
The Crucial Role of NLRP3 Inflammasome in Viral Infection-Associated Fibrosing Interstitial Lung Diseases


{kind=link}
{kind=link}
Abstract
1. Introduction
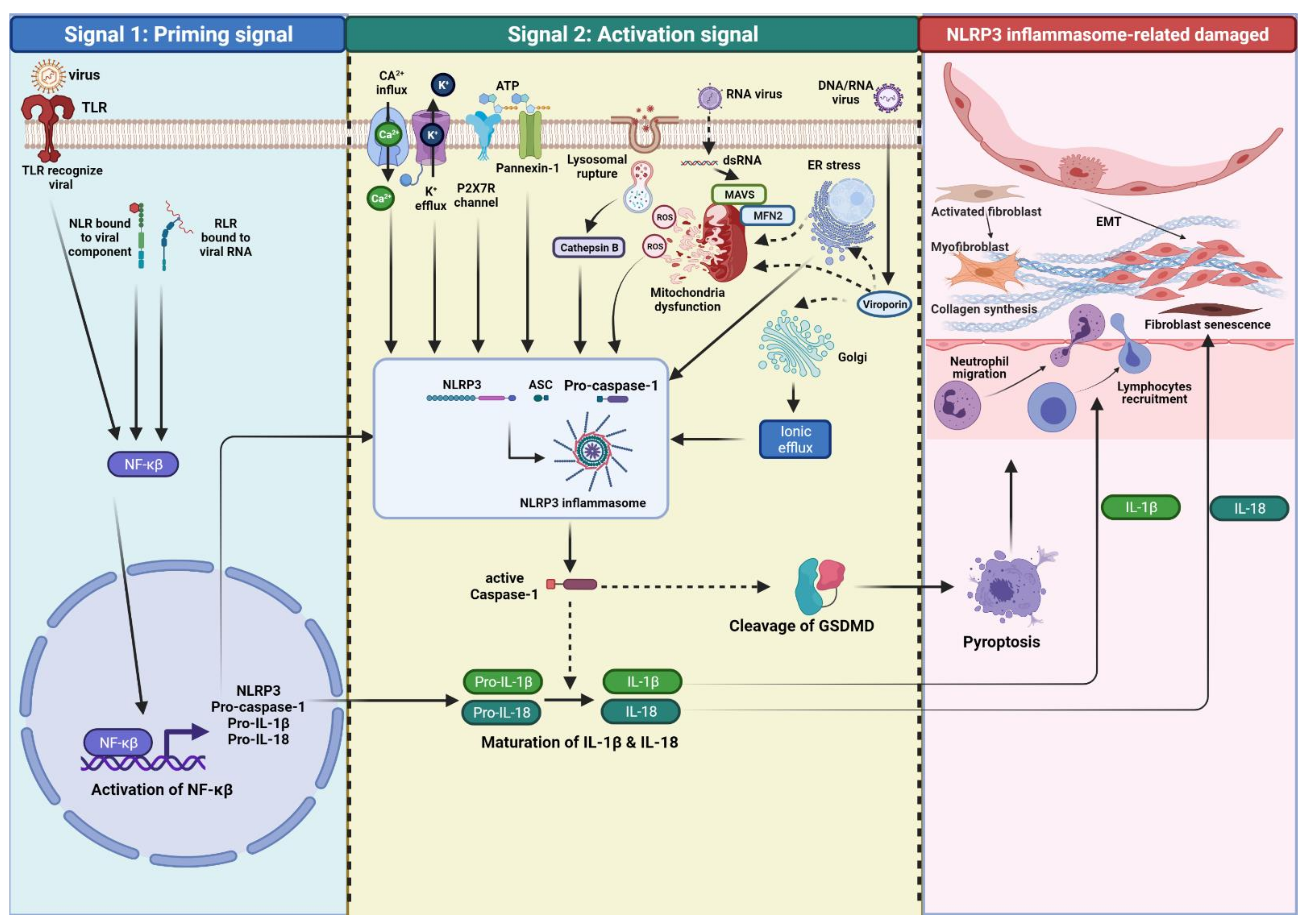
2. NLRP3 Inflammasome
3. The Activation of NLRP3 Inflammasome
4. Viral Infection Triggered The Activation of NLRP3 Inflammasome
4.1. Viral DNA-Induced NLRP3 Inflammasome Activation
4.2. Viral RNA-Induced NLRP3 Inflammasome Activation
4.3. Viroporin-Induced NLRP3 Inflammasome Activation
4.4. Ionic Flux Disturbance-Induced NLRP3 Inflammasome Activation
4.5. Intracellular ROS and Mitochondria Dysfunction-Induced NLRP3 Inflammasome Activation
4.6. ER Stress-Induced NLRP3 Inflammasome Activation
4.7. Lysosomal Damage-Induced NLRP3 Inflammasome Activation
5. Potential Mechanism of Virus Infection-Induced Damage That Associated with The NLRP3 Inflammasome
5.1. Inflammasome-Derived Inflammatory Cytokines
5.2. Pyroptosis and Necroptosis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Bendstrup, E.; Dron, L.; Langley, J.; Smith, G.; Khalid, J.M.; Patel, H.; Kreuter, M. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir. Res. 2021, 22, 197. [Google Scholar] [CrossRef] [PubMed]
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27, 180033. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, M.; Kurniawan, N.A. Mechanical and physical regulation of fibroblast–myofibroblast transition: From cellular mechanoresponse to tissue pathology. Front. Bioeng. Biotechnol. 2020, 8, 1459. [Google Scholar]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Kropski, J.A.; Blackwell, T.S.; Loyd, J.E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1717–1727. [Google Scholar] [CrossRef]
- Guiot, J.; Struman, I.; Chavez, V.; Henket, M.; Herzog, M.; Scoubeau, K.; Hardat, N.; Bondue, B.; Corhay, J.L.; Moermans, C.; et al. Altered epigenetic features in circulating nucleosomes in idiopathic pulmonary fibrosis. Clin. Epigenet. 2017, 9, 84. [Google Scholar] [CrossRef]
- Sack, C.; Raghu, G. Idiopathic pulmonary fibrosis: Unmasking cryptogenic environmental factors. Eur. Respir. J. 2019, 53, 1801699. [Google Scholar] [CrossRef]
- Racanelli, A.C.; Kikkers, S.A.; Choi, A.M.K.; Cloonan, S.M. Autophagy and inflammation in chronic respiratory disease. Autophagy 2018, 14, 221–232. [Google Scholar] [CrossRef]
- Balestro, E.; Calabrese, F.; Turato, G.; Lunardi, F.; Bazzan, E.; Marulli, G.; Biondini, D.; Rossi, E.; Sanduzzi, A.; Rea, F.; et al. Immune inflammation and disease progression in idiopathic pulmonary fibrosis. PLoS ONE 2016, 11, e0154516. [Google Scholar] [CrossRef]
- Desai, O.; Winkler, J.; Minasyan, M.; Herzog, E.L. The role of immune and inflammatory cells in idiopathic pulmonary fibrosis. Front. Med. 2018, 5, 43. [Google Scholar] [CrossRef]
- Calabrese, F.; Kipar, A.; Lunardi, F.; Balestro, E.; Perissinotto, E.; Rossi, E.; Nannini, N.; Marulli, G.; Stewart, J.P.; Rea, F. Herpes virus infection is associated with vascular remodeling and pulmonary hypertension in idiopathic pulmonary fibrosis. PLoS ONE 2013, 8, e55715. [Google Scholar] [CrossRef]
- Folcik, V.A.; Garofalo, M.; Coleman, J.; Donegan, J.J.; Rabbani, E.; Suster, S.; Nuovo, A.; Magro, C.M.; Di Leva, G.; Nuovo, G.J. Idiopathic pulmonary fibrosis is strongly associated with productive infection by herpesvirus saimiri. Mod. Pathol. 2014, 27, 851–862. [Google Scholar] [CrossRef][Green Version]
- Kikkert, M. Innate immune evasion by human respiratory RNA viruses. J. Innate Immun. 2020, 12, 4–20. [Google Scholar] [CrossRef]
- Land, W.G. Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome—With a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun. 2021, 22, 141–160. [Google Scholar] [CrossRef]
- Effendi, W.I.; Nagano, T.; Hasan, H.; Yudhawati, R. Immunoregulatory property of C-type lectin-like receptors in fibrosing interstitial lung diseases. Int. J. Mol. Sci. 2020, 21, 3665. [Google Scholar] [CrossRef]
- Krishnaswamy, J.K.; Chu, T.; Eisenbarth, S.C. Beyond pattern recognition: NOD-like receptors in dendritic cells. Trends Immunol. 2013, 34, 224–233. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Jo, E.-K.; Kim, J.K.; Shin, D.-M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef]
- Zhang, W.-J.; Fang, Z.-M.; Liu, W.-Q. NLRP3 inflammasome activation from Kupffer cells is involved in liver fibrosis of Schistosoma japonicum-infected mice via NF-κB. Parasit. Vectors 2019, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Wei, X.; Ji, S.; Gui, S.-Y.; Zhang, S.-M. In vivo effects of the NLRP1/NLRP3 inflammasome pathway on latent respiratory virus infection. Int. J. Mol. Med. 2018, 41, 3620–3628. [Google Scholar] [CrossRef] [PubMed]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.Y.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef] [PubMed]
- Stout-Delgado, H.W.; Cho, S.J.; Chu, S.G.; Mitzel, D.N.; Villalba, J.; El-Chemaly, S.; Ryter, S.W.; Choi, A.M.K.; Rosas, I.O. Age-dependent susceptibility to pulmonary fibrosis is associated with NLRP3 inflammasome activation. Am. J. Respir. Cell Mol. Biol. 2016, 55, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Lupfer, C.; Malik, A.; Kanneganti, T.-D. Inflammasome control of viral infection. Curr. Opin. Virol. 2015, 12, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, Y.; Shen, W.; Oladejo, A.O.; Yang, J.; Jiang, W.; Imam, B.H.; Wu, X.; Ding, X.; Yang, Y.; et al. LPS mediates bovine endometrial epithelial cell pyroptosis directly through both NLRP3 classical and non-classical inflammasome pathways. Front. Immunol. 2021, 12, 1935. [Google Scholar] [CrossRef]
- Stoilova, B.; Vyas, P. The inflammasome: More than a protective innate immune mechanism. Immunity 2019, 51, 3–5. [Google Scholar] [CrossRef]
- Amir, M.; Czaja, M.J. Inflammasome-mediated inflammation and fibrosis: It is more than just the IL-1β. Hepatology 2018, 67, 479–481. [Google Scholar] [CrossRef]
- Sharma, D.; Kanneganti, T.-D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.-D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef]
- Zhong, Y.; Kinio, A.; Saleh, M. Functions of NOD-like receptors in human diseases. Front. Immunol. 2013, 4, 333. [Google Scholar] [CrossRef]
- Chen, I.-Y.; Ichinohe, T. Response of host inflammasomes to viral infection. Trends Microbiol. 2015, 23, 55–63. [Google Scholar] [CrossRef]
- Danis, J.; Mellett, M. Nod-like receptors in host defence and disease at the epidermal barrier. Int. J. Mol. Sci. 2021, 22, 4677. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Kelly, P.; Meade, K.G.; O’Farrelly, C. Non-canonical inflammasome-mediated IL-1β production by primary endometrial epithelial and stromal fibroblast cells is NLRP3 and caspase-4 dependent. Front. Immunol. 2019, 10, 102. [Google Scholar] [CrossRef]
- Lasithiotaki, I.; Giannarakis, I.; Tsitoura, E.; Samara, K.D.; Margaritopoulos, G.A.; Choulaki, C.; Vasarmidi, E.; Tzanakis, N.; Voloudaki, A.; Sidiropoulos, P.; et al. NLRP3 inflammasome expression in idiopathic pulmonary fibrosis and rheumatoid lung. Eur. Respir. J. 2016, 47, 910–918. [Google Scholar] [CrossRef]
- Looi, C.K.; Hii, L.-W.; Chung, F.F.-L.; Mai, C.-W.; Lim, W.-M.; Leong, C.-O. Roles of inflammasomes in epstein-barr virus-associated nasopharyngeal cancer. Cancers 2021, 13, 1786. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef]
- Yuk, J.-M.; Silwal, P.; Jo, E.-K. Inflammasome and mitophagy connection in health and disease. Int. J. Mol. Sci. 2020, 21, 4714. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Kang, S.; Anderson, C.; Sagara, J.; Fitzgerald, K.A.; Alnemri, E.S. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J. Immunol. 2013, 191, 3995–3999. [Google Scholar] [CrossRef]
- Lin, K.-M.; Hu, W.; Troutman, T.D.; Jennings, M.; Brewer, T.; Li, X.; Nanda, S.; Cohen, P.; Thomas, J.A.; Pasare, C. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. USA 2014, 111, 775–780. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Karki, R.; Berwin, B.; Yamamoto, M.; Kanneganti, T.-D. Guanylate binding proteins facilitate caspase-11-dependent pyroptosis in response to type 3 secretion system-negative Pseudomonas aeruginosa. Cell Death Discov. 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.J.; Boucher, D.; Bierschenk, D.; Tebartz, C.; Whitney, P.G.; D’Silva, D.B.; Tanzer, M.C.; Monteleone, M.; Robertson, A.A.B.; Cooper, M.A.; et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur. J. Immunol. 2015, 45, 2918–2926. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Vanaja, S.K.; Waggoner, L.; Sokolovska, A.; Becker, C.; Stuart, L.M.; Leong, J.M.; Fitzgerald, K.A. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 2012, 150, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Hornung, V. Alternative inflammasome activation enables IL-1β release from living cells. Curr. Opin. Immunol. 2017, 44, 7–13. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.B.; Cooper, M.A.; Graf, T.; Hornung, V. Human monocytes engage an alternative inflammasome pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef]
- Zhao, C.; Zhao, W. NLRP3 inflammasome—A key player in antiviral responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef]
- Storek, K.M.; Monack, D.M. Bacterial recognition pathways that lead to inflammasome activation. Immunol. Rev. 2015, 265, 112–129. [Google Scholar] [CrossRef]
- Hayward, J.A.; Mathur, A.; Ngo, C.; Man, S.M. Cytosolic recognition of microbes and pathogens: Inflammasomes in action. Microbiol. Mol. Biol. Rev. 2018, 82, e00015-18. [Google Scholar] [CrossRef]
- Jacobs, S.R.; Damania, B. NLRs, inflammasomes, and viral infection. J. Leukoc. Biol. 2012, 92, 469–477. [Google Scholar] [CrossRef]
- Shrivastava, G.; León-Juárez, M.; García-Cordero, J.; Meza-Sánchez, D.E.; Cedillo-Barrón, L. Inflammasomes and its importance in viral infections. Immunol. Res. 2016, 64, 1101–1117. [Google Scholar] [CrossRef]
- Wang, W.; Hu, D.; Wu, C.; Feng, Y.; Li, A.; Liu, W.; Wang, Y.; Chen, K.; Tian, M.; Xiao, F.; et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLOS Pathog. 2020, 16, e1008335. [Google Scholar] [CrossRef]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef]
- Johnson, K.E.; Chikoti, L.; Chandran, C. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J. Virol. 2013, 87, 5005–5018. [Google Scholar] [CrossRef] [PubMed]
- Spel, L.; Martinon, F. Detection of viruses by inflammasomes. Curr. Opin. Virol. 2021, 46, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hu, L.; Liu, Y.; Huang, L.; Mu, Y.; Cai, X.; Weng, C. DDX19A senses viral RNA and mediates NLRP3-dependent inflammasome activation. J. Immunol. 2015, 195, 5732–5749. [Google Scholar] [CrossRef] [PubMed]
- Silwal, P.; Kim, J.K.; Jeon, S.M.; Lee, J.-Y.; Kim, Y.J.; Kim, Y.S.; Seo, Y.; Kim, J.; Kim, S.Y.; Lee, M.J.; et al. Mitofusin-2 boosts innate immunity through the maintenance of aerobic glycolysis and activation of xenophagy in mice. Commun. Biol. 2021, 4, 548. [Google Scholar] [CrossRef]
- Choudhury, S.K.M.; Ma, X.; Abdullah, S.W.; Zheng, H. Activation and inhibition of the NLRP3 inflammasome by RNA viruses. J. Inflamm. Res. 2021, 14, 1145–1163. [Google Scholar] [CrossRef]
- Farag, N.S.; Breitinger, U.; Breitinger, H.G.; El Azizi, M.A. Viroporins and inflammasomes: A key to understand virus-induced inflammation. Int. J. Biochem. Cell Biol. 2020, 122, 105738. [Google Scholar] [CrossRef]
- Triantafilou, K.; Triantafilou, M. Ion flux in the lung: Virus-induced inflammasome activation. Trends Microbiol. 2014, 22, 580–588. [Google Scholar] [CrossRef]
- Da Costa, L.S.; Outlioua, A.; Anginot, A.; Akarid, K.; Arnoult, D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death Dis. 2019, 10, 346. [Google Scholar] [CrossRef]
- Zhi, X.; Zhang, Y.; Sun, S.; Zhang, Z.; Dong, H.; Luo, X.; Wei, Y.; Lu, Z.; Dou, Y.; Wu, R.; et al. NLRP3 inflammasome activation by Foot-and-mouth disease virus infection mainly induced by viral RNA and non-structural protein 2B. RNA Biol. 2020, 17, 335–349. [Google Scholar] [CrossRef]
- Negash, A.A.; Olson, R.M.; Griffin, S.; Gale, M., Jr. Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PLOS Pathog. 2019, 15, e1007593. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.-K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell. Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Tang, T.; Lang, X.; Xu, C.; Wang, X.; Gong, T.; Yang, Y.; Cui, J.; Bai, L.; Wang, J.; Jiang, W.; et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 2017, 8, 202. [Google Scholar] [CrossRef]
- Gong, T.; Yang, Y.; Jin, T.; Jiang, W.; Zhou, R. Orchestration of NLRP3 inflammasome activation by Ion fluxes. Trends Immunol. 2018, 39, 393–406. [Google Scholar] [CrossRef]
- Ren, Z.; Zhang, X.; Ding, T.; Zhong, Z.; Hu, H.; Xu, Z.; Deng, J. Mitochondrial dynamics imbalance: A strategy for promoting viral infection. Front. Microbiol. 2020, 11, 1992. [Google Scholar] [CrossRef]
- Xu, M.; Wang, L.; Wang, M.; Wang, H.; Zhang, H.; Chen, Y.; Wang, X.; Gong, J.; Zhang, J.J.; Adcock, I.M.; et al. Mitochondrial ROS and NLRP3 inflammasome in acute ozone-induced murine model of airway inflammation and bronchial hyperresponsiveness. Free Radic. Res. 2019, 53, 780–790. [Google Scholar] [CrossRef]
- De Nardo, D.; De Nardo, C.M.; Latz, E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am. J. Pathol. 2014, 184, 42–54. [Google Scholar] [CrossRef]
- Holley, C.L.; Schroder, K. The rOX-stars of inflammation: Links between the inflammasome and mitochondrial meltdown. Clin. Transl. Immunol. 2020, 9, e01109. [Google Scholar] [CrossRef]
- Seok, J.K.; Kang, H.C.; Cho, Y.-Y.; Lee, H.S.; Lee, J.Y. Regulation of the NLRP3 inflammasome by post-translational modifications and small molecules. Front. Immunol. 2021, 11, 3877. [Google Scholar] [CrossRef]
- So, J.-S. Roles of endoplasmic reticulum stress in immune responses. Mol. Cells 2018, 41, 705–716. [Google Scholar]
- Choi, J.-A.; Song, C.-H. Insights into the role of endoplasmic reticulum stress in infectious diseases. Front. Immunol. 2020, 10, 3147. [Google Scholar] [CrossRef]
- Gao, P.; Chai, Y.; Song, J.; Liu, T.; Chen, P.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Yang, H. Reprogramming the unfolded protein response for replication by porcine reproductive and respiratory syndrome virus. PLOS Pathog. 2019, 15, e1008169. [Google Scholar] [CrossRef]
- Tao, L.; Lin, H.; Wen, J.; Sun, Q.; Gao, Y.; Xu, X.; Wang, J.; Zhang, J.; Weng, D. The kinase receptor-interacting protein 1 is required for inflammasome activation induced by endoplasmic reticulum stress. Cell Death Dis. 2018, 9, 641. [Google Scholar] [CrossRef]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nuñez, G.; He, Y.; Yin, X.-M.; O’Riordan, M.X.D. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef]
- Kim, S.; Joe, Y.; Jeong, S.O.; Zheng, M.; Back, S.H.; Park, S.W.; Ryter, S.W.; Chung, H.T. Endoplasmic reticulum stress is sufficient for the induction of IL-1β production via activation of the NF-κB and inflammasome pathways. Innate Immun. 2013, 20, 799–815. [Google Scholar] [CrossRef]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012, 3, e261. [Google Scholar] [CrossRef]
- Duckworth, A.; Longhurst, H.J.; Paxton, J.K.; Scotton, C.J. The role of herpes viruses in pulmonary fibrosis. Front. Med. 2021, 8, 1137. [Google Scholar] [CrossRef]
- Jheng, J.-R.; Ho, J.-Y.; Horng, J.-T. ER stress, autophagy, and RNA viruses. Front. Microbiol. 2014, 5, 388. [Google Scholar] [CrossRef]
- Chong, W.C.; Shastri, M.D.; Peterson, G.M.; Patel, R.P.; Pathinayake, P.S.; Dua, K.; Hansbro, N.G.; Hsu, A.C.; Wark, P.A.; Shukla, S.D.; et al. The complex interplay between endoplasmic reticulum stress and the NLRP3 inflammasome: A potential therapeutic target for inflammatory disorders. Clin. Transl. Immunol. 2021, 10, e1247. [Google Scholar] [CrossRef]
- Mostafaei, S.; Sayad, B.; Azar, M.E.F.; Doroudian, M.; Hadifar, S.; Behrouzi, A.; Riahi, P.; Hussen, B.M.; Bayat, B.; Nahand, J.S.; et al. The role of viral and bacterial infections in the pathogenesis of IPF: A systematic review and meta-analysis. Respir. Res. 2021, 22, 53. [Google Scholar] [CrossRef]
- Spandole, S.; Cimponeriu, D.; Berca, L.M.; Mihăescu, G. Human anelloviruses: An update of molecular, epidemiological and clinical aspects. Arch. Virol. 2015, 160, 893–908. [Google Scholar] [CrossRef]
- Arase, Y.; Suzuki, F.; Suzuki, Y.; Akuta, N.; Kobayashi, M.; Kawamura, Y.; Yatsuji, H.; Sezaki, H.; Hosaka, T.; Hirakawa, M.; et al. Hepatitis C virus enhances incidence of idiopathic pulmonary fibrosis. World J. Gastroenterol. 2008, 14, 5880–5886. [Google Scholar] [CrossRef] [PubMed]
- Aliannejad, R.; Ghanei, M. Hepatitis C and pulmonary fibrosis: Hepatitis C and pulmonary fibrosis. Hepat. Mon. 2011, 11, 71–73. [Google Scholar] [PubMed]
- Samir, A.; El-Beheiry, A.A.; Gharraf, H.S.; Khalifa, M.H. Viral hepatitis and interstitial lung diseases: Can HRCT assess their relation and characterize its pattern? Egypt. J. Radiol. Nucl. Med. 2020, 51, 163. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, T.; Bozkanat, M.; Ibe, J.C.F.; Christman, J.W.; Raj, J.U.; Zhou, G. Intratracheal instillation of high dose adenoviral vectors is sufficient to induce lung injury and fibrosis in mice. PLoS ONE 2015, 9, e116142. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Moore, T.A. Viruses in idiopathic pulmonary fibrosis—Etiology and exacerbation. Ann. Am. Thorac. Soc. 2015, 12, S186–S192. [Google Scholar] [PubMed]
- Jafarian, A.H.; Mohamadian Roshan, N.; Ayatollahi, H.; Omidi, A.A.; Ghaznavi, M.; Gharib, M. Epstein-barr virus and human herpesvirus 8 in idiopathic pulmonary fibrosis. Iran. J. Pathol. 2020, 15, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J. Gammaherpesviruses and pulmonary fibrosis: Evidence from humans, horses, and rodents. Vet. Pathol. 2014, 51, 372–384. [Google Scholar] [CrossRef]
- Li, Y.; Gao, J.; Wang, G.; Fei, G. Latent cytomegalovirus infection exacerbates experimental pulmonary fibrosis by activating TGF-β1. Mol. Med. Rep. 2016, 14, 1297–1301. [Google Scholar] [CrossRef]
- Lasithiotaki, I.; Antoniou, K.M.; Vlahava, V.-M.; Karagiannis, K.; Spandidos, D.A.; Siafakas, N.M.; Sourvinos, G. Detection of herpes simplex virus type-1 in patients with fibrotic lung diseases. PLoS ONE 2011, 6, e27800. [Google Scholar] [CrossRef]
- Karaba, A.H.; Figueroa, A.; Massaccesi, G.; Botto, S.; DeFilippis, V.R.; Cox, A.L. Herpes simplex virus type 1 inflammasome activation in proinflammatory human macrophages is dependent on NLRP3, ASC, and caspase-1. PLoS ONE 2020, 15, e0229570. [Google Scholar] [CrossRef]
- Peng, L.; Wen, L.; Shi, Q.-F.; Gao, F.; Huang, B.; Meng, J.; Hu, C.-P.; Wang, C.-M. Scutellarin ameliorates pulmonary fibrosis through inhibiting NF-κB/NLRP3-mediated epithelial–mesenchymal transition and inflammation. Cell Death Dis. 2020, 11, 978. [Google Scholar] [CrossRef]
- Sheng, G.; Chen, P.; Wei, Y.; Yue, H.; Chu, J.; Zhao, J.; Wang, Y.; Zhang, W.; Zhang, H.-L. Viral infection increases the risk of idiopathic pulmonary fibrosis: A meta-analysis. Chest 2020, 157, 1175–1187. [Google Scholar] [CrossRef]
- Wu, Y.-S.; Chen, S.-N. Apoptotic cell: Linkage of inflammation and wound healing. Front. Pharmacol. 2014, 5, 1. [Google Scholar] [CrossRef]
- Pinar, A.A.; Yuferov, A.; Gaspari, T.A.; Samuel, C.S. Relaxin can mediate its anti-fibrotic effects by targeting the myofibroblast NLRP3 inflammasome at the level of caspase-1. Front. Pharmacol. 2020, 11, 1201. [Google Scholar] [CrossRef]
- Artlett, C.M. Inflammasomes in wound healing and fibrosis. J. Pathol. 2013, 229, 157–167. [Google Scholar] [CrossRef]
- Cheon, S.Y.; Koo, B.-N. Inflammatory response in COVID-19 patients resulting from the interaction of the inflammasome and SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 7914. [Google Scholar] [CrossRef]
- Van den Berg, D.F.; Te Velde, A.A. Severe COVID-19: NLRP3 inflammasome dysregulated. Front. Immunol. 2020, 11, 1580. [Google Scholar] [CrossRef]
- Lara, P.C.; Macías-Verde, D.; Burgos-Burgos, J. Age-induced NLRP3 inflammasome over-activation increases lethality of SARS-CoV-2 pneumonia in elderly patients. Aging Dis. 2020, 11, 756–762. [Google Scholar] [CrossRef]
- Zhang, H.; Tang, Y.; Tao, J. Sex-related overactivation of NLRP3 inflammasome increases lethality of the male COVID-19 patients. Front. Mol. Biosci. 2021, 8, 516. [Google Scholar] [CrossRef]
- She, Y.X.; Yu, Q.Y.; Tang, X.X. Role of interleukins in the pathogenesis of pulmonary fibrosis. Cell Death Discov. 2021, 7, 52. [Google Scholar] [CrossRef]
- Biswas, R.; Bunderson-Schelvan, M.; Holian, A. Potential role of the inflammasome-derived inflammatory cytokines in pulmonary fibrosis. Pulm. Med. 2011, 2011, 105707. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Wang, S.-F.; Weng, I.-C.; Hong, M.-H.; Lo, T.-H.; Jan, J.-T.; Hsu, L.-C.; Chen, H.-Y.; Liu, F.-T. Galectin-3 Enhances avian H5N1 influenza A virus–induced pulmonary inflammation by promoting NLRP3 inflammasome activation. Am. J. Pathol. 2018, 188, 1031–1042. [Google Scholar] [CrossRef]
- Hirani, N.; MacKinnon, A.C.; Nicol, L.; Ford, P.; Schambye, H.; Pedersen, A.; Nilsson, U.J.; Leffler, H.; Sethi, T.; Tantawi, S.; et al. Target inhibition of galectin-3 by inhaled TD139 in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2021, 57, 2002559. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; Peaper, D.R.; Ho, J.H.; Murray, T.S.; Iwasaki, A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5354–5359. [Google Scholar] [CrossRef]
- Lee, S.; Ishitsuka, A.; Noguchi, M.; Hirohama, M.; Fujiyasu, Y.; Petric, P.P.; Schwemmle, M.; Staeheli, P.; Nagata, K.; Kawaguchi, A. Influenza restriction factor MxA functions as inflammasome sensor in the respiratory epithelium. Sci. Immunol. 2019, 4, aau4643. [Google Scholar] [CrossRef]
- Lee, K.-H.; Kang, T.-B. The molecular links between cell death and inflammasome. Cells 2019, 8, 1057. [Google Scholar] [CrossRef]
- Drakopanagiotakis, F.; Xifteri, A.; Polychronopoulos, V.; Bouros, D. Apoptosis in lung injury and fibrosis. Eur. Respir. J. 2008, 32, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Yue, R.; Zheng, Z.; Luo, Y.; Wang, X.; Lv, M.; Qin, D.; Tan, Q.; Zhang, Y.; Wang, T.; Hu, H. NLRP3-mediated pyroptosis aggravates pressure overload-induced cardiac hypertrophy, fibrosis, and dysfunction in mice: Cardioprotective role of irisin. Cell Death Discov. 2021, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hao, Q.; Florence, J.M.; Jung, B.-G.; Kurdowska, A.K.; Samten, B.; Idell, S.; Tang, H. Influenza virus infection induces ZBP1 expression and necroptosis in mouse lungs. Front. Cell. Infect. Microbiol. 2019, 9, 286. [Google Scholar] [CrossRef]
- Murtha, L.A.; Morten, M.; Schuliga, M.J.; Mabotuwana, N.S.; Hardy, S.A.; Waters, D.W.; Burgess, J.K.; Ngo, D.T.; Sverdlov, A.L.; Knight, D.A.; et al. The role of pathological aging in cardiac and pulmonary fibrosis. Aging Dis. 2019, 10, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Takezaki, A.; Tsukumo, S.; Setoguchi, Y.; Ledford, J.G.; Goto, H.; Hosomichi, K.; Uehara, H.; Nishioka, Y.; Yasutomo, K. A homozygous SFTPA1 mutation drives necroptosis of type II alveolar epithelial cells in patients with idiopathic pulmonary fibrosis. J. Exp. Med. 2019, 216, 2724–2735. [Google Scholar] [CrossRef] [PubMed]
- Colunga Biancatelli, R.M.L.; Solopov, P.; Dimitropoulou, C.; Catravas, J.D. Age-dependent chronic lung injury and pulmonary fibrosis following single exposure to hydrochloric acid. Int. J. Mol. Sci. 2021, 22, 8833. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Effendi, W.I.; Nagano, T. The Crucial Role of NLRP3 Inflammasome in Viral Infection-Associated Fibrosing Interstitial Lung Diseases. Int. J. Mol. Sci. 2021, 22, 10447. https://doi.org/10.3390/ijms221910447
Effendi WI, Nagano T. The Crucial Role of NLRP3 Inflammasome in Viral Infection-Associated Fibrosing Interstitial Lung Diseases. International Journal of Molecular Sciences. 2021; 22(19):10447. https://doi.org/10.3390/ijms221910447
Chicago/Turabian StyleEffendi, Wiwin Is, and Tatsuya Nagano. 2021. "The Crucial Role of NLRP3 Inflammasome in Viral Infection-Associated Fibrosing Interstitial Lung Diseases" International Journal of Molecular Sciences 22, no. 19: 10447. https://doi.org/10.3390/ijms221910447
APA StyleEffendi, W. I., & Nagano, T. (2021). The Crucial Role of NLRP3 Inflammasome in Viral Infection-Associated Fibrosing Interstitial Lung Diseases. International Journal of Molecular Sciences, 22(19), 10447. https://doi.org/10.3390/ijms221910447