
N-Acyl Dopamines Induce Apoptosis in Endometrial Stromal Cells from Patients with Endometriosis

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
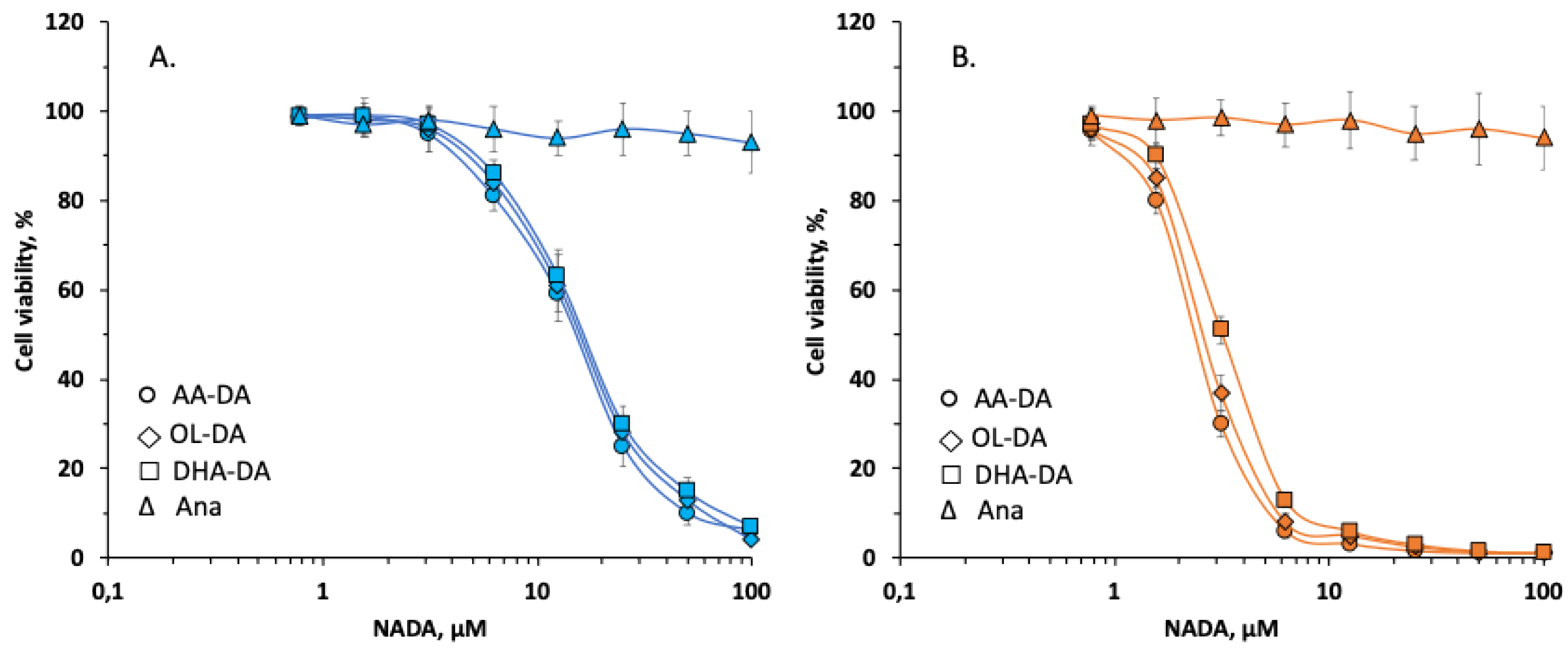
2.1. Effect of N-Acyl Dopamines on the Viability of Endometriotic Stromal Cells
2.2. The Mechanism of Cytotoxic Effect of N-Acyl Dopamines
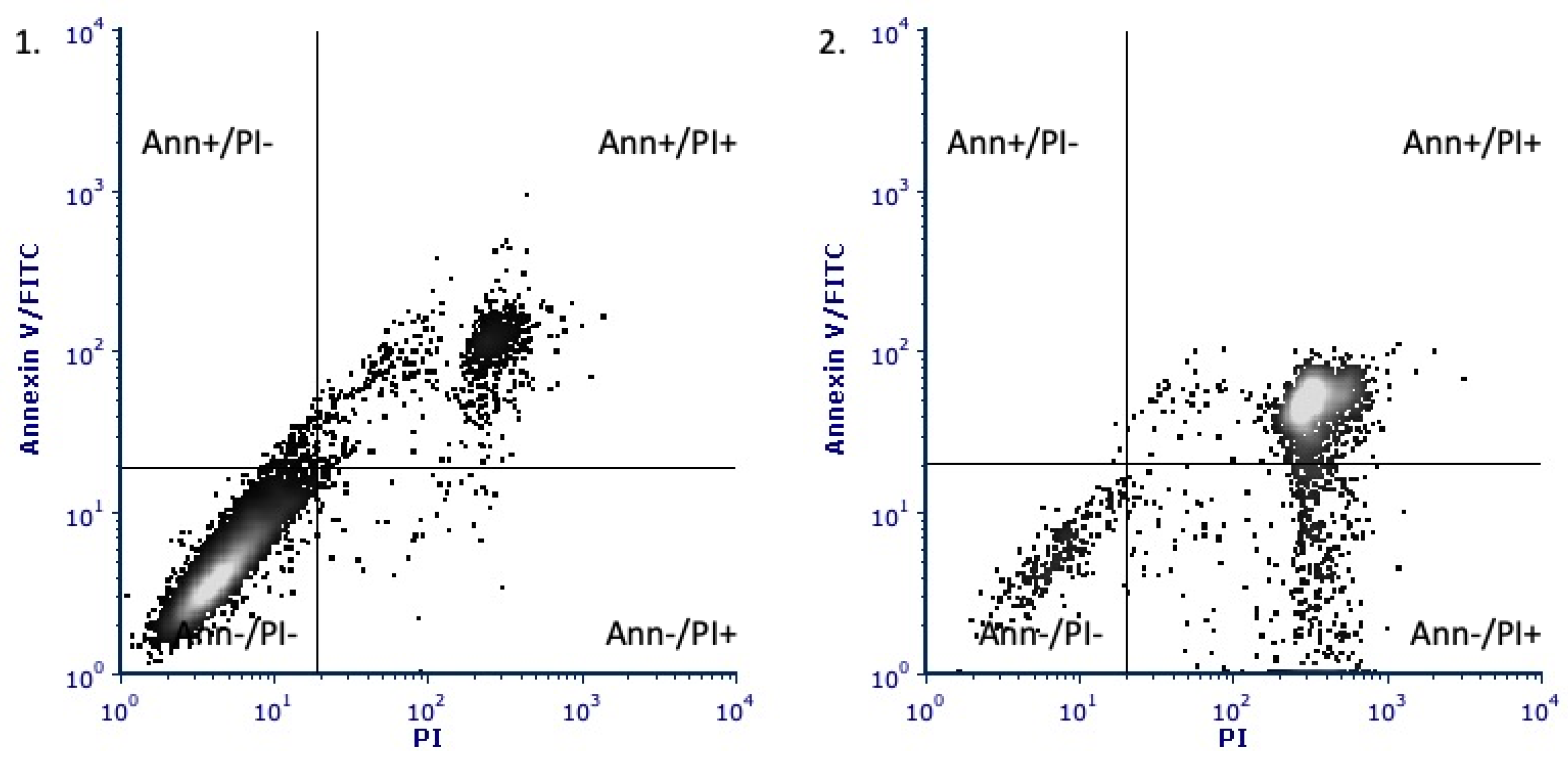
2.3. Annexin-Propidium Iodide Assay
2.4. Caspase Activity Assay
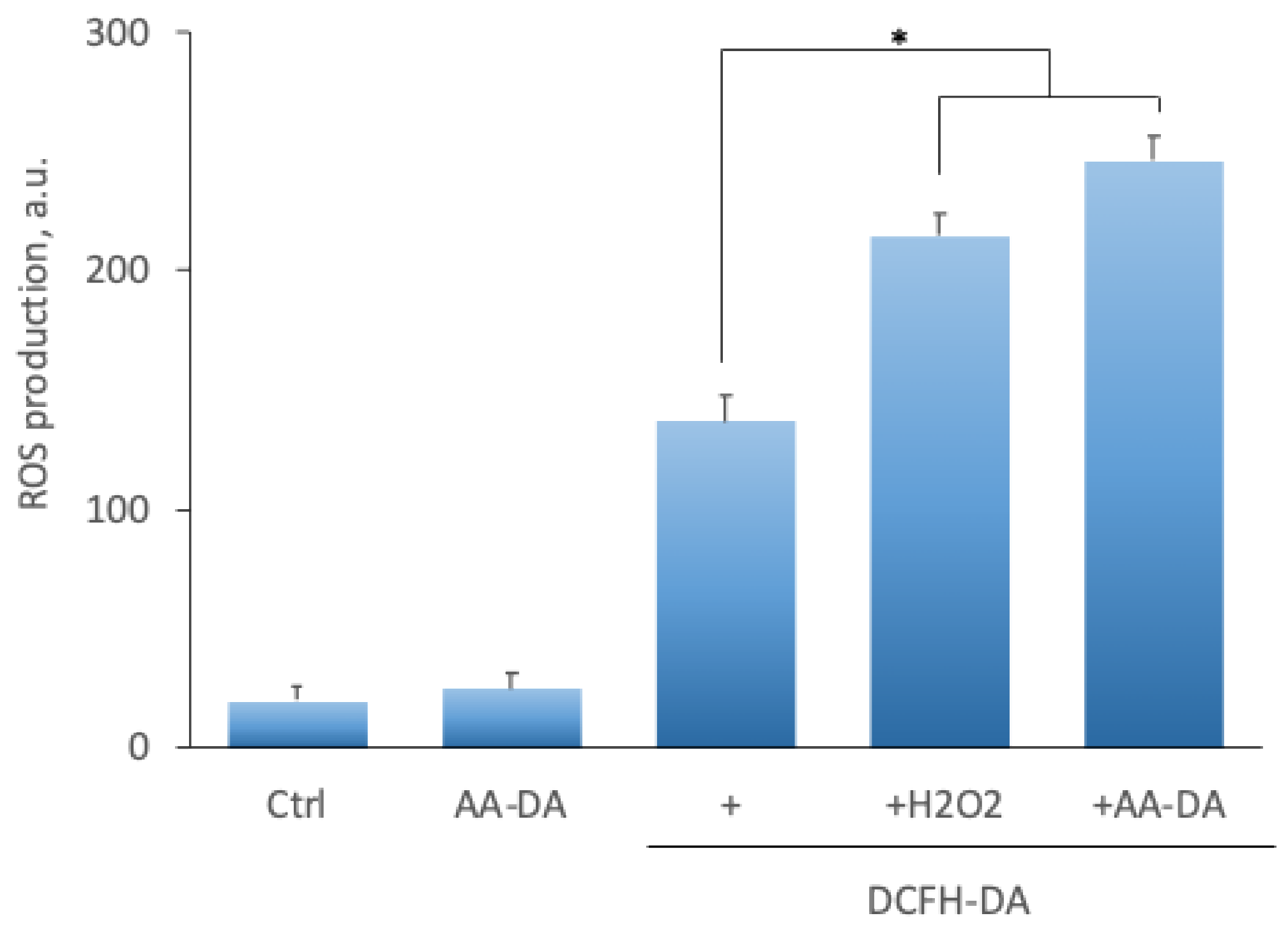
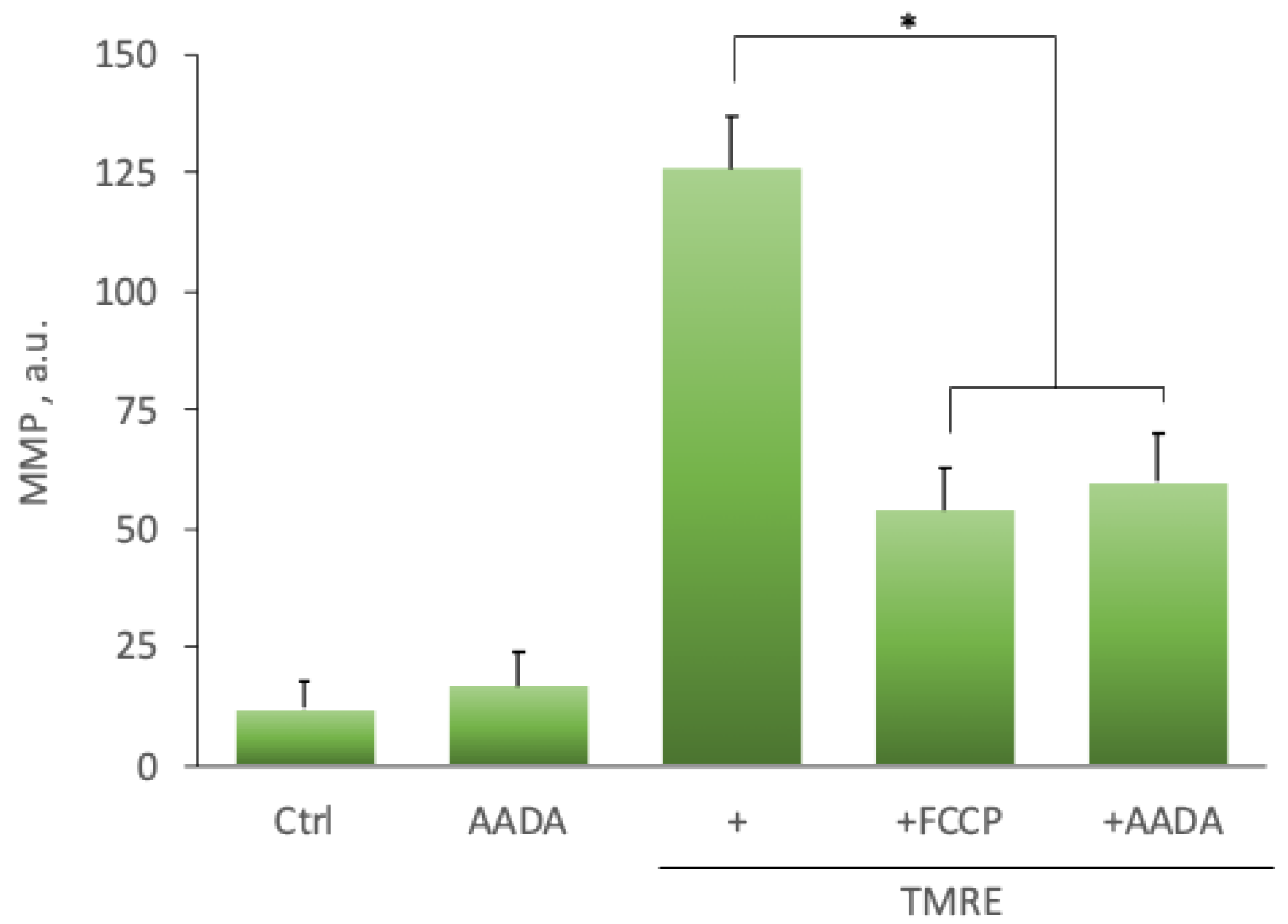
2.5. Effect of N-Acyl Dopamines on ROS Production and Mitochondrial Membrane Potential
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Cell Isolation and Culture
4.3. Cell Viability Assays
4.4. Inhibitory Analysis of N-Acyl Dopamines Potential Targets and Effectors
4.5. Cell Death Annexin V-FITC/PI Assay
4.6. Caspase-3 Activity and Caspase-9 Activity Measurement
4.7. DCFH-DA Reactive Oxygen Species ROS Assay
4.8. Measuring Mitochondrial Transmembrane Potential by TMRE Staining
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giudice, L.C.; Kao, L.C. Endometriosis. Lancet 2004, 364, 1789–1799. [Google Scholar] [CrossRef]
- Moradi, M.; Parker, M.; Sneddon, A.; Lopez, V.; Ellwood, D. Impact of endometriosis on women’s lives: A qualitative study. BMC Womens Health 2014, 14, 123. [Google Scholar] [CrossRef] [Green Version]
- Moreno, E.; Cavic, M.; Krivokuca, A.; Canela, E.I. The Interplay between Cancer Biology and the Endocannabinoid System-Significance for Cancer Risk, Prognosis and Response to Treatment. Cancers 2020, 12, 3275. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Gupta, A.S.; Kumar, P. Emerging role of cannabinoids and synthetic cannabinoid receptor 1/cannabinoid receptor 2 receptor agonists in cancer treatment and chemotherapy-associated cancer management. J. Cancer Res. Ther. 2021, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.H.; Abbas, M.S.; Habiba, M.A.; Konje, J.C. Histomorphometric evaluation of cannabinoid receptor and anandamide modulating enzyme expression in the human endometrium through the menstrual cycle. Histochem. Cell Biol. 2010, 133, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.; Fonseca, B.M.; Teixeira, N.; Correia-da-Silva, G. The fundamental role of the endocannabinoid system in endometrium and placenta: Implications in pathophysiological aspects of uterine and pregnancy disorders. Hum. Reprod. Update 2020, 26, 586–602. [Google Scholar] [CrossRef]
- Sanchez, A.M.; Vigano, P.; Mugione, A.; Panina-Bordignon, P.; Candiani, M. The molecular connections between the cannabinoid system and endometriosis. Mol. Hum. Reprod. 2012, 18, 563–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biringer, R.G. The rise and fall of anandamide: Processes that control synthesis, degradation, and storage. Mol. Cell. Biochem. 2021, 476, 2753–2775. [Google Scholar] [CrossRef]
- Fezza, F.; Bari, M.; Florio, R.; Talamonti, E.; Feole, M.; Maccarrone, M. Endocannabinoids, Related Compounds and Their Metabolic Routes. Molecules 2014, 19, 17078–17106. [Google Scholar] [CrossRef]
- Haspula, D.; Clark, M.A. Cannabinoid Receptors: An Update on Cell Signaling, Pathophysiological Roles and Therapeutic Opportunities in Neurological, Cardiovascular, and Inflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 7693. [Google Scholar] [CrossRef]
- Lu, H.-C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [Green Version]
- Verhoeckx, K.C.; Voortman, T.; Balvers, M.G.; Hendriks, H.F.; Wortelboer, H.M.; Witkamp, R.F. Presence, formation and putative biological activities of N-acyl serotonins, a novel class of fatty-acid derived mediators, in the intestinal tract. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2011, 1811, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Bisogno, T.; Petros, T.J.; Chang, S.Y.; Zavitsanos, P.A.; Zipkin, R.E.; Sivakumar, R.; Coop, A.; Maeda, D.Y.; De Petrocellis, L.; et al. Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J. Biol. Chem. 2001, 276, 42639–42644. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.M.; Bisogno, T.; Trevisani, M.; Al-Hayani, A.; De Petrocellis, L.; Fezza, F.; Tognetto, M.; Petros, T.J.; Krey, J.F.; Chu, C.J.; et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 8400–8405. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Kaul, K.; Mishra, S.; Charan, M.; Ganju, R.K. Cannabinoid Signaling in Cancer. Recent Adv. Cannabinoid Physiol. Pathol. 2019, 1162, 51–61. [Google Scholar] [CrossRef]
- Leconte, M.; Nicco, C.; Ngô, C.; Arkwright, S.; Chéreau, C.; Guibourdenche, J.; Weill, B.; Chapron, C.; Dousset, B.; Batteux, F. Antiproliferative effects of cannabinoid agonists on deep infiltrating endometriosis. Am. J. Pathol. 2010, 177, 2963–2970. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.M.; Fernandes, R.; Almada, M.; Santos, M.; Carvalho, F.; Teixeira, N.A.; Correia-da-Silva, G. Synthetic cannabinoids and endometrial stromal cell fate: Dissimilar effects of JWH-122, UR-144 and WIN55,212-2. Toxicology 2019, 413, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Ashba, A.M.; Yushina, M.N.; Fedorova-Gogolina, I.A.; Gretskaya, N.M.; Bezuglov, V.V.; Melkumyan, A.G.; Pavlovich, S.V.; Bobrov, M.Y. Selective Action of N-Arachidonoyl Dopamine on Viability and Proliferation of Stromal Cells from Eutopic and Ectopic Endometrium. Bull. Exp. Biol. Med. 2019, 167, 43–46. [Google Scholar] [CrossRef]
- Grabiec, U.; Dehghani, F. N-Arachidonoyl Dopamine: A Novel Endocannabinoid and Endovanilloid with Widespread Physiological and Pharmacological Activities. Cannabis Cannabinoid Res. 2017, 2, 183–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisogno, T.; Melck, D.; Bobrov, M.Y.; Gretskaya, N.M.; Bezuglov, V.V.; De Petrocellis, L.; Di Marzo, V. N-acyl-dopamines: Novel synthetic CB1 cannabinoid-receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo. Biochem. J. 2000, 351, 817–824. [Google Scholar] [CrossRef]
- Akimov, M.G.; Gretskaya, N.M.; Zinchenko, G.N.; Bezuglov, V.V. Cytotoxicity of Endogenous Lipids N-acyl Dopamines and their Possible Metabolic Derivatives for Human Cancer Cell Lines of Different Histological Origin. Anticancer Res. 2015, 35, 2657–2661. [Google Scholar] [PubMed]
- Visnyei, K.; Onodera, H.; Damoiseaux, R.; Saigusa, K.; Petrosyan, S.; De Vries, D.; Ferrari, D.; Saxe, J.; Panosya, E.H.; Masterman-Smith, M.; et al. A molecular screening approach to identify and characterize inhibitors of glioblastoma stem cells. Mol. Cancer Ther. 2011, 10, 1818–1828. [Google Scholar] [CrossRef] [Green Version]
- Laganà, A.S.; Garzon, S.; Götte, M.; Viganò, P.; Franchi, M.; Ghezzi, F.; Martin, D.C. The Pathogenesis of Endometriosis: Molecular and Cell Biology Insights. Int. J. Mol. Sci. 2019, 20, 5615. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, X.; Duan, K.; Zhang, Y.; Guo, S.W. The expression and functionality of transient receptor potential vanilloid 1 in ovarian endometriomas. Reprod. Sci. 2012, 19, 1110–1124. [Google Scholar] [CrossRef]
- Dmitrieva, N.; Nagabukuro, H.; Resuehr, D.; Zhang, G.; McAllister, S.L.; McGinty, K.A.; Mackie, K.; Berkley, K.J. Endocannabinoid involvement in endometriosis. PAIN 2010, 151, 703–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resuehr, D.; Glore, D.R.; Taylor, H.S.; Bruner-Tran, K.L.; Osteen, K.G. Progesterone-dependent regulation of endometrial cannabinoid receptor type 1 (CB1-R) expression is disrupted in women with endometriosis and in isolated stromal cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Fertil. Steril. 2012, 98, 948–956. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Duan, H.; Wang, S.; Gan, L.; Xu, Q.; Li, J.J. Decreased Expression of Cannabinoid Receptors in the Eutopic and Ectopic Endometrium of Patients with Adenomyosis. BioMed Res. Int. 2019, 2019, 5468954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilgic, E.; Guzel, E.; Kose, S.; Aydin, M.C.; Karaismailoglu, E.; Akar, I.; Usubutun, A.; Korkusuz, P. Endocannabinoids modulate apoptosis in endometriosis and adenomyosis. Acta Histochem. 2017, 119, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Almada, M.; Fonseca, B.M.; Amaral, C.; Diniz-da-Costa, M.; Correia-da-Silva, G.; Teixeira, N. Anandamide oxidative metabolism-induced endoplasmic reticulum stress and apoptosis. Apoptosis 2017, 22, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Huang, J.; Zhang, J.; Benes, C.; Jiao, B.; Ren, R. N-Arachidonoyl Dopamine Inhibits NRAS Neoplastic Transformation by Suppressing Its Plasma Membrane Translocation. Mol. Cancer Ther. 2017, 16, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Galve-Roperh, I.; Sánchez, C.; Cortés, M.L.; Gómez del Pulgar, T.; Izquierdo, M.; Guzmán, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, C.; Rueda, D.; Ségui, B.; Galve-Roperh, I.; Levade, T.; Guzmán, M. The CB(1) cannabinoid receptor of astrocytes is coupled to sphingomyelin hydrolysis through the adaptor protein fan. Mol. Pharmacol. 2001, 59, 955–959. [Google Scholar] [CrossRef] [Green Version]
- Gómez del Pulgar, T.; Velasco, G.; Sánchez, C.; Haro, A.; Guzmán, M. De novo-synthesized ceramide is involved in cannabinoid-induced apoptosis. Biochem. J. 2002, 363 Pt 1, 183–188. [Google Scholar] [CrossRef]
- Fonseca, B.M.; Correia-da-Silva, G.; Teixeira, N.A. The endocannabinoid anandamide induces apoptosis of rat decidual cells through a mechanism involving ceramide synthesis and p38 MAPK activation. Apoptosis 2013, 18, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Sarker, K.P.; Biswas, K.K.; Yamakuchi, M.; Lee, K.Y.; Hahiguchi, T.; Kracht, M.; Kitajima, I.; Maruyama, I. ASK1-p38 MAPK/JNK signaling cascade mediates anandamide-induced PC12 cell death. J. Neurochem. 2003, 85, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Bari, M.; Battista, N.; Fezza, F.; Finazzi-Agrò, A.; Maccarrone, M. Lipid rafts control signaling of type-1 cannabinoid receptors in neuronal cells. Implications for anandamide-induced apoptosis. J. Biol. Chem. 2005, 280, 12212–12220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasiou, A.; Clarke, A.B.; Turner, A.E.; Kumaran, N.M.; Vakilpour, S.; Smith, P.A.; Bagiokou, D.; Bradshaw, T.D.; Westwell, A.D.; Fang, L.; et al. Cannabinoid receptor agonists are mitochondrial inhibitors: A unified hypothesis of how cannabinoids modulate mitochondrial function and induce cell death. Biochem. Biophys. Res. Commun. 2007, 364, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, S.V.; Qian, T.; de Minicis, S.; Harvey-White, J.; Kunos, G.; Vinod, K.Y.; Hungund, B.; Schwabe, R.F. The endocannabinoid 2-arachidonoyl glycerol induces death of hepatic stellate cells via mitochondrial reactive oxygen species. FASEB J. 2007, 21, 2798–2806. [Google Scholar] [CrossRef] [PubMed]
- Zaccagnino, P.; Saltarella, M.; D’Oria, S.; Corcelli, A.; Saponetti, M.S.; Lorusso, M. N-arachidonylglycine causes ROS production and cytochrome c release in liver mitochondria. Free Radic. Biol. Med. 2009, 47, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Almada, M.; Alves, P.; Fonseca, B.M.; Carvalho, F.; Queirós, C.R.; Gaspar, H.; Amaral, C.; Teixeira, N.A.; Correia-da-Silva, G. Synthetic cannabinoids JWH-018, JWH-122, UR-144 and the phytocannabinoid THC activate apoptosis in placental cells. Toxicol. Lett. 2020, 319, 129–137. [Google Scholar] [CrossRef]
- Bobrov, M.Y.; Lizhin, A.A.; Andrianova, E.L.; Gretskaya, N.M.; Frumkina, L.E.; Khaspekov, L.G.; Bezuglov, V.V. Antioxidant and neuroprotective properties of N-arachidonoyldopamine. Neurosci. Lett. 2008, 431, 6–11. [Google Scholar] [CrossRef]
- Van Laar, V.S.; Mishizen, A.J.; Cascio, M.; Hastings, T.G. Proteomic identification of dopamine-conjugated proteins from isolated rat brain mitochondria and SH-SY5Y cells. Neurobiol. Dis. 2009, 34, 487–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimov, M.G.; Gamisonia, A.M.; Dudina, P.V.; Gretskaya, N.M.; Gaydaryova, A.A.; Kuznetsov, A.S.; Zinchenko, G.N.; Bezuglov, V.V. GPR55 Receptor Activation by the N-Acyl Dopamine Family Lipids Induces Apoptosis in Cancer Cells via the Nitric Oxide Synthase (nNOS) Over-Stimulation. Int. J. Mol. Sci. 2021, 22, 622. [Google Scholar] [CrossRef]
- Fonseca, B.M.; Correia-da-Silva, G.; Almada, M.; Costa, M.A.; Teixeira, N.A. The Endocannabinoid System in the Postimplantation Period: A Role during Decidualization and Placentation. Int. J. Endocrinol. 2013, 2013, 510540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, W.R.; Carnevale, L.N.; Xie, Z.; Baylon, J.L.; Tajkhorshid, E.; Hu, H.; Das, A. Anti-inflammatory dopamine- and serotonin-based endocannabinoid epoxides reciprocally regulate cannabinoid receptors and the TRPV1 channel. Nat. Commun. 2021, 12, 926. [Google Scholar] [CrossRef] [PubMed]
- Ryan, I.P.; Schriock, E.D.; Taylor, R.N. Isolation, characterization, and comparison of human endometrial and endometriosis cells in vitro. J. Clin. Endocrinol. Metab. 1994, 78, 642–649. [Google Scholar] [CrossRef]
- Bezuglov, V.; Bobrov, M.; Gretskaya, N.; Gonchar, A.; Zinchenko, G.; Melck, D.; Bisogno, T.; Di Marzo, V.; Kuklev, D.; Rossi, J.C.; et al. Synthesis and biological evaluation of novel amides of polyunsaturated fatty acids with dopamine. Bioorg. Med. Chem. Lett. 2001, 11, 447–449. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | EuSC (LC50, µM) | EcSC (LC50, µM) |
|---|---|---|
| OL-DA | 15.03 ± 0.97 | 2.72 ± 0.55 |
| AA-DA | 14.06 ± 0.98 | 2.45 ± 0.45 |
| DHA-DA | 16.02 ± 0.99 | 3.24 ± 0.50 |
| Ana | - | - |
| Compounds | OL-DA | AA-DA | DHA-DA | |
|---|---|---|---|---|
| NADA | 21.4 ± 2.9 | 22.9 ± 1.9 | 29.1 ± 2.3 | |
| NADA + SR141716A | CB1 | 70.2 ± 3.4 * | 73.2 ± 3.0 * | 75.1 ± 1.4 * |
| NADA + SR144528 | CB2 | 19.4 ± 2.4 | 18.2 ± 1.4 | 23.3 ± 3.4 |
| NADA + Capsazepine | TRPV1 | 24.1 ± 1.3 | 27.1 ± 1.6 | 27.9 ± 2.9 |
| NADA + Z-VAD-FMK | Casp | 86.2 ± 3.9 * | 81.8 ± 1.9 * | 81.2 ± 1.3 * |
| NADA + N-acetyl-L-cysteine | ROS | 72.2 ± 2.6 * | 70.3 ± 2.9 * | 74.4 ± 1.4 * |
| NADA + Ascorbate | ROS | 74.8 ± 2.7 * | 76.4 ± 2.1 * | 79.3 ± 2.6 * |
| NADA + Imideazopyridine | SPT | 78.4 ± 2.3 * | 78.6 ± 2.7 * | 74.2 ± 2.4 * |
| NADA + L-NAME | NOS | 42.1 ± 1.9 | 36.2 ± 4.7 | 39.4 ± 2.1 |
| Cell Populations | OL-DA | AA-DA | DHA-DA | Control |
|---|---|---|---|---|
| Annexin V−/PI−, vital cells | 12.5 ± 3.1 | 10.6 ± 2.7 | 16.2 ± 2.2 | 88.4 ± 2.6 |
| Annexin V+/PI−, early apoptosis | 2.1 ± 0.4 | 1.4 ± 0.6 | 1.2 ± 0.4 | 1.4 ± 0.7 |
| Annexin V+/PI+, late apoptosis | 68.2 ± 6.4 | 66.5 ± 4.2 | 62.3 ± 4.8 | 9.6 ± 2.5 |
| Annexin V−/PI+, necrosis | 18.4 ± 3.2 | 22.8 ± 2.4 | 21.6 ± 2.1 | 1.3 ± 0.4 |
| OL-DA | AA-DA | DHA-DA | ||||
|---|---|---|---|---|---|---|
| casp-3 | casp-9 | casp-3 | casp-9 | casp-3 | casp-9 | |
| Control | 134.2 ± 6.4 | 142.4 ± 4.2 | 121.3 ± 5.1 | 128.7 ± 5.1 | 137.6 ± 7.2 | 144.3 ± 3.9 |
| Z-VAD-FMK | 141.6 ± 6.3 | 142.1 ± 3.8 | 134.2 ± 3.3 | 132.2 ± 4.9 | 147.5 ± 5.3 | 144.8 ± 4.4 |
| NADA | 434.1 ± 5.9 * | 450.8 ± 6.9 * | 432.4 ± 4.2 * | 465.2 ± 8.4 * | 439.2 ± 7.9 * | 470.8 ± 5.5 * |
| NADA+Z-VAD-FMK | 137.4 ± 5.2 | 145.9 ± 4.3 | 129.1 ± 3.2 | 139.4 ± 5.8 | 138.8 ± 6.2 | 143.2 ± 6.4 |
| OL-DA | AA-DA | DHA-DA | |
|---|---|---|---|
| Control | 19.3 ± 5.4 | 20.1 ± 5.9 | 21.9 ± 7.2 |
| NADA | 24.2 ± 7.8 | 25.4 ± 6.7 | 27.3 ± 5.3 |
| DCFH-DA | 134.3 ± 9.2 | 136.8 ± 10.8 | 138.5 ± 6.4 |
| DCFH-DA + H2O2 | 220.2 ± 6.4 * | 215.3 ± 9.4 * | 221.4 ± 7.9 * |
| DCFH-DA + NADA | 241.4 ± 8.9 * | 246.1 ± 9.7 * | 250.5 ± 7.1 * |
| OL-DA | AA-DA | DHA-DA | |
|---|---|---|---|
| Control | 10.4 ± 4.4 | 12.1 ± 3.3 | 12.8 ± 6.2 |
| NADA | 15.2 ± 3.2 | 16.8 ± 3.2 | 18.3 ± 4.7 |
| TMRE | 125.3 ± 3.4 | 125.9 ± 5.8 | 128.2 ± 4.7 |
| TMRE + FCCP | 51.4 ± 5.2 * | 54.3 ± 4.4 * | 55.9 ± 4.3 * |
| TMRE + NADA | 47.1 ± 4.3 * | 59.8 ± 5.1 * | 48.7 ± 3.4 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamisonia, A.M.; Yushina, M.N.; Fedorova-Gogolina, I.A.; Akimov, M.G.; Eldarov, C.M.; Pavlovich, S.V.; Bezuglov, V.V.; Gretskaya, N.M.; Sukhikh, G.T.; Bobrov, M.Y. N-Acyl Dopamines Induce Apoptosis in Endometrial Stromal Cells from Patients with Endometriosis. Int. J. Mol. Sci. 2021, 22, 10648. https://doi.org/10.3390/ijms221910648
Gamisonia AM, Yushina MN, Fedorova-Gogolina IA, Akimov MG, Eldarov CM, Pavlovich SV, Bezuglov VV, Gretskaya NM, Sukhikh GT, Bobrov MY. N-Acyl Dopamines Induce Apoptosis in Endometrial Stromal Cells from Patients with Endometriosis. International Journal of Molecular Sciences. 2021; 22(19):10648. https://doi.org/10.3390/ijms221910648
Chicago/Turabian StyleGamisonia, Alina M., Marina N. Yushina, Irina A. Fedorova-Gogolina, Mikhail G. Akimov, Chupalav M. Eldarov, Stanislav V. Pavlovich, Vladimir V. Bezuglov, Natalia M. Gretskaya, Gennady T. Sukhikh, and Mikhail Yu. Bobrov. 2021. "N-Acyl Dopamines Induce Apoptosis in Endometrial Stromal Cells from Patients with Endometriosis" International Journal of Molecular Sciences 22, no. 19: 10648. https://doi.org/10.3390/ijms221910648
APA StyleGamisonia, A. M., Yushina, M. N., Fedorova-Gogolina, I. A., Akimov, M. G., Eldarov, C. M., Pavlovich, S. V., Bezuglov, V. V., Gretskaya, N. M., Sukhikh, G. T., & Bobrov, M. Y. (2021). N-Acyl Dopamines Induce Apoptosis in Endometrial Stromal Cells from Patients with Endometriosis. International Journal of Molecular Sciences, 22(19), 10648. https://doi.org/10.3390/ijms221910648