
Host Defense Peptides: Dual Antimicrobial and Immunomodulatory Action

 , , , and
, , , and 
Abstract
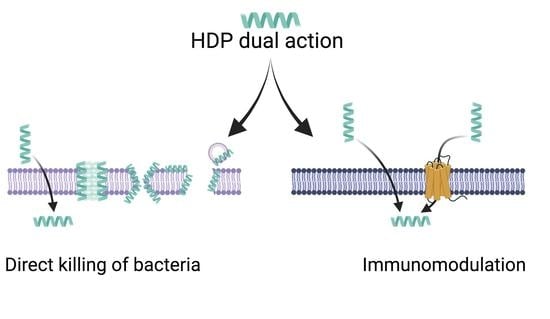
:
1. Antibiotic Resistance
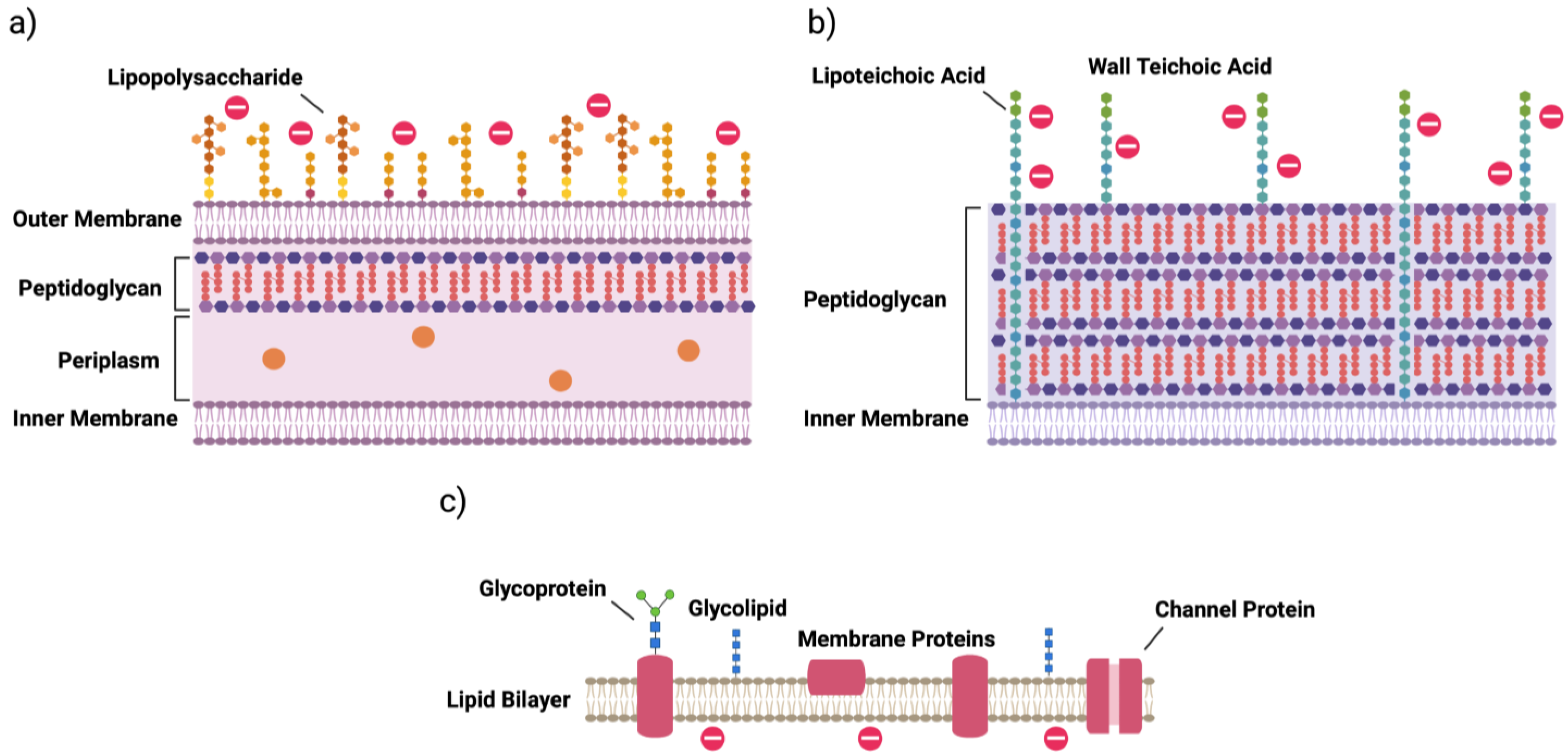
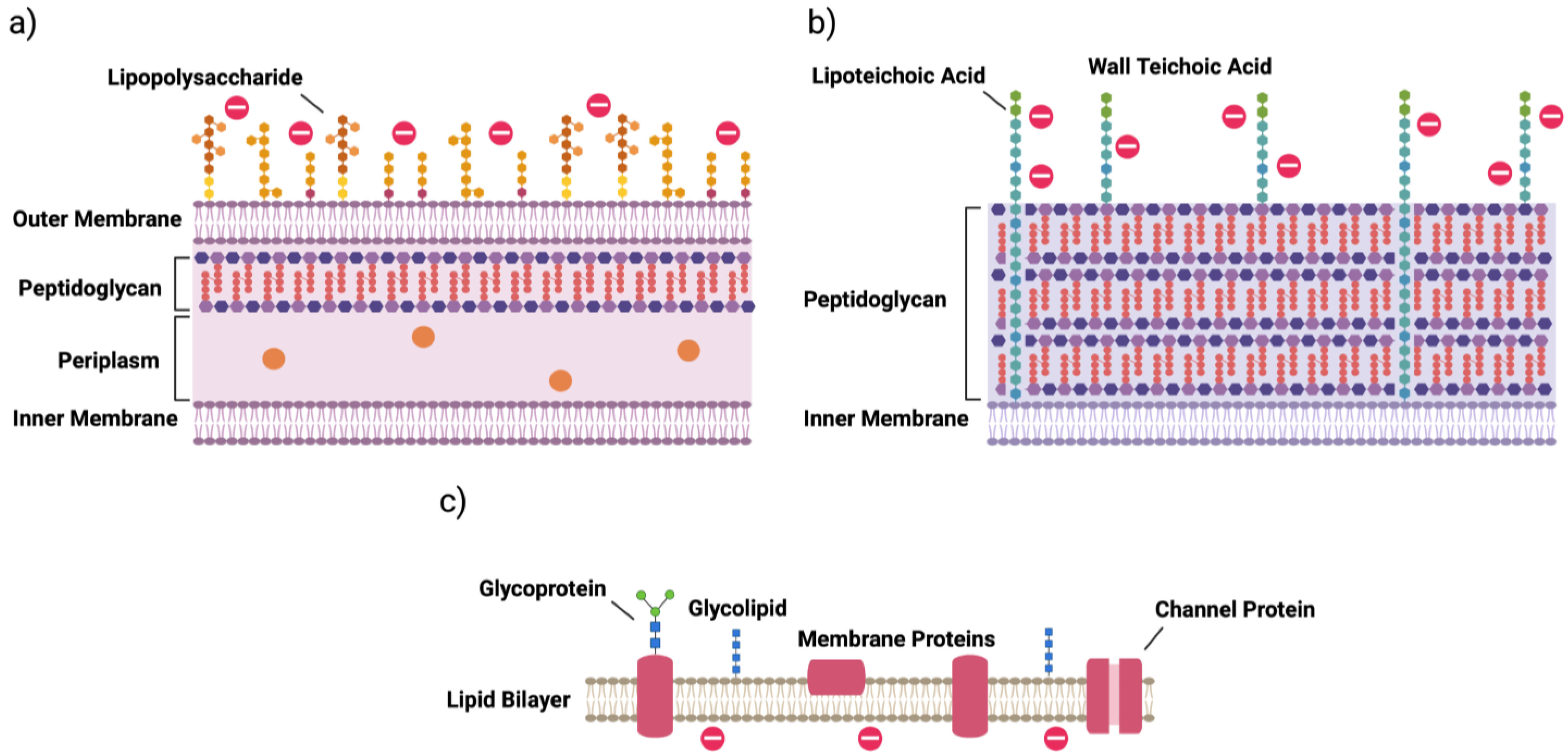
2. Bacterial Cells
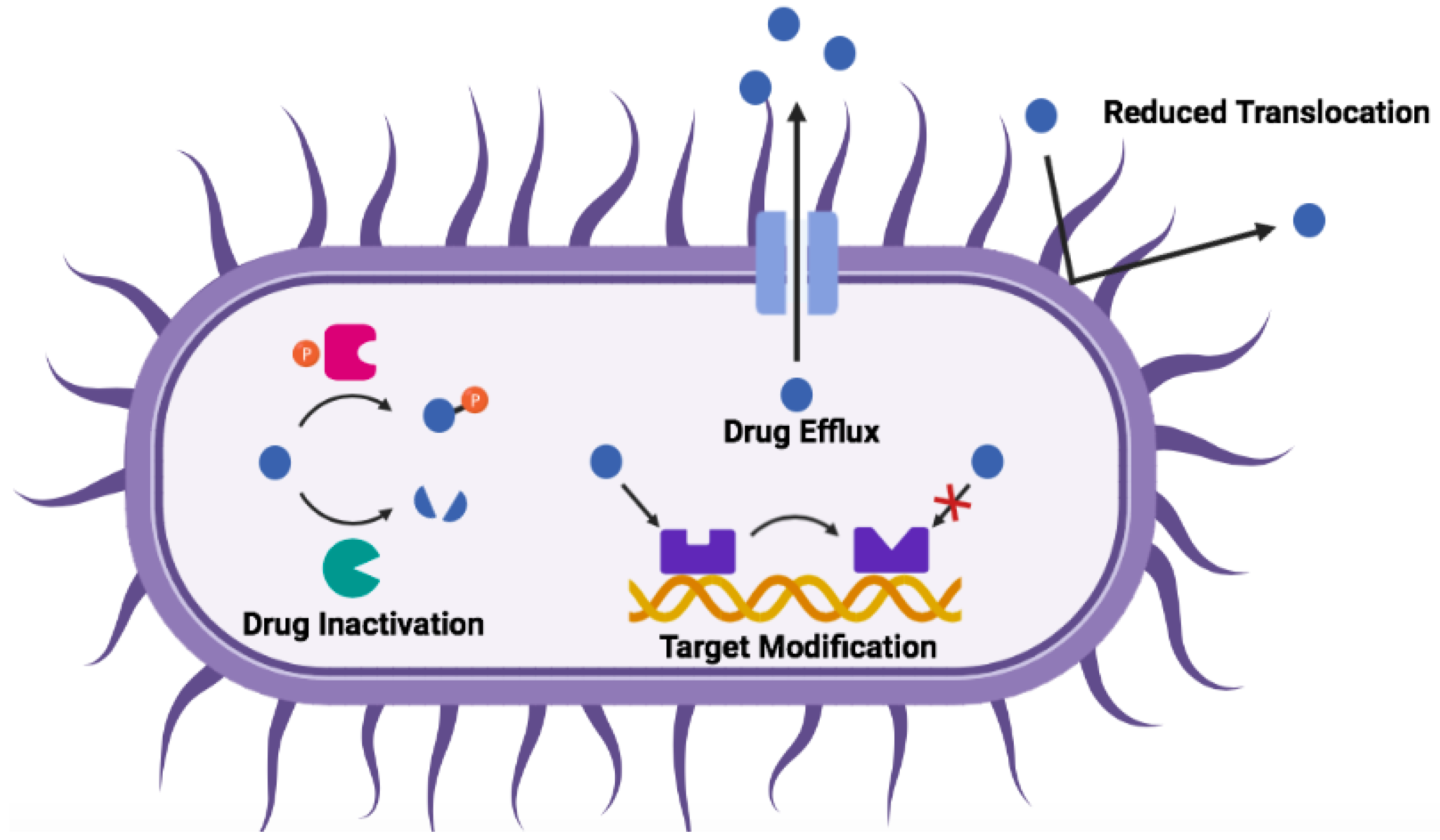
3. Mechanisms of Antibiotic Resistance
4. Host Defense Peptides
4.1. Structures and Classes
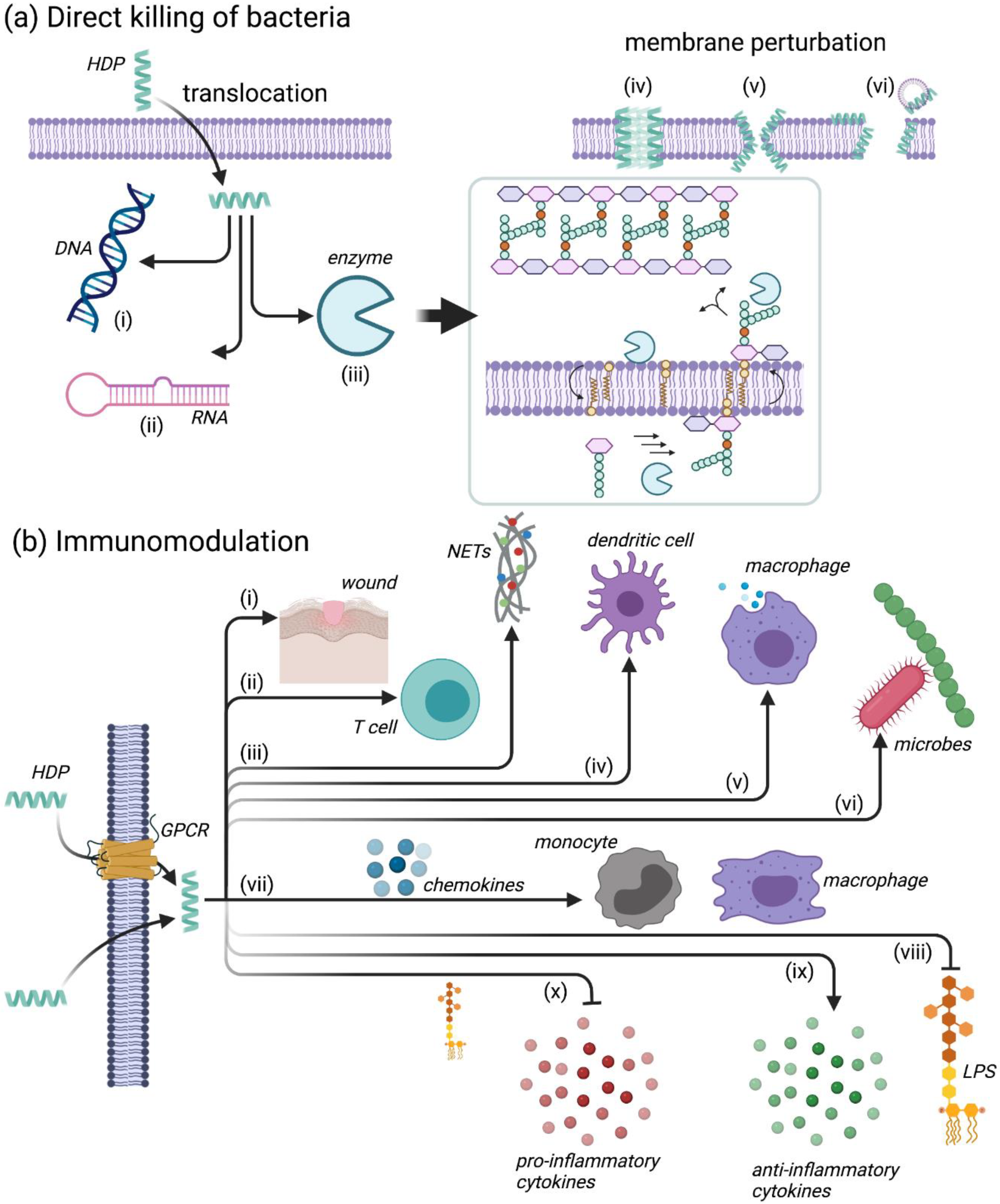
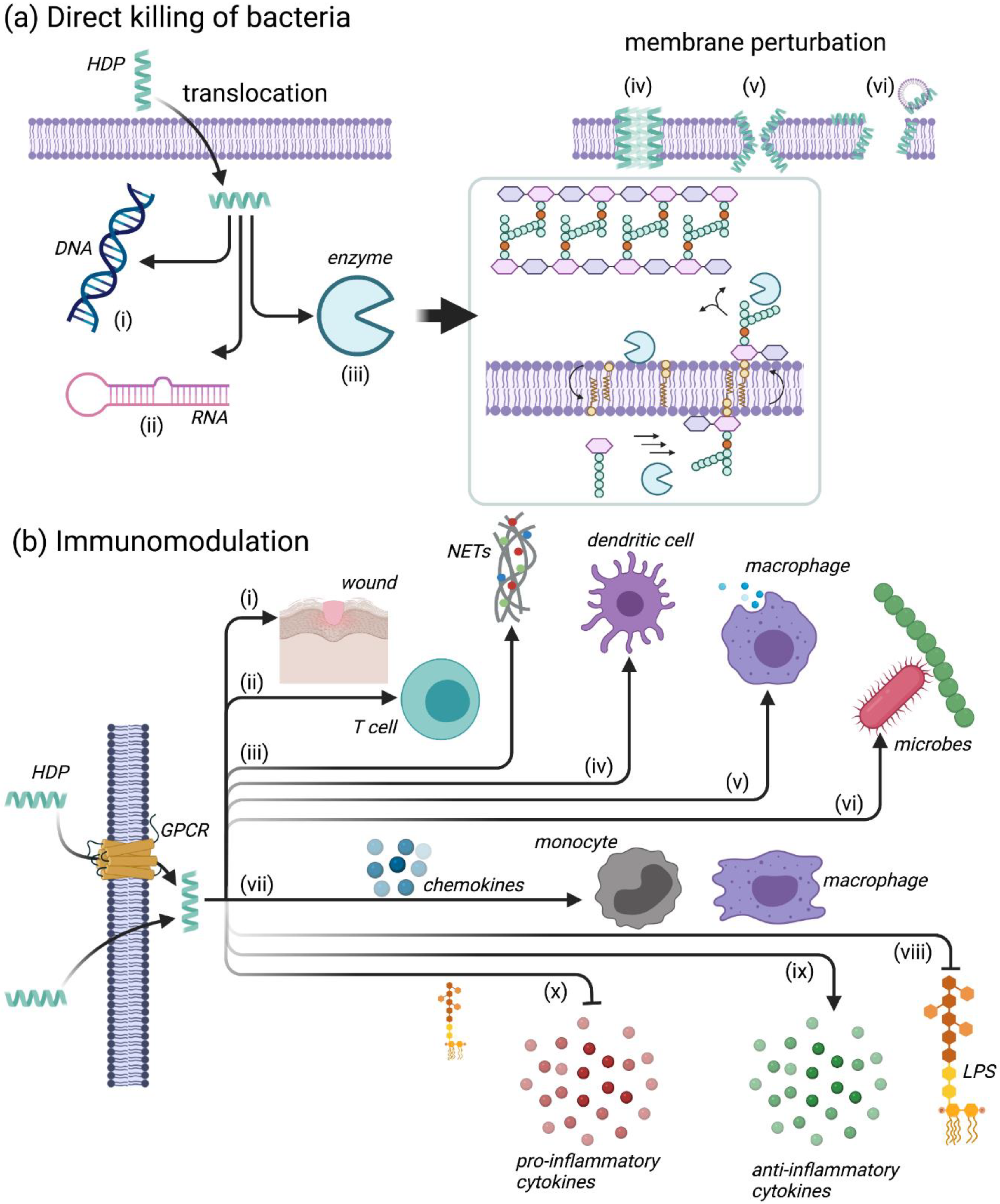
4.2. Mechanisms of Action for HDPs
4.2.1. Leukocyte Recruitment
4.2.2. Modulation of Neutrophil Function
4.2.3. Regulation of Inflammation
4.2.4. Additional Immunomodulatory Functions
4.2.5. Illustration of the Diversity in HDP Function: From a Natural Peptide to Synthetic Analogues
4.3. Shortcomings of HDPs and Mitigation Strategies
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aminov, R.I. A brief history of the antibiotic era: Lessons learned and challenges for the future. Front. Microbiol. 2010, 1, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ting, D.S.J.; Beuerman, R.W.; Dua, H.S.; Lakshminarayanan, R.; Mohammed, I. Strategies in Translating the Therapeutic Potentials of Host Defense Peptides. Front. Immunol. 2020, 11, 983. [Google Scholar] [CrossRef]
- Davies, J. Origins and evolution of antibiotic resistance. Microbiologia 1996, 12, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Ventola, C.L. The antibiotic resistance crisis: Causes and threats. P T J. 2015, 40, 277–283. [Google Scholar]
- Shallcross, L.J.; Howard, S.J.; Fowler, T.; Davies, S.C. Tackling the threat of antimicrobial resistance: From policy to sustainable action. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140082. [Google Scholar] [CrossRef] [PubMed]
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karlén, A. The global preclinical antibacterial pipeline. Nat. Rev. Microbiol. 2019, 18, 275–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrei, S.; Valeanu, L.; Chirvasuta, R.; Stefan, M.-G. New FDA approved antibacterial drugs: 2015–2017. Discoveries 2018, 6, e81. [Google Scholar] [CrossRef] [PubMed]
- Andrei, S.; Droc, G.; Stefan, G. FDA approved antibacterial drugs: 2018–2019. Discoveries 2019, 7, e102. [Google Scholar] [CrossRef]
- WHO. Antimicrobial resistance. Bull. World Health Organ. 2014, 61, 383–394. [Google Scholar]
- Matsunaga, N.; Hayakawa, K. Estimating the impact of antimicrobial resistance. Lancet Glob. Health 2018, 6, e934–e935. [Google Scholar] [CrossRef] [Green Version]
- Langford, B.J.; So, M.; Raybardhan, S.; Leung, V.; Westwood, D.; MacFadden, D.R.; Soucy, J.-P.R.; Daneman, N. Bacterial co-infection and secondary infection in patients with COVID-19: A living rapid review and meta-analysis. Clin. Microbiol. Infect. 2020, 26, 1622–1629. [Google Scholar] [CrossRef]
- Strathdee, S.A.; Davies, S.C.; Marcelin, J.R. Confronting antimicrobial resistance beyond the COVID-19 pandemic and the 2020 US election. Lancet 2020, 396, 1050–1053. [Google Scholar] [CrossRef]
- Hsu, J. How covid-19 is accelerating the threat of antimicrobial resistance. BMJ 2020, 369, 1–2. [Google Scholar]
- Coico, R. Gram staining. Curr. Protoc. Microbiol. 2005, 3, A.3C.1–A.3C.2. [Google Scholar] [CrossRef]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- Opal, S.M. Endotoxins and Other Sepsis Triggers. Endotoxemia Endotoxin Shock: Dis. Diagn. Ther. 2010, 167, 14–24. [Google Scholar] [CrossRef]
- Swoboda, J.G.; Campbell, J.; Meredith, T.C.; Walker, S. Wall teichoic acid function, biosynthesis, and inhibition. ChemBioChem 2010, 11, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sani, M.A.; Henriques, S.T.; Weber, D.; Separovic, F. Bacteria may cope differently from similar membrane damage caused by the Australian tree frog antimicrobial peptide maculatin 1.1. J. Biol. Chem. 2015, 290, 19853–19862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, Y.; Shai, Y. Lipopolysaccharide (Endotoxin)-host defense antibacterial peptides interactions: Role in bacterial resistance and prevention of sepsis. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1513–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, D.C. Mechanisms of action of antimicrobials: Focus on fluoroquinolones. Clin. Infect. Dis. 2001, 32, 9–15. [Google Scholar] [CrossRef]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Reygaert, C.W. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.; Harper, D.; et al. Alternatives to antibiotics—a pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Theuretzbacher, U.; Piddock, L.J.V. Non-traditional Antibacterial Therapeutic Options and Challenges. Cell Host Microbe 2019, 26, 61–72. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Falla, T.J. Antimicrobial peptides: Broad-spectrum antibiotics from nature. Clin. Microbiol. Infect. 1996, 1, 226–229. [Google Scholar] [CrossRef] [Green Version]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Alford, M.A.; Baquir, B.; Santana, F.L.; Haney, E.F.; Hancock, R.E.W. Cathelicidin Host Defense Peptides and Inflammatory Signaling: Striking a Balance. Front. Microbiol. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Joo, H.S.; Fu, C.I.; Otto, M. Bacterial strategies of resistance to antimicrobial peptides. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150292. [Google Scholar] [CrossRef] [Green Version]
- Koo, H.B.; Seo, J. Antimicrobial peptides under clinical investigation. Pept. Sci. 2019, 111, e24122. [Google Scholar] [CrossRef]
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- de la Fuente-Núñez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Broad-Spectrum Anti-biofilm Peptide that Targets a Cellular Stress Response. PLoS Pathog. 2014, 10, e1004152. [Google Scholar] [CrossRef] [Green Version]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial peptides: An emerging category of therapeutic agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliani, A.; Pirri, G.; Nicoletto, S.F. Antimicrobial peptides: An overview of a promising class of therapeutics. Cent. Eur. J. Biol. 2007, 2, 1–33. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with Dual Antimicrobial and Anticancer Activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleury, Y.; Dayem, M.A.; Montagne, J.J.; Chaboisseau, E.; Le Caer, J.P.; Nicolas, P.; Delfour, A. Covalent structure, synthesis, and structure-function studies of mesentericin Y 10537, a defensive peptide from gram-positive bacteria leuconostoc mesenteroides. J. Biol. Chem. 1996, 271, 14421–14429. [Google Scholar] [CrossRef] [Green Version]
- Breukink, E.; De Kruijff, B. The lantibiotic nisin, a special case or not? Biochim. Biophys. Acta Biomembr. 1999, 1462, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Fillion, M.; Valois-Paillard, G.; Lorin, A.; Noël, M.; Voyer, N.; Auger, M. Membrane Interactions of Synthetic Peptides with Antimicrobial Potential: Effect of Electrostatic Interactions and Amphiphilicity. Probiotics Antimicrob. Proteins 2014, 7, 66–74. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.G.; Dullaghan, E.; Mookherjee, N.; Glavas, N.; Waldbrook, M.; Thompson, A.; Wang, A.; Lee, K.; Doria, S.; Hamill, P.; et al. An anti-infective peptide that selectively modulates the innate immune response. Nat. Biotechnol. 2007, 25, 465–472. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Nijnik, A.; Philpott, D.J. Modulating immunity as a therapy for bacterial infections. Nat. Rev. Microbiol. 2012, 10, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Madera, L.; Ma, S.; Waldbrook, M.; Elliott, M.R.; Easton, D.M.; Mayer, M.L.; Mullaly, S.C.; Kindrachuk, J.; Jenssen, H.; et al. Synthetic Cationic Peptide IDR-1002 Provides Protection against Bacterial Infections through Chemokine Induction and Enhanced Leukocyte Recruitment. J. Immunol. 2010, 184, 2539–2550. [Google Scholar] [CrossRef] [Green Version]
- Holly, M.K.; Diaz, K.; Smith, J.G. Defensins in Viral Infection and Pathogenesis. Annu. Rev. Virol. 2017, 4, 369–391. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: Beyond antimicrobial activity. Nat. Publ. Gr. 2016, 16, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Pundir, P.; Catalli, A.; Leggiadro, C.; Douglas, S.E.; Kulka, M. Pleurocidin, a novel antimicrobial peptide, induces human mast cell activation through the FPRL1 receptor. Mucosal Immunol. 2014, 7, 177–187. [Google Scholar] [CrossRef]
- Di Nardo, A.; Yamasaki, K.; Dorschner, R.A.; Lai, Y.; Gallo, R.L. Mast Cell Cathelicidin Antimicrobial Peptide Prevents Invasive Group A Streptococcus Infection of the Skin. J. Immunol. 2008, 180, 7565–7573. [Google Scholar] [CrossRef] [Green Version]
- Hazlett, L.; Wu, M. Defensins in innate immunity. Cell Tissue Res. 2011, 343, 175–188. [Google Scholar] [CrossRef]
- Semple, F.; Dorin, J.R. β-Defensins: Multifunctional modulators of infection, inflammation and more? J. Innate Immun. 2012, 4, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Funderburg, N.; Lederman, M.M.; Feng, Z.; Drage, M.G.; Jadlowsky, J.; Harding, C.V.; Weinberg, A.; Sieg, S.F. Human β-defensin-3 activates professional antigen-presenting cells via Toll-like receptors 1 and 2. Proc. Natl. Acad. Sci. USA 2007, 104, 18631–18635. [Google Scholar] [CrossRef] [Green Version]
- Hemshekhar, M.; Anaparti, V.; Mookherjee, N. Functions of cationic host defense peptides in immunity. Pharmaceuticals 2016, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Niyonsaba, F.; Ushio, H.; Nagaoka, I.; Ikeda, S.; Okumura, K.; Ogawa, H. Cathelicidin LL-37 induces the generation of reactive oxygen species and release of human α-defensins from neutrophils. Br. J. Dermatol. 2007, 157, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Wang, G.; White, M.; Rynkiewicz, M.; Seaton, B.; Hartshorn, K. Identifying the critical domain of LL-37 involved in mediating neutrophil activation in the presence of influenza virus: Functional and structural analysis. PLoS ONE 2015, 10, e0133454. [Google Scholar] [CrossRef]
- Lande, R.; Pietraforte, I.; Mennella, A.; Palazzo, R.; Spinelli, F.; Giannakakis, K.; Spadaro, F.; Falchi, M.; Riccieri, V.; Stefanantoni, K.; et al. Complementary effects of carbamylated and citrullinated ll37 in autoimmunity and inflammation in systemic lupus erythematosus. Int. J. Mol. Sci. 2021, 22, 1650. [Google Scholar] [CrossRef]
- Pena, O.M.; Afacan, N.; Pistolic, J.; Chen, C.; Madera, L.; Falsafi, R.; Fjell, C.; Hancock, R. Synthetic Cationic Peptide IDR-1018 Modulates Human Macrophage Differentiation. PLoS ONE 2013, 8, e52449. [Google Scholar] [CrossRef] [Green Version]
- Bevins, C.L.; Salzman, N.H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 2011, 9, 356–368. [Google Scholar] [CrossRef]
- Cai, J.; Cui, X.; Wang, X.; You, L.; Ji, C.; Cao, Y. A Novel Anti-Infective Peptide BCCY-1 With Immunomodulatory Activities. Front. Immunol. 2021, 12, 713960. [Google Scholar] [CrossRef]
- He, X.; Yang, Y.; Mu, L.; Zhou, Y.; Chen, Y.; Wu, J.; Wang, Y.; Yang, H.; Li, M.; Xu, W.; et al. A frog-derived immunomodulatory peptide promotes cutaneous wound healing by regulating cellular response. Front. Immunol. 2019, 10, 2421. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Hu, Q.; Zhang, R.; Zhou, B.; Xie, D.; Wang, Y.; Zhang, X.; Yang, L. Rational design of innate defense regulator peptides as tumor vaccine adjuvants. NPJ Vaccines 2021, 6, 1–13. [Google Scholar] [CrossRef]
- Peng, L.; Scheenstra, M.R.; van Harten, R.M.; Haagsman, H.P.; Veldhuizen, E.J.A. The immunomodulatory effect of cathelicidin-B1 on chicken macrophages. Vet. Res. 2020, 51, 122. [Google Scholar] [CrossRef]
- Xiang, X.-W.; Zheng, H.-Z.; Wang, R.; Chen, H.; Xiao, J.-X.; Zheng, B.; Liu, S.-L.; Ding, Y.-T. Ameliorative Effects of Peptides Derived from Oyster (Crassostrea gigas) on Immunomodulatory Function and Gut Microbiota Structure in Cyclophosphamide-Treated Mice. Mar. Drugs 2021, 19, 456. [Google Scholar] [CrossRef]
- Bhattacharjya, S.; Straus, S.K. Design, engineering and discovery of novel α-helical and β-boomerang antimicrobial peptides against drug resistant bacteria. Int. J. Mol. Sci. 2020, 21, 5773. [Google Scholar] [CrossRef] [PubMed]
- Raheem, N.; Kumar, P.; Lee, E.; Cheng, J.T.; Hancock, R.; Straus, S.K. Insights into the mechanism of action of two analogues of aurein 2.2. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183262. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.-L.; Cheng, J.T.-J.; Hale, J.; Pan, J.; Hancock, R.; Straus, S.K. Characterization of the Structure and Membrane Interaction of the Antimicrobial Peptides Aurein 2.2 and 2.3 from Australian Southern Bell Frogs. Biophys. J. 2007, 92, 2854–2864. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.T.J.; Hale, J.D.; Elliot, M.; Hancock, R.E.W.; Straus, S.K. Effect of membrane composition on antimicrobial peptides aurein 2.2 and 2.3 from australian southern bell frogs. Biophys. J. 2009, 96, 552–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.T.J.; Hale, J.D.; Elliott, M.; Hancock, R.E.W.; Straus, S.K. The importance of bacterial membrane composition in the structure and function of aurein 2.2 and selected variants. Biochim. Biophys. Acta Biomembr. 2011, 1808, 622–633. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, M.; Senges, C.H.R.; Zhang, J.; Suleman, S.; Nguyen, M.; Kumar, P.; Chiriac, A.I.; Stepanek, J.J.; Raatschen, N.; May, C.; et al. Antimicrobial Peptides from the Aurein Family Form Ion-Selective Pores inBacillus subtilis. ChemBioChem 2015, 16, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Grein, F.; Müller, A.; Scherer, K.M.; Liu, X.; Ludwig, K.C.; Klöckner, A.; Strach, M.; Sahl, H.-G.; Kubitscheck, U.; Schneider, T. Ca2+-Daptomycin targets cell wall biosynthesis by forming a tripartite complex with undecaprenyl-coupled intermediates and membrane lipids. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Scoten, K.; Straus, S.K. Daptomycin Leakage Is Selective. ACS Infect. Dis. 2016, 2, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.T.; Hale, J.D.; Kindrachuk, J.; Jessen, H.; Elliott, M.; Hancock, R.E.; Straus, S.K. Importance of Residue 13 and the C-Terminus for the Structure and Activity of the Antimicrobial Peptide Aurein 2.2. Biophys. J. 2010, 99, 2926–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Takayesu, A.; Abbasi, U.; Kalathottukaren, M.T.; Abbina, S.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptide-Polymer Conjugates with High Activity: Influence of Polymer Molecular Weight and Peptide Sequence on Antimicrobial Activity, Proteolysis, and Biocompatibility. ACS Appl. Mater. Interfaces 2017, 9, 37575–37586. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Nguyen, L.T.; Schibli, D.J.; Vogel, H.J. Design of a novel tryptophan-rich membrane-active antimicrobial peptide from the membrane-proximal region of the HIV glycoprotein, gp41. Beilstein J. Org. Chem. 2012, 8, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, W.; Demcoe, A.R.; Vogel, H.J. Conformation of a Bactericidal Domain of Puroindoline a: Structure and Mechanism of Action of a 13-Residue Antimicrobial Peptide. J. Bacteriol. 2003, 185, 4938–4947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascher, T.; Zimmer, S.L.; Smith, T.-A.; Helmann, J.D. Antibiotic-Inducible Promoter Regulated by the Cell Envelope Stress-Sensing Two-Component System LiaRS of Bacillus subtilis. Antimicrob. Agents Chemother. 2004, 48, 2888–2896. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, M.; Resende, J.M.; Moraes, C.M.; Marquette, A.; Chich, J.; Metz-Boutigue, M.; Bechinger, B. Membrane structure and interactions of human catestatin by multidimensional solution and solid-state NMR spectroscopy. FASEB J. 2010, 24, 1737–1746. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- Marchand, C.; Krajewski, K.; Lee, H.-F.; Antony, S.; Johnson, A.A.; Amin, R.; Roller, P.; Kvaratskhelia, M.; Pommier, Y. Covalent binding of the natural antimicrobial peptide indolicidin to DNA abasic sites. Nucleic Acids Res. 2006, 34, 5157–5165. [Google Scholar] [CrossRef] [PubMed]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.W.; Rehm, B.H.A.; Hancock, R.E.W. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khafagy, E.-S.; Morishita, M.; Ida, N.; Nishio, R.; Isowa, K.; Takayama, K. Structural requirements of penetratin absorption enhancement efficiency for insulin delivery. J. Control. Release 2010, 143, 302–310. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide Antimicrobial Agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [Green Version]
- Kosikowska, P.; Lesner, A. Antimicrobial peptides (AMPs) as drug candidates: A patent review (2003–2015). Expert Opin. Ther. Pat. 2016, 26, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef]
- Haney, E.F.; Hancock, R.E.W. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 2013, 100, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, C.; Zhang, M.Z.; Zhang, S. Beta-defensin derived cationic antimicrobial peptides with potent killing activity against gram negative and gram positive bacteria. BMC Microbiol. 2018, 18, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehbach, J.; Gani, J.; Hilpert, K.; Craik, D.J. Comparison of a short linear antimicrobial peptide with its disulfide-cyclized and cyclotide-grafted variants against clinically relevant pathogens. Microorganisms 2021, 9, 1249. [Google Scholar] [CrossRef] [PubMed]
- Perron, G.G.; Zasloff, M.; Bell, G. Experimental evolution of resistance to an antimicrobial peptide. Proc. R. Soc. B Biol. Sci. 2006, 273, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Avila, L.U.; Loyola-Cruz, M.A.; Hernández-Cortez, C.; Bello-López, J.M.; Castro-Escarpulli, G. Colistin resistance in aeromonas spp. Int. J. Mol. Sci. 2021, 22, 5974. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Gorr, S.U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starr, C.G.; He, J.; Wimley, W.C. Host Cell Interactions are a Significant Barrier to the Clinical Utility of Peptide Antibiotics. ACS Chem Biol. 2016, 11, 3391–3399. [Google Scholar] [CrossRef] [Green Version]
- Amponnawarat, A.; Chompunud Na Ayudhya, C.; Ali, H. Murepavadin, a Small Molecule Host Defense Peptide Mimetic, Activates Mast Cells via MRGPRX2 and MrgprB2. Front. Immunol. 2021, 12, 689410. [Google Scholar] [CrossRef]
- Lin, S.; Wade, J.D.; Liu, S. De Novo Design of Flavonoid-Based Mimetics of Cationic Antimicrobial Peptides: Discovery, Development, and Applications. Acc. Chem. Res. 2021, 54, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Luo, Q.; Bannon, M.S.; Gray, V.P.; Bloom, T.G.; Clore, M.F.; Hughes, M.A.; Crawford, M.A.; Letteri, R.A. Molecular engineering of antimicrobial peptide (AMP)–polymer conjugates. Biomater. Sci. 2021, 9, 5069–5091. [Google Scholar] [CrossRef]
- Drayton, M.; Kizhakkedathu, J.N.; Straus, S.K. Towards Robust Delivery of Antimicrobial Peptides to Combat Bacterial Resistance. Molecules 2020, 25, 3048. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, M.; Qiu, S.; Wang, J.; Peng, J.; Zhao, P.; Zhu, R.; Wang, H.; Li, Y.; Wang, K.; et al. Antimicrobial activity and stability of the d-amino acid substituted derivatives of antimicrobial peptide polybia-MPI. AMB Express 2016, 6, 122. [Google Scholar] [CrossRef] [Green Version]
- Jia, F.; Wang, J.; Peng, J.; Zhao, P.; Kong, Z.; Wang, K.; Yan, W.; Wang, R. D-amino acid substitution enhances the stability of antimicrobial peptide polybia-CP. Acta Biochim. Biophys. Sin. 2017, 49, 916–925. [Google Scholar] [CrossRef] [Green Version]
- Spokoyny, A.M.; Zou, Y.; Ling, J.J.; Yu, H.; Lin, Y.-S.; Pentelute, B.L. A Perfluoroaryl-Cysteine SNAr Chemistry Approach to Unprotected Peptide Stapling. J. Am. Chem. Soc. 2013, 135, 5946–5949. [Google Scholar] [CrossRef] [Green Version]
- Lautrette, G.; Touti, F.; Lee, H.G.; Dai, P.; Pentelute, B.L. Nitrogen Arylation for Macrocyclization of Unprotected Peptides. J. Am. Chem. Soc. 2016, 138, 8340–8343. [Google Scholar] [CrossRef]
- Etayash, H.; Pletzer, D.; Kumar, P.; Straus, S.K.; Hancock, R.E.W. Cyclic Derivative of Host-Defense Peptide IDR-1018 Improves Proteolytic Stability, Suppresses Inflammation, and Enhances in Vivo Activity. J. Med. Chem. 2020, 63, 9228–9236. [Google Scholar] [CrossRef]
- Mourtada, R.; Herce, H.D.; Yin, D.J.; Moroco, J.A.; Wales, T.E.; Engen, J.R.; Walensky, L.D. Design of stapled antimicrobial peptides that are stable, nontoxic and kill antibiotic-resistant bacteria in mice. Nat. Biotechnol. 2019, 37, 1186–1197. [Google Scholar] [CrossRef]
- Migoń, D.; Neubauer, D.; Kamysz, W. Hydrocarbon Stapled Antimicrobial Peptides. Protein J. 2018, 37, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Rounds, T.; Straus, S.K. Lipidation of antimicrobial peptides as a design strategy for future alternatives to antibiotics. Int. J. Mol. Sci. 2020, 21, 9692. [Google Scholar] [CrossRef]
- Lombardi, L.; Shi, Y.; Falanga, A.; Galdiero, E.; de Alteriis, E.; Franci, G.; Chourpa, I.; Azevedo, H.S.; Galdiero, S. Enhancing the Potency of Antimicrobial Peptides through Molecular Engineering and Self-Assembly. Biomacromolecules 2019, 20, 1362–1374. [Google Scholar] [CrossRef]
- Molchanova, N.; Hansen, P.R.; Franzyk, H. Advances in development of antimicrobial peptidomimetics as potential drugs. Molecules 2017, 22, 1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, C.; Sarkar, P.; Issa, R.; Haldar, J. Alternatives to Conventional Antibiotics in the Era of Antimicrobial Resistance. Trends Microbiol. 2019, 27, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, R.P.; Romanowski, E.G.; Yates, K.A.; Mah, F.S. An independent evaluation of a novel peptide mimetic, Brilacidin (PMX30063), for ocular anti-infective. J. Ocul. Pharmacol. Ther. 2016, 32, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, R.; Willcox, M.; Black, D.S.C.; Kumar, N. Short cationic peptidomimetic antimicrobials. Antibiotics 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luther, A.; Urfer, M.; Zahn, M.; Müller, M.; Wang, S.-Y.; Mondal, M.; Vitale, A.; Hartmann, J.-B.; Sharpe, T.; Monte, F.L.; et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 2019, 576, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Molchanova, N.; Nielsen, J.E.; Sørensen, K.B.; Prabhala, B.K.; Hansen, P.R.; Lund, R.; Barron, A.E.; Jenssen, H. Halogenation as a tool to tune antimicrobial activity of peptoids. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, Á.; Gómez, R.; Ortega, P.; De La Mata, F.J.D. Nanosystems as vehicles for the delivery of antimicrobial peptides (Amps). Pharmaceutics 2019, 11, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makowski, M.; Silva, Í.C.; Do Amaral, C.P.; Gonçalves, S.; Santos, N.C. Advances in lipid and metal nanoparticles for antimicrobial peptide delivery. Pharmaceutics 2019, 11, 588. [Google Scholar] [CrossRef] [Green Version]
- Rajchakit, U.; Sarojini, V. Recent Developments in Antimicrobial-Peptide-Conjugated Gold Nanoparticles. Bioconjug. Chem. 2017, 28, 2673–2686. [Google Scholar] [CrossRef]
- Liu, L.; Yang, J.; Xie, J.; Luo, Z.; Jiang, J.; Yang, Y.Y.; Liu, S. The potent antimicrobial properties of cell penetrating peptide-conjugated silver nanoparticles with excellent selectivity for Gram-positive bacteria over erythrocytes. Nanoscale 2013, 5, 3834–3840. [Google Scholar] [CrossRef]
- Gao, J.; Na, H.; Zhong, R.; Yuan, M.; Guo, J.; Zhao, L.; Wang, Y.; Wang, L.; Zhang, F. One step synthesis of antimicrobial peptide protected silver nanoparticles: The core-shell mutual enhancement of antibacterial activity. Colloids Surf. B Biointerfaces 2020, 186, 110704. [Google Scholar] [CrossRef]
- Lee, B.; Park, J.; Ryu, M.; Kim, S.; Joo, M.; Yeom, J.-H.; Kim, S.; Park, Y.; Lee, K.; Bae, J. Antimicrobial peptide-loaded gold nanoparticle-DNA aptamer conjugates as highly effective antibacterial therapeutics against Vibrio vulnificus. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer–drug conjugate therapeutics: Advances, insights and prospects. Nat. Rev. Drug Discov. 2019, 18, 273–294. [Google Scholar] [CrossRef]
- Imura, Y.; Nishida, M.; Ogawa, Y.; Takakura, Y.; Matsuzaki, K. Action mechanism of tachyplesin I and effects of PEGylation. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1160–1169. [Google Scholar] [CrossRef] [Green Version]
- Kaur, N.; Dilawari, R.; Kaur, A.; Sahni, G.; Rishi, P. Recombinant expression, purification and PEGylation of Paneth cell peptide (cryptdin-2) with value added attributes against Staphylococcus aureus. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Shenoi, R.A.; Lai, B.F.L.; Nguyen, M.; Kizhakkedathu, J.N.; Straus, S. Conjugation of Aurein 2.2 to HPG Yields an Antimicrobial with Better Properties. Biomacromolecules 2015, 16, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Almaaytah, A.; Mohammed, G.K.; Abualhaijaa, A.; Al-Balas, Q. Development of novel ultrashort antimicrobial peptide nanoparticles with potent antimicrobial and antibiofilm activities against multidrug-resistant bacteria. Drug Des. Dev. Ther. 2017, 11, 3159–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahariah, P.; Sørensen, K.K.; Hjálmarsdóttir, M.; Sigurjónsson, E.; Jensen, K.J.; Másson, M.; Thygesen, M.B. Antimicrobial peptide shows enhanced activity and reduced toxicity upon grafting to chitosan polymers. Chem. Commun. 2015, 51, 11611–11614. [Google Scholar] [CrossRef]
- Hou, Z.; Shankar, Y.V.; Liu, Y.; Ding, F.; Subramanion, J.L.; Ravikumar, V.; Zamudio-Vázquez, R.; Keogh, D.; Lim, H.; Tay, M.Y.F.; et al. Nanoparticles of Short Cationic Peptidopolysaccharide Self-Assembled by Hydrogen Bonding with Antibacterial Effect against Multidrug-Resistant Bacteria. ACS Appl. Mater. Interfaces 2017, 9, 38288–38303. [Google Scholar] [CrossRef]
- Piras, A.M.; Emaisetta, G.; Esandreschi, S.; Egazzarri, M.; Ebartoli, C.; Grassi, L.; Eesin, S.; Chiellini, F.; Ebatoni, G. Chitosan nanoparticles loaded with the antimicrobial peptide temporin B exert a long-term antibacterial activity in vitro against clinical isolates of Staphylococcus epidermidis. Front. Microbiol. 2015, 6, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrin, T.H.C.; Fadel, V.; Martins, D.B.; Dias, S.A.; Cruz, A.; Sergio, L.M.; Arcisio-Miranda, M.; Castanho, M.A.R.B.; Cabrera, M.P.D.S. Synthesis and Characterization of Peptide-Chitosan Conjugates (PepChis) with Lipid Bilayer Affinity and Antibacterial Activity. Biomacromolecules 2019, 20, 2743–2753. [Google Scholar] [CrossRef]
- Lee, H.; Lim, S.I.; Shin, S.-H.; Lim, Y.; Koh, J.W.; Yang, S. Conjugation of Cell-Penetrating Peptides to Antimicrobial Peptides Enhances Antibacterial Activity. ACS Omega 2019, 4, 15694–15701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.L.; Ong, H.K.; Teo, J.; Ow, D.S.W.; Chao, S.H. HEXIM1 peptide exhibits antimicrobial activity against antibiotic resistant bacteria through guidance of cell penetrating peptide. Front. Microbiol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; De Palatis, L.; Raab, H.; Hazenbos, W.L.; Morisaki, J.H.; et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Mela, I.; Endo, M.; Sugiyama, H.; Henderson, R.M.; Kaminski, C.F. DNA Origami as a Tool in the Targeted Destruction of Bacteria. Biophys. J. 2019, 116, 324a. [Google Scholar] [CrossRef] [Green Version]
- Obuobi, S.; Tay, H.K.-L.; Tram, N.D.T.; Selvarajan, V.; Khara, J.S.; Wang, Y.; Ee, P.L.R. Facile and efficient encapsulation of antimicrobial peptides via crosslinked DNA nanostructures and their application in wound therapy. J. Control. Release 2019, 313, 120–130. [Google Scholar] [CrossRef]
- Dijksteel, G.S.; Ulrich, M.M.W.; Middelkoop, E.; Boekema, B.K.H.L. Review: Lessons Learned from Clinical Trials Using Antimicrobial Peptides (AMPs). Front. Microbiol. 2021, 12, 616979. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism | Gram-Negative | Gram-Positive |
|---|---|---|
| protease degradation | metalloproteinases (e.g., ZapA, ZmpA, ZmpB); aspartate proteases (e.g., OmpT, PgtE, Pla) | metalloproteinases (e.g., aureolysin, SepA); serine endopeptidases (e.g., V8); cysteine proteases (e.g., SpeB) |
| sequestration | extracellular proteins; biofilm matrix (e.g., alginate, polysialic acid) | extracellular proteins; biofilm matrix (e.g., poly-N-acetyl glucosamine; poly-gamma-glutamic acid) |
| surface modification | lipid A phosphate modification; lipid A acylation; O-antigen of LPS | d-alanylation of teichoic acids (TA, Figure 1); l-rhamnosylation of WTA; lipid II modification |
| membrane modification | phosphatidyl glycerol (PG) acylation | PG amino-acylation |
| efflux pumps | RND family | ABC transporters |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drayton, M.; Deisinger, J.P.; Ludwig, K.C.; Raheem, N.; Müller, A.; Schneider, T.; Straus, S.K. Host Defense Peptides: Dual Antimicrobial and Immunomodulatory Action. Int. J. Mol. Sci. 2021, 22, 11172. https://doi.org/10.3390/ijms222011172
Drayton M, Deisinger JP, Ludwig KC, Raheem N, Müller A, Schneider T, Straus SK. Host Defense Peptides: Dual Antimicrobial and Immunomodulatory Action. International Journal of Molecular Sciences. 2021; 22(20):11172. https://doi.org/10.3390/ijms222011172
Chicago/Turabian StyleDrayton, Matthew, Julia P. Deisinger, Kevin C. Ludwig, Nigare Raheem, Anna Müller, Tanja Schneider, and Suzana K. Straus. 2021. "Host Defense Peptides: Dual Antimicrobial and Immunomodulatory Action" International Journal of Molecular Sciences 22, no. 20: 11172. https://doi.org/10.3390/ijms222011172
APA StyleDrayton, M., Deisinger, J. P., Ludwig, K. C., Raheem, N., Müller, A., Schneider, T., & Straus, S. K. (2021). Host Defense Peptides: Dual Antimicrobial and Immunomodulatory Action. International Journal of Molecular Sciences, 22(20), 11172. https://doi.org/10.3390/ijms222011172