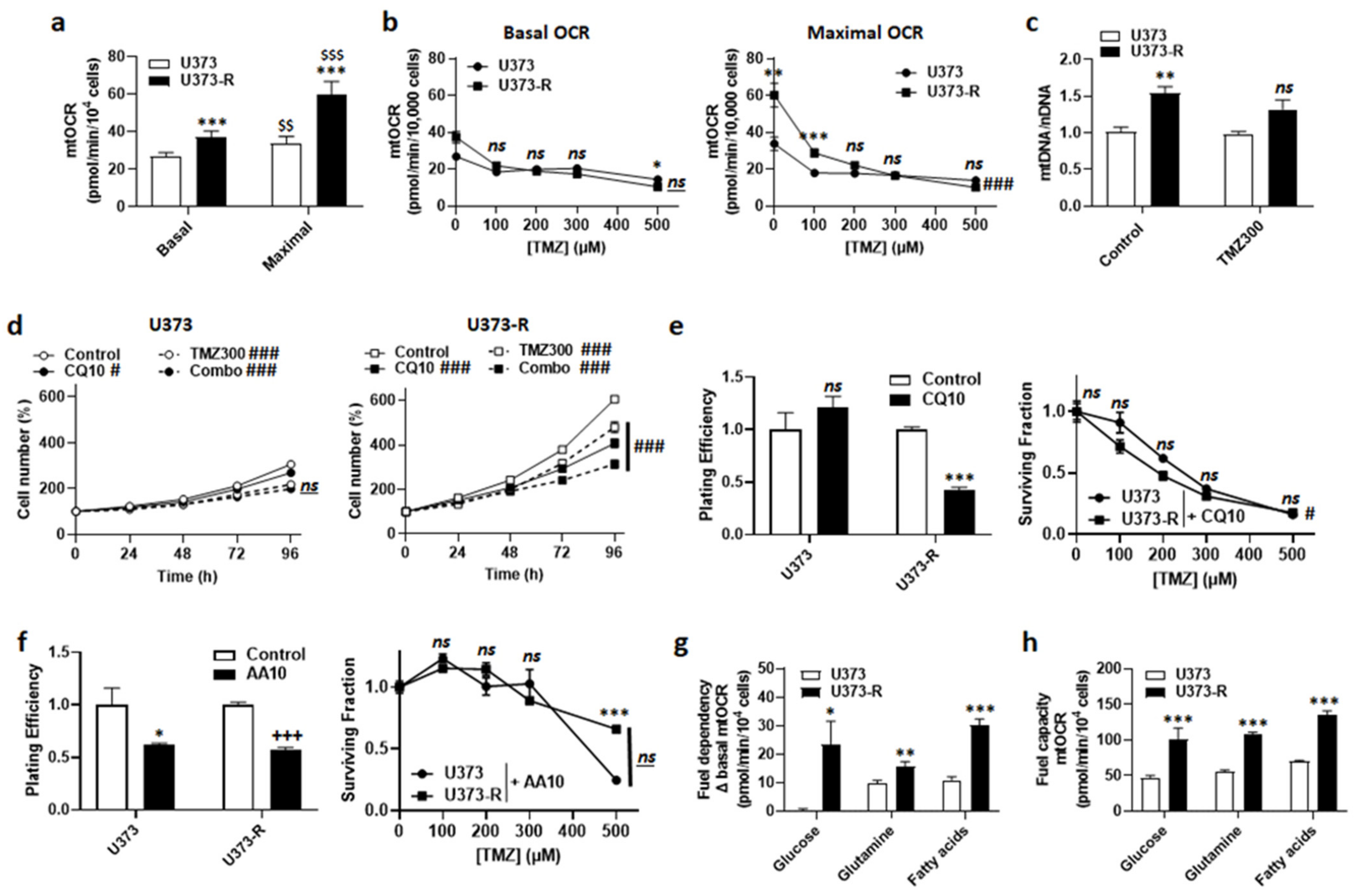
2.4. Maintenance of Fitter Mitochondria with Enhanced Oxidative Activity Participates in the Resistance of Human U373-R Glioblastoma Cells to TMZ
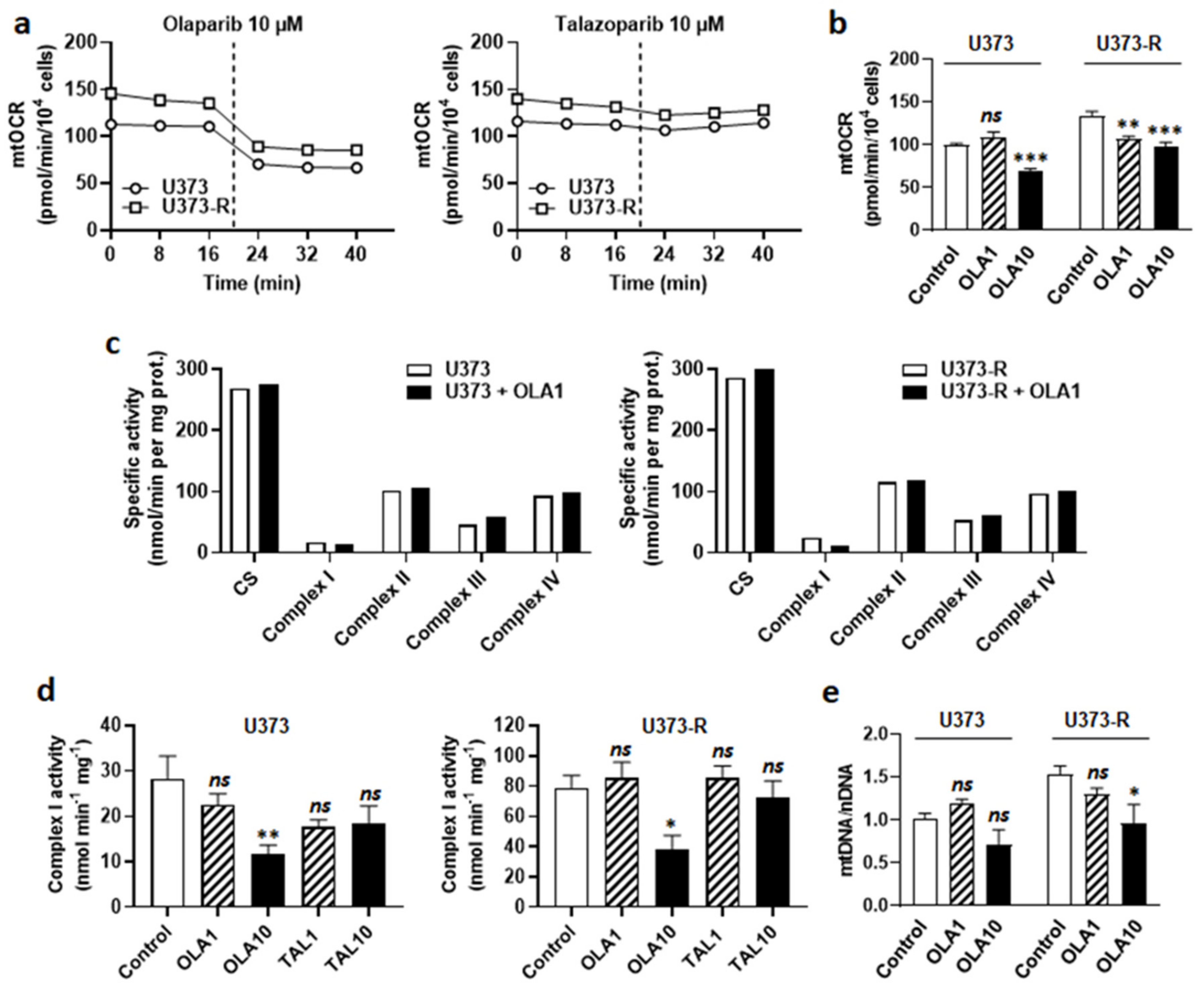
2.5. Olaparib Is an ETC Complex I Inhibitor That Sensitizes Human U373-R Glioblastoma Cells to TMZ
2.6. Targeting Either Autophagy or Oxidative Metabolism Sensitizes Intrinsically Resistant Human T98G Glioblastoma Cells to TMZ
3. Discussion
4. Materials and Methods
4.1. Cells and Cell Culture
4.2. Drugs and Treatments
4.3. Cell Number
4.4. Clonogenicity
4.5. Western Blotting
4.6. Seahorse Oximetry and pH-Metry
4.7. Glycolytic Efficiency
4.8. Mitochondrial DNA Content
4.9. Activities of Electron Transport Chain Complexes
4.10. Nuclear Magnetic Resonance
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tan, A.C.; Ashley, D.M.; Lopez, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Lefranc, F.; Brotchi, J.; Kiss, R. Possible future issues in the treatment of glioblastomas: Special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J. Clin. Oncol. 2005, 23, 2411–2422. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Sadeghi, N.; Camby, I.; Metens, T.; Dewitte, O.; Kiss, R. Present and potential future issues in glioblastoma treatment. Expert Rev. Anticancer Ther. 2006, 6, 719–732. [Google Scholar] [CrossRef]
- Lara-Velazquez, M.; Al-Kharboosh, R.; Jeanneret, S.; Vazquez-Ramos, C.; Mahato, D.; Tavanaiepour, D.; Rahmathulla, G.; Quinones-Hinojosa, A. Advances in brain tumor surgery for glioblastoma in adults. Brain Sci. 2017, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.H.; Hsueh, W.T.; Chuang, J.Y.; Chang, K.Y. Dissecting the mechanism of temozolomide resistance and its association with the regulatory roles of intracellular reactive oxygen species in glioblastoma. J. Biomed. Sci. 2021, 28, 18. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S.M.; Newlands, E.S.; Glaser, M.G.; Brampton, M.; Rice-Edwards, J.M.; Illingworth, R.D.; Richards, P.G.; Kennard, C.; Colquhoun, I.R.; Lewis, P.; et al. Temozolomide: A new oral cytotoxic chemotherapeutic agent with promising activity against primary brain tumours. Eur. J. Cancer 1993, 29A, 940–942. [Google Scholar] [CrossRef]
- Agarwala, S.S.; Kirkwood, J.M. Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. Oncologist 2000, 5, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Denny, B.J.; Wheelhouse, R.T.; Stevens, M.F.; Tsang, L.L.; Slack, J.A. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 1994, 33, 9045–9051. [Google Scholar] [CrossRef] [PubMed]
- Saleem, A.; Brown, G.D.; Brady, F.; Aboagye, E.O.; Osman, S.; Luthra, S.K.; Ranicar, A.S.; Brock, C.S.; Stevens, M.F.; Newlands, E.; et al. Metabolic activation of temozolomide measured in vivo using positron emission tomography. Cancer Res. 2003, 63, 2409–2415. [Google Scholar] [PubMed]
- Zhang, J.; Stevens, M.F.; Hummersone, M.; Madhusudan, S.; Laughton, C.A.; Bradshaw, T.D. Certain imidazotetrazines escape O6-methylguanine-DNA methyltransferase and mismatch repair. Oncology 2011, 80, 195–207. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Godard, S.; Dietrich, P.Y.; Regli, L.; Ostermann, S.; Otten, P.; Van Melle, G.; de Tribolet, N.; Stupp, R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004, 10, 1871–1874. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Preusser, M. Clinical neuropathology practice guide 06–2012: MGMT testing in elderly glioblastoma patients--yes, but how? Clin. Neuropathol. 2012, 31, 405–408. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weller, M.; Tabatabai, G.; Kastner, B.; Felsberg, J.; Steinbach, J.P.; Wick, A.; Schnell, O.; Hau, P.; Herrlinger, U.; Sabel, M.C.; et al. MGMT promoter methylation is a strong prognostic biomarker for benefit from dose-intensified temozolomide rechallenge in progressive glioblastoma: The DIRECTOR trial. Clin. Cancer Res. 2015, 21, 2057–2064. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W.; et al. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.A.; Jiang, S.X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; Sampson, J.H.; et al. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J. Clin. Oncol. 2009, 27, 1262–1267. [Google Scholar] [CrossRef]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro. Oncol. 2016, 18, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Le Calve, B.; Rynkowski, M.; Le Mercier, M.; Bruyere, C.; Lonez, C.; Gras, T.; Haibe-Kains, B.; Bontempi, G.; Decaestecker, C.; Ruysschaert, J.M.; et al. Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia 2010, 12, 727–739. [Google Scholar] [CrossRef]
- Tano, K.; Shiota, S.; Collier, J.; Foote, R.S.; Mitra, S. Isolation and structural characterization of a cDNA clone encoding the human DNA repair protein for O6-alkylguanine. Proc. Natl. Acad. Sci. USA 1990, 87, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Broniscer, A.; Gururangan, S.; MacDonald, T.J.; Goldman, S.; Packer, R.J.; Stewart, C.F.; Wallace, D.; Danks, M.K.; Friedman, H.S.; Poussaint, T.Y.; et al. Phase I trial of single-dose temozolomide and continuous administration of o6-benzylguanine in children with brain tumors: A pediatric brain tumor consortium report. Clin. Cancer Res. 2007, 13, 6712–6718. [Google Scholar] [CrossRef] [PubMed]
- Grasso, D.; Medeiros, H.C.D.; Zampieri, L.X.; Bol, V.; Danhier, P.; van Gisbergen, M.W.; Bouzin, C.; Brusa, D.; Gregoire, V.; Smeets, H.; et al. Fitter mitochondria are associated with radioresistance in human head and neck SQD9 cancer cells. Front. Pharmacol. 2020, 11, 263. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, L.X.; Grasso, D.; Bouzin, C.; Brusa, D.; Rossignol, R.; Sonveaux, P. Mitochondria participate in chemoresistance to cisplatin in human ovarian cancer cells. Mol. Cancer Res. 2020, 18, 1379–1391. [Google Scholar] [CrossRef]
- Hardy, L.; Frison, M.; Campanella, M. Breast cancer cells exploit mitophagy to exert therapy resistance. Oncotarget 2018, 9, 14040–14041. [Google Scholar] [CrossRef]
- Garcia-Heredia, J.M.; Carnero, A. Role of mitochondria in cancer stem cell resistance. Cells 2020, 9, 1693. [Google Scholar] [CrossRef]
- Yan, C.; Luo, L.; Guo, C.Y.; Goto, S.; Urata, Y.; Shao, J.H.; Li, T.S. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017, 388, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Atkins, R.J.; Ng, W.; Stylli, S.S.; Hovens, C.M.; Kaye, A.H. Repair mechanisms help glioblastoma resist treatment. J. Clin. Neurosci. 2015, 22, 14–20. [Google Scholar] [CrossRef]
- Gupta, S.K.; Smith, E.J.; Mladek, A.C.; Tian, S.; Decker, P.A.; Kizilbash, S.H.; Kitange, G.J.; Sarkaria, J.N. PARP inhibitors for sensitization of alkylation chemotherapy in glioblastoma: Impact of blood-brain barrier and molecular heterogeneity. Front. Oncol. 2018, 8, 670. [Google Scholar] [CrossRef]
- Gupta, S.K.; Mladek, A.C.; Carlson, B.L.; Boakye-Agyeman, F.; Bakken, K.K.; Kizilbash, S.H.; Schroeder, M.A.; Reid, J.; Sarkaria, J.N. Discordant in vitro and in vivo chemopotentiating effects of the PARP inhibitor veliparib in temozolomide-sensitive versus -resistant glioblastoma multiforme xenografts. Clin. Cancer Res. 2014, 20, 3730–3741. [Google Scholar] [CrossRef]
- Clarke, M.J.; Mulligan, E.A.; Grogan, P.T.; Mladek, A.C.; Carlson, B.L.; Schroeder, M.A.; Curtin, N.J.; Lou, Z.; Decker, P.A.; Wu, W.; et al. Effective sensitization of temozolomide by ABT-888 is lost with development of temozolomide resistance in glioblastoma xenograft lines. Mol. Cancer Ther. 2009, 8, 407–414. [Google Scholar] [CrossRef]
- Erice, O.; Smith, M.P.; White, R.; Goicoechea, I.; Barriuso, J.; Jones, C.; Margison, G.P.; Acosta, J.C.; Wellbrock, C.; Arozarena, I. MGMT expression predicts PARP-mediated resistance to temozolomide. Mol. Cancer Ther. 2015, 14, 1236–1246. [Google Scholar] [CrossRef]
- Hanna, C.; Kurian, K.M.; Williams, K.; Watts, C.; Jackson, A.; Carruthers, R.; Strathdee, K.; Cruickshank, G.; Dunn, L.; Erridge, S.; et al. Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: Results of the phase I OPARATIC trial. Neuro. Oncol. 2020, 22, 1840–1850. [Google Scholar] [CrossRef]
- Majd, N.K.; Yap, T.A.; Koul, D.; Balasubramaniyan, V.; Li, X.; Khan, S.; Gandy, K.S.; Yung, W.K.A.; de Groot, J.F. The promise of DNA damage response inhibitors for the treatment of glioblastoma. Neuro-Oncol. Adv. 2021, 3, vdab015. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.C.; Van Meir, E.G. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef]
- Cao, V.T.; Jung, T.Y.; Jung, S.; Jin, S.G.; Moon, K.S.; Kim, I.Y.; Kang, S.S.; Park, C.S.; Lee, K.H.; Chae, H.J. The correlation and prognostic significance of MGMT promoter methylation and MGMT protein in glioblastomas. Neurosurgery 2009, 65, 866–875. [Google Scholar] [CrossRef]
- Pan, Y.; Cao, M.; Liu, J.; Yang, Q.; Miao, X.; Go, V.L.W.; Lee, P.W.N.; Xiao, G.G. Metabolic regulation in mitochondria and drug resistance. Adv. Exp. Med. Biol. 2017, 1038, 149–171. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, M.; van Niekerk, G.; Fourie, C.; du Plessis, M.; Engelbrecht, A.M. The impact of mitochondria on cancer treatment resistance. Cell Oncol. 2021, 44, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.R.; Nozell, S.E.; Diers, A.; McClugage, S.G., 3rd; Sarkaria, J.N.; Markert, J.M.; Darley-Usmar, V.M.; Bailey, S.M.; Gillespie, G.Y.; Landar, A.; et al. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J. Biol. Chem. 2010, 285, 39759–39767. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Lee, C.C.; Shih, Y.L.; Lin, C.H.; Wang, S.H.; Chen, T.H.; Shih, C.M. Inhibition of mitochondria- and endoplasmic reticulum stress-mediated autophagy augments temozolomide-induced apoptosis in glioma cells. PLoS ONE 2012, 7, e38706. [Google Scholar] [CrossRef]
- Khan, I.; Baig, M.H.; Mahfooz, S.; Rahim, M.; Karacam, B.; Elbasan, E.B.; Ulasov, I.; Dong, J.J.; Hatiboglu, M.A. Deciphering the role of autophagy in treatment of resistance mechanisms in glioblastoma. Int. J. Mol. Sci. 2021, 22, 1318. [Google Scholar] [CrossRef]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef]
- He, C.; Lu, S.; Wang, X.Z.; Wang, C.C.; Wang, L.; Liang, S.P.; Luo, T.F.; Wang, Z.C.; Piao, M.H.; Chi, G.F.; et al. FOXO3a protects glioma cells against temozolomide-induced DNA double strand breaks via promotion of BNIP3-mediated mitophagy. Acta Pharmacol. Sin. 2021, 42, 1324–1337. [Google Scholar] [CrossRef]
- Nagy, A.; Eder, K.; Selak, M.A.; Kalman, B. Mitochondrial energy metabolism and apoptosis regulation in glioblastoma. Brain Res. 2015, 1595, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, S.; Lo Dico, A.; Raccagni, I.; Gaglio, D.; Belloli, S.; Politi, L.S.; Martelli, C.; Diceglie, C.; Bonanomi, M.; Ercoli, G.; et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget 2017, 8, 113090–113104. [Google Scholar] [CrossRef] [PubMed]
- Kizilbash, S.H.; Gupta, S.K.; Chang, K.; Kawashima, R.; Parrish, K.E.; Carlson, B.L.; Bakken, K.K.; Mladek, A.C.; Schroeder, M.A.; Decker, P.A.; et al. Restricted delivery of talazoparib across the blood-brain barrier limits the sensitizing effects of PARP inhibition on temozolomide therapy in glioblastoma. Mol. Cancer Ther. 2017, 16, 2735–2746. [Google Scholar] [CrossRef]
- Van Hee, V.F.; Perez-Escuredo, J.; Cacace, A.; Copetti, T.; Sonveaux, P. Lactate does not activate NF-kappaB in oxidative tumor cells. Front. Pharmacol. 2015, 6, 228. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrowsf-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Aleardi, A.M.; Benard, G.; Augereau, O.; Malgat, M.; Talbot, J.C.; Mazat, J.P.; Letellier, T.; Dachary-Prigent, J.; Solaini, G.C.; Rossignol, R. Gradual alteration of mitochondrial structure and function by beta-amyloids: Importance of membrane viscosity changes, energy deprivation, reactive oxygen species production, and cytochrome c release. J. Bioenerg. Biomembr. 2005, 37, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Takeshima, H.; Kataoka, H. Anti-glioma therapy with temozolomide and status of the DNA-repair gene MGMT. Anticancer Res. 2009, 29, 4845–4854. [Google Scholar] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zampieri, L.X.; Sboarina, M.; Cacace, A.; Grasso, D.; Thabault, L.; Hamelin, L.; Vazeille, T.; Dumon, E.; Rossignol, R.; Frédérick, R.; et al. Olaparib Is a Mitochondrial Complex I Inhibitor That Kills Temozolomide-Resistant Human Glioblastoma Cells. Int. J. Mol. Sci. 2021, 22, 11938. https://doi.org/10.3390/ijms222111938
Zampieri LX, Sboarina M, Cacace A, Grasso D, Thabault L, Hamelin L, Vazeille T, Dumon E, Rossignol R, Frédérick R, et al. Olaparib Is a Mitochondrial Complex I Inhibitor That Kills Temozolomide-Resistant Human Glioblastoma Cells. International Journal of Molecular Sciences. 2021; 22(21):11938. https://doi.org/10.3390/ijms222111938
Chicago/Turabian StyleZampieri, Luca X., Martina Sboarina, Andrea Cacace, Debora Grasso, Léopold Thabault, Loïc Hamelin, Thibaut Vazeille, Elodie Dumon, Rodrigue Rossignol, Raphaël Frédérick, and et al. 2021. "Olaparib Is a Mitochondrial Complex I Inhibitor That Kills Temozolomide-Resistant Human Glioblastoma Cells" International Journal of Molecular Sciences 22, no. 21: 11938. https://doi.org/10.3390/ijms222111938
APA StyleZampieri, L. X., Sboarina, M., Cacace, A., Grasso, D., Thabault, L., Hamelin, L., Vazeille, T., Dumon, E., Rossignol, R., Frédérick, R., Sonveaux, E., Lefranc, F., & Sonveaux, P. (2021). Olaparib Is a Mitochondrial Complex I Inhibitor That Kills Temozolomide-Resistant Human Glioblastoma Cells. International Journal of Molecular Sciences, 22(21), 11938. https://doi.org/10.3390/ijms222111938