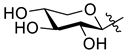
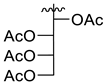
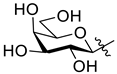
5.1. Synthesis
5.1.1. General Methods
Optical rotations were measured on a Jasco P-2000 polarimeter (Jasco, Easton, MD, USA) at r.t., and the data referred to the average of three parallel measurements. NMR spectra were recorded with DRX360 (360/90 MHz for
1H/
13C) or DRX400 (400/100 MHz for
1H/
13C) spectrometers (Bruker, Karlsruhe, Germany). Chemical shifts are referenced to Me
4Si (
1H-NMR) or to the residual solvent signals (
13C-NMR). Assigments of the proton and carbon signals of the new compounds were based on COSY and HSQC correlations. MS spectra were obtained using a Bruker maXis II (ESI-HRMS) spectrometer. TLC analysis were carried out by using DC Kieselgel 60 F
254 plates (Sigma-Aldrich, Saint Louis, MO, USA), and the spots were visualized under UV light and by gentle heating. For column chromatographic purification, Kieselgel 60 (Molar Chemicals, Halásztelek, Hungary, particle size 0.063–0.2 mm) silica gel was applied. Anhydrous solvents were obtained by using standard distillation procedures. Anhydrous CH
2Cl
2, CHCl
3, and toluene were produced by distillation from P
4O
10 and then stored over 4 Å molecular sieves (CH
2Cl
2, CHCl
3) or sodium wires (toluene). MeOH was dried by distillation over Mg turnings and iodine. The 2-ethynylpyridine (TCI Chemicals, Zwijndrecht, The Netherlands) and the dichloro(η
6-
p-cymene)ruthenium(II) dimer (
Ru-dimer, Strem Chemicals, Newburyport, MA, USA) were purchased from the indicated suppliers. The 2,3,4,6-tetra-
O-acetyl-β-
d-glucopyranosyl-azide [
28,
29] (
1), the 5-(2′,3′,4′,6′-tetra-
O-benzoyl-β-
d-glucopyranosyl)-tetrazole [
58,
59] (
2), the 5-(2′,3′,4′-tri-
O-benzoyl-β-
d-xylopyranosyl)-tetrazole [
60] (
3), the 5-(2′,3′,4′,6′-tetra-
O-acetyl-β-
d-galactopyranosyl)-tetrazole [
61] (
4), the 2-(
l-
arabino-1′,2′,3′,4′-tetraacetoxybutyl)-tetrazole [
33] (
5), the 2-ethynylquinoline [
62], 1-phenyl-4-(pyridine-2-yl)-1,2,3-triazole [
34] (
L-13), and the 2-phenyl-5-(pyridine-2-yl)-1,3,4-oxadiazole [
35,
36] (
L-14) were synthesized according to procedures in the literature.
5.1.2. General Procedure I for the Preparation of 1-(2′,3′,4′,6′-Tetra-O-acetyl-β-d-glucopyranosyl)-4-hetaryl-1,2,3-triazoles
To a solution of 1-(2,3,4,6-tetra-
O-acetyl-β-
d-glucopyranosyl)-azide [
28,
29] (
1) in CH
2Cl
2 (1 mL/50 mg azide), the appropriate 2-ethynylated heterocycle (1 eq.) and the C
3H
7COOCu(PPh
3)
2 catalyst [
30] (3 mol.%) were added. The reaction mixture was stirred at r.t. for 1 day, and the completion of the reaction was judged by TLC (1:1 EtOAc–hexane). The solvent was then removed under diminished pressure, and the residue was purified by column chromatography.
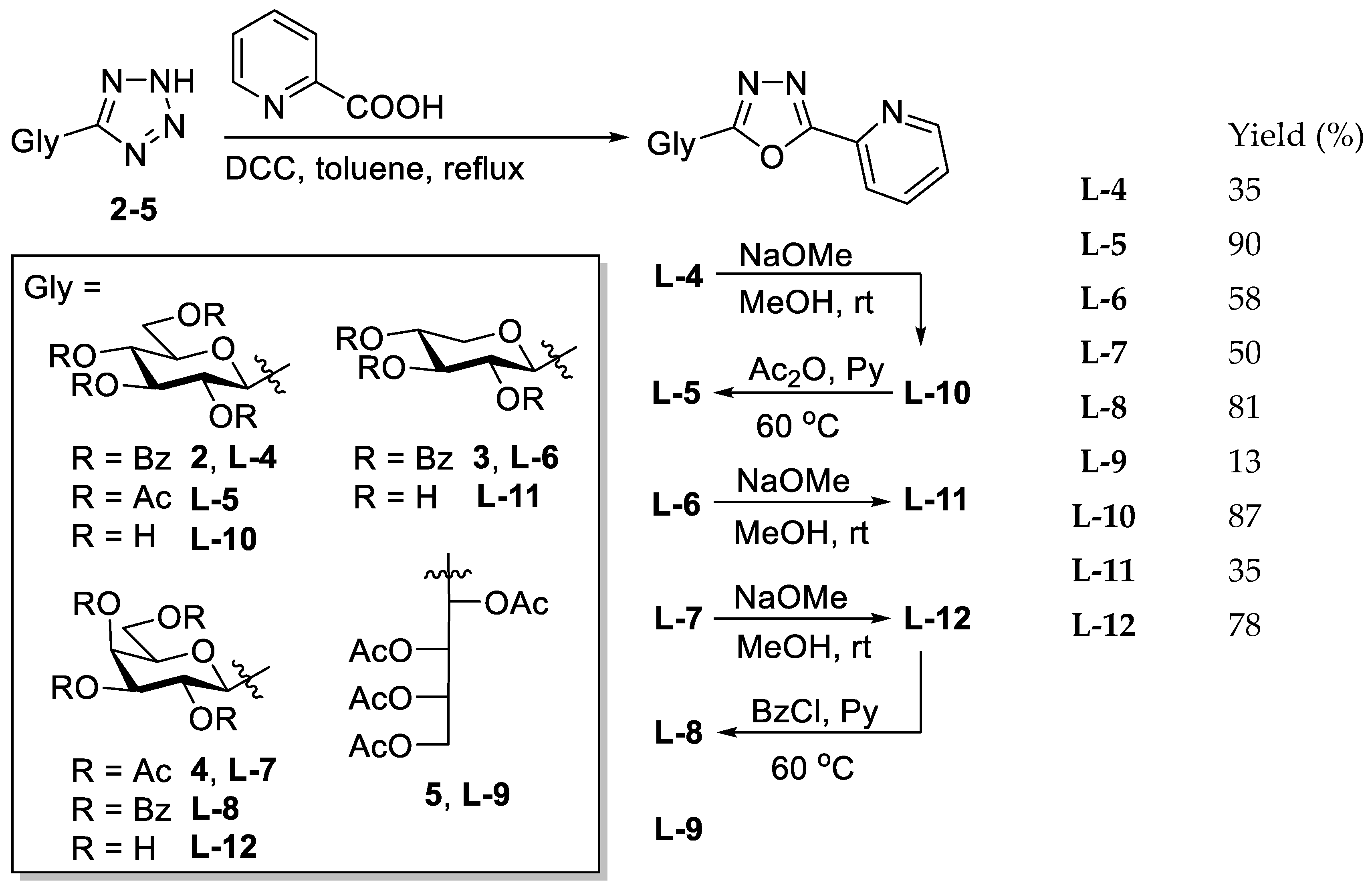
5.1.3. General Procedure II for the Synthesis of O-Peracylated 2-glycosyl-5-(pyridin-2-yl)-1,3,4-oxadiazoles
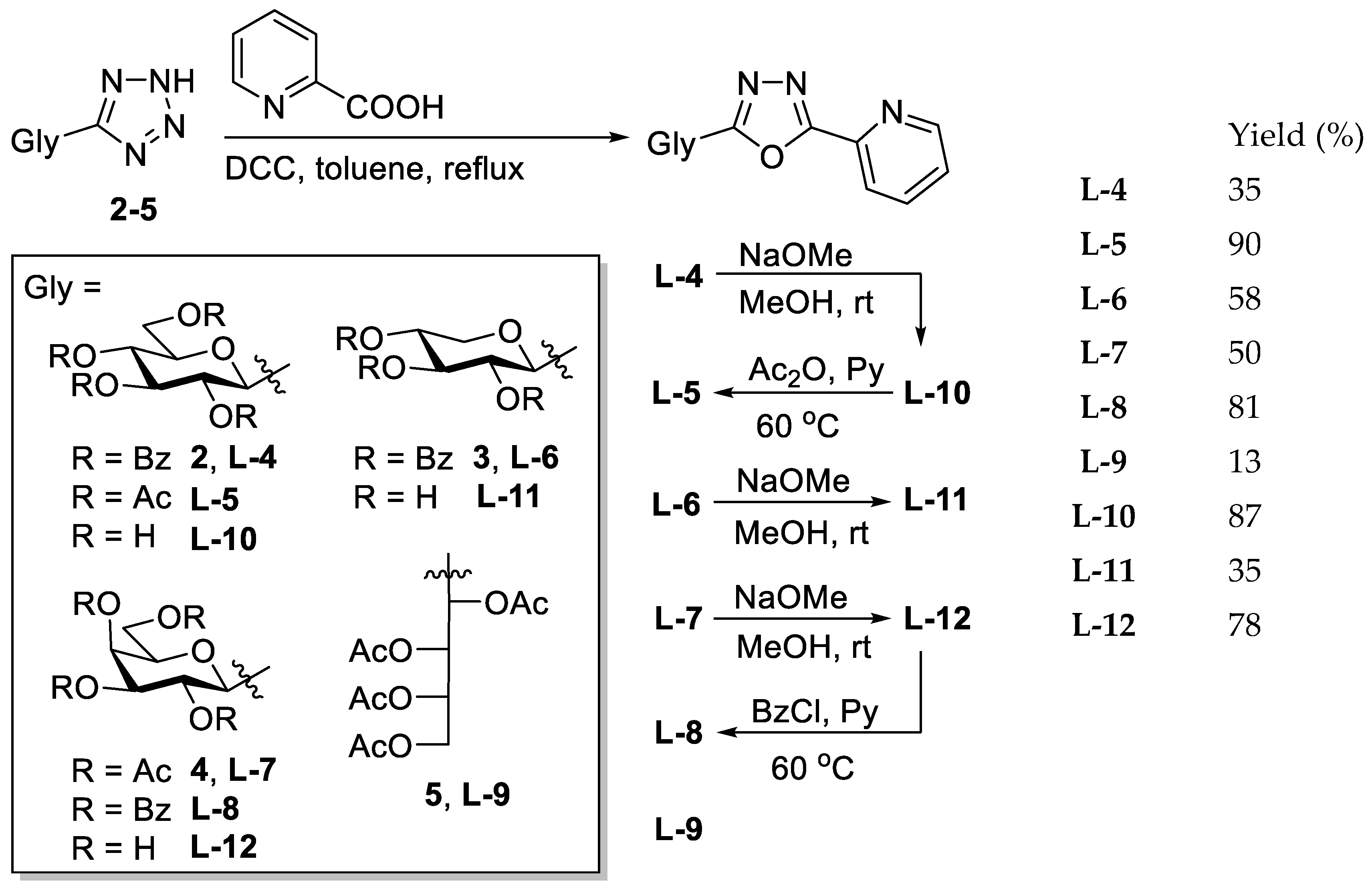
The corresponding O-peracylated 5-glycosyl-tetrazole (2–5) was dissolved in dry toluene (1 mL/100 mg substrate), and then 2-picolinic acid (2 eq.) and DCC (2 eq.) were added. The reaction mixture was stirred at boiling temperature until the TLC showed total consumption of the tetrazole. The insoluble materials were filtered off and washed with CH2Cl2, and the filtrate was evaporated under reduced pressure. The crude product was purified by column chromatography and crystallisation.
5.1.4. General Procedure III for the Zemplén Deacylation
An O-acyl protected glycosyl-azole was dissolved in a 1:1 mixture of dry MeOH and dry CHCl3 (1 mL/25 mg substrate), and a few drops of a ~1 M solution of NaOMe in MeOH was added (pH = 8–9). The reaction mixture was left at r.t. until the TLC indicated total conversion of the starting material. The neutralization of the solution was carried out by the addition of a cation exchange resin (Amberlyst 15, H+ form). The resin was then filtered off, and the solution was evaporated under reduced pressure. The crude product was purified by column chromatography or crystallization.
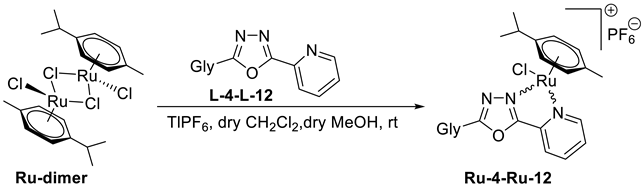
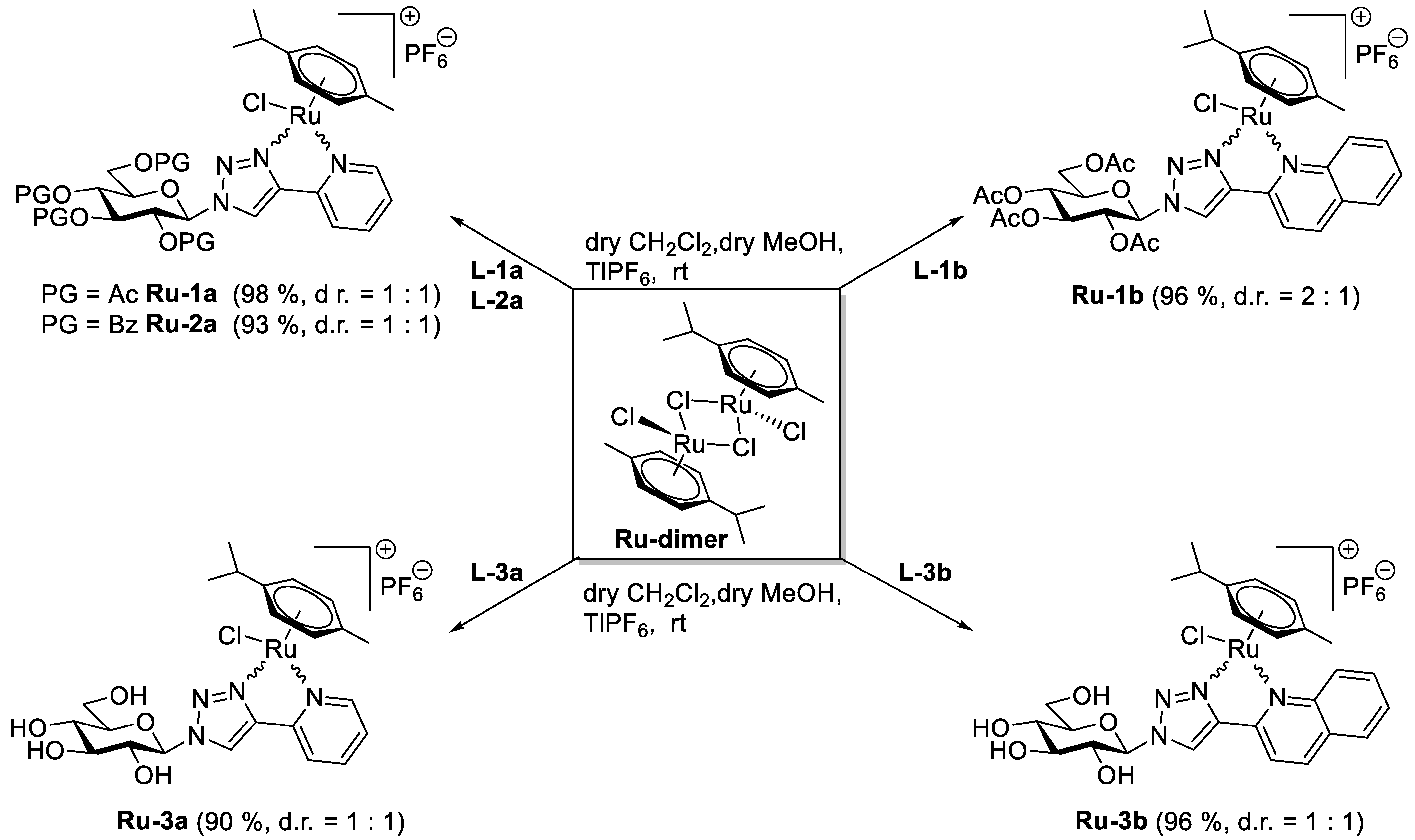
5.1.5. General Procedure IV for the Formation of the [(η6-p-cym)RuII(N-N))Cl]PF6 Type Complexes Containing O-peracylated Glycosyl Azole Ligands
To a solution of ([(η6-p-cym)RuCl2]2) dimer (Ru-dimer) in CH2Cl2 (1 mL/10 mg dimer), the corresponding O-peracylated glycosyl azole (2–2.3 eq.) and TlPF6 (2 eq.) were added. Under stirring, 1 mL of methanol was added to this reaction mixture in order to accelerate the precipitation of the TlCl. The initial red solution turned to yellow, indicating the formation of the [(η6-p-cym)RuII(N-N)Cl]PF6 type complex. The reaction mixture was then stirred at r.t. for an additional hour, and the total disappearance of the Ru-dimer was judged by TLC (9:1 CHCl3-MeOH). After completion of the complexation reaction, the precipitated TlCl was filtered off, and the solution was evaporated in vacuo. The crude complex was purified by column chromatography or crystallization.
5.1.6. General Procedure V for the Formation of the [(η6-p-cym)RuII(N-N)Cl]PF6 Type Complexes Containing Unprotected Glycosyl Azole Ligands
In a solution of ([(η6-p-cym)RuCl2]2) dimer (Ru-dimer) in CH2Cl2 (1 mL/10 mg dimer), the corresponding unprotected glycosyl azole (2 eq.) and TlPF6 (2 eq.) were suspended. Under stirring, 1 mL of methanol was added to this reaction mixture in order to dissolve the heterocyclic sugar derivative and accelerate the precipitation of the TlCl. The initial red solution turned to yellow, indicating the formation of the [(η6-p-cym)RuII(N-N)Cl]PF6 type complex. The reaction mixture was then stirred at r.t. for an additional hour, and the total disappearance of the Ru-dimer was judged by TLC (9:1 CHCl3-MeOH). After completion of the complexation reaction, the precipitated TlCl was filtered off, and the solution was evaporated in vacuo. The crude complex was purified by crystallization.
5.1.7. 1-(2′,3′,4′,6′-Tetra-O-acetyl-β-d-glucopyranosyl)-4-(pyridin-2-yl)-1,2,3-triazole (L-1a)
Prepared from azide [
28,
29]
1 (100 mg, 0.27 mmol) and 2-ethynylpyridine (28 µL, 0.27 mmol) according to general procedure I. Purified by column chromatography (1:1 → 2:1 EtOAc–hexane) to give 121 mg white amorphous solid (95%). R
f = 0.18 (1:1 EtOAc–hexane); [α]
D = −55 (c 0.21, CHCl
3).
1H-NMR (360 MHz, CDCl
3) δ (ppm): 8.61 (1H, d,
J = 4.8 Hz, Py-H-6), 8.42 (1H, s, Tria-H-5), 8.15 (1H, d,
J = 7.8 Hz, Py-H-3), 7.78 (1H, dt,
J = 7.8, 1.6 Hz, Py-H-4), 7.25 (1H, m, Py-H-5), 5.94 (1H, d,
J = 8.8 Hz, H-1′), 5.51 (1H, pt,
J = 9.5, 9.5 Hz, H-2′), 5.46 (1H, pt,
J = 9.5, 9.5 Hz, H-3′), 5.28 (1H, pt,
J = 9.5, 9.5 Hz, H-4′), 4.32 (1H, dd,
J = 12.6, 4.8 Hz, H-6′a), 4.17 (1H, dd,
J = 12.6, 1.7 Hz, H-6′b), 4.04 (1H, ddd,
J = 9.5, 4.8, 1.7 Hz, H-5′), 2.10, 2.08, 2.05, 1.90 (4 × 3H, 4 s, 4 × CH
3);
13C-NMR (90 MHz, CDCl
3) δ (ppm): 170.6, 170.1, 169.4, 168.9 (4 × C=O), 149.8, 149.2 (Tria-C-4, Py-C-2), 149.7 (Py-C-6), 137.0 (Py-C-4), 123.3 (Py-C-5), 120.5 (Py-C-3), 120.7 (Tria-C-5), 86.0 (C-1′), 75.3 (C-5′), 72.8 (C-3′), 70.7 (C-2′), 67.8 (C-4′), 61.7 (C-6′), 20.8, 20.7 (2), 20.3 (4 × CH
3). The
1H and
13C NMR data are in good agreement with the reported ones [
63]. ESI-HRMS positive mode (
m/
z): calculated for C
21H
25N
4O
9+ [M + H]
+ 477.1616; C
21H
24N
4NaO
9+ [M + Na]
+ 499.1435. Found: [M + H]
+ 477.1614; [M + Na]
+ 499.1432.
5.1.8. 1-(2′,3′,4′,6′-Tetra-O-acetyl-β-d-glucopyranosyl)-4-(quinolin-2-yl)-1,2,3-triazole (L-1b)
Prepared from azide [
28,
29]
1 (1.00 g, 2.68 mmol) and 2-ethynylquinoline [
62] (0.41 g, 2.68 mmol) according to general procedure I. Purified by column chromatography (1:1 EtOAc–hexane) to yield 1.20 g white amorphous solid (85%). R
f = 0.23 (1:1 EtOAc–hexane); [α]
D = −90 (c 0.20, CHCl
3).
1H-NMR (360 MHz, CDCl
3) δ (ppm): 8.63 (1H, s, Tria-H-5), 8.31 (1H, d,
J = 8.6 Hz, Qu-H-3), 8.24 (1H, d,
J = 8.6 Hz, Qu-H-4), 8.08 (1H, d,
J = 8.5 Hz, Qu-H-5 or Qu-H-8), 7.82 (1H, d,
J = 8.0 Hz, Qu-H-5 or Qu-H-8), 7.72 (1H, pt,
J = 7.9, 7.6 Hz, Qu-H-6 or Qu-H-7), 7.53 (1H, pt,
J = 7.5, 7.4 Hz, Qu-H-6 or Qu-H-7), 5.99 (1H, d,
J = 9.2 Hz, H-1′), 5.59 (1H, pt,
J = 9.4, 9.2 Hz, H-2′), 5.48 (1H, pt,
J = 9.5, 9.4 Hz, H-3′), 5.31 (1H, pt,
J = 9.7, 9.5 Hz, H-4′), 4.35 (1H, dd,
J = 12.6, 4.8 Hz, H-6′a), 4.18 (1H, dd,
J = 12.6, 2.1 Hz, H-6′b), 4.07 (1H, ddd,
J = 9.7, 4.8, 2.1 Hz, H-5′), 2.11, 2.09, 2.05, 1.91 (4 × 3H, 4 s, 4 × CH
3);
13C-NMR (90 MHz, CDCl
3) δ (ppm): 170.6, 170.1, 169.4, 169.0 (4 × C=O), 149.8, 149.4, 148.2 (Tria-C-4, Qu-C-2, Qu-C-8a), 137.0 (Qu-C-4), 129.9 (Qu-C-6 or Qu-C-7), 129.3 (Qu-C-5 or Qu-C-8), 128.0 (Qu-C-4a), 127.8 (Qu-C-5 or Qu-C-8), 126.6 (Qu-C-6 or Qu-C-7), 121.4 (Tria-C-5), 118.7 (Qu-C-3), 86.0 (C-1′), 75.3 (C-5′), 72.9 (C-3′), 70.7 (C-2′), 67.8 (C-4′), 61.7 (C-6′), 20.8, 20.6 (2), 20.3 (4 × CH
3). The
1H and
13C NMR data are in good agreement with the reported ones [
64]. ESI-HRMS positive mode (
m/
z): calculated for C
25H
27N
4O
9+ [M + H]
+ 527.1773; C
25H
26N
4NaO
9+ [M + Na]
+ 549.1592. Found: [M + H]
+ 527.1773; [M + Na]
+ 549.1593.
5.1.9. 1-(2′,3′,4′,6′-Tetra-O-benzoyl-β-d-glucopyranosyl)-4-(pyridin-2-yl)-1,2,3-triazole (L-2a)
The 1-(β-d-glucopyranosyl)-4-(pyridin-2-yl)-1,2,3-triazole (L-3a, 20.0 mg, 0.065 mmol) was suspended in dry pyridine (0.5 mL), and benzoyl chloride (36 µL, 0.310 mmol) was added. The reaction mixture was stirred at 60 °C until the TLC (3:2 EtOAc–hexane) showed complete disappearance of the starting material (1 h). The solvent was removed under diminished pressure. The residue was dissolved in CH2Cl2 (20 mL) and extracted with sat. aq. NaHCO3 (10 mL) and then with water (10 mL). The organic phase was dried over MgSO4, filtered, and evaporated. Purification by column chromatography (3:2 EtOAc–hexane) yielded 41 mg of white amorphous solid (87%). Rf = 0.30 (3:2 EtOAc–hexane); [α]D = −75 (c 0.20, CHCl3). 1H-NMR (400 MHz, CDCl3) δ (ppm): 8.61 (2H, 1 signal, Tria-H-5, Py-H-6), 8.17–7.21 (23H, m, Ar, Py-H-3–Py-H-5), 6.34 (1H, d, J = 9.2 Hz, H-1′), 6.16 (1H, pt, J = 9.6, 9.5 Hz, H-3′) 6.07 (1H, pt, J = 9.5, 9.2 Hz, H-2′), 5.90 (1H, pt, J = 9.6, 9.6 Hz, H-4′), 4.69–4.49 (3H, m, H-5′, H-6′a, H-6′b); 13C-NMR (100 MHz, CDCl3) δ (ppm): 166.2, 165.8, 165.2, 164.7 (4 × C=O), 149.7, 149.0 (Tria-C-4, Py-C-2), 149.6 (Py-C-6), 137.1 (Py-C-4), 133.8, 133.7, 133.6, 133.5, 133.4, 130.2–128.2 (Ar), 123.3 (Py-C-5), 121.0 (Tria-C-5), 120.6 (Py-C-3), 86.4 (C-1′), 75.7 (C-5′), 73.2 (C-3′), 71.3 (C-2′), 69.0 (C-4′), 62.8 (C-6′). ESI-HRMS positive mode (m/z): calculated for C41H32N4NaO9+ [M + Na]+ 747.2061. Found: 747.2041.
5.1.10. 1-(β-d-Glucopyranosyl)-4-(pyridin-2-yl)-1,2,3-triazole (L-3a)
Prepared from compound
L-1a (0.81 g, 1.70 mmol) according to general procedure III. Purified by column chromatography (7:2 CHCl
3–MeOH) to give 0.40 g white amorphous solid (76%). R
f = 0.26 (7:2 CHCl
3–MeOH); [α]
D = −12 (c 0.20, MeOH).
1H-NMR (400 MHz, CD
3OD) δ (ppm): 8.63 (1H, s, Tria-H-5), 8.59 (1H, d,
J = 4.3 Hz, Py-H-6), 8.09 (1H, d,
J = 7.9 Hz, Py-H-3), 7.92 (1H, pt,
J = 7.9, 7.8 Hz, Py-H-4), 7.38 (1H, m, Py-H-5), 5.70 (1H, d,
J = 9.2 Hz, H-1′), 3.96 (1H, pt,
J = 9.1, 9.0 Hz, H-2′), 3.90 (1H, dd,
J = 12.2, 1.3 Hz, H-6′a), 3.74 (1H, dd,
J = 12.2, 5.3 Hz, H-6′b), 3.64–3.58 (2H, m, H-3′ or H-4′, H-5′), 3.54 (1H, pt,
J = 9.2, 9.1 Hz, H-3′ or H-4′);
13C-NMR (90 MHz, CD
3OD) δ (ppm): 150.9, 148.6 (Tria-C-4, Py-C-2), 150.5 (Py-C-6), 138.9 (Py-C-4), 124.6 (Py-C-5), 123.4 (Tria-C-5), 121.7 (Py-C-3), 89.8 (C-1′), 81.2 (C-5′), 78.5 (C-3′ or C-4′), 74.1 (C-2′), 70.9 (C-3′ or C-4′), 62.4 (C-6′). The
1H and
13C NMR data are in good agreement with the reported ones [
65]. ESI-HRMS positive mode (
m/
z): calculated for C
13H
16N
4NaO
5+ [M + Na]
+ 331.1013; C
26H
32N
8NaO
10+ [2M + Na]
+ 639.2134. Found: [M + Na]
+ 331.1012; [2M + Na]
+ 639.2135.
5.1.11. 1-(β-d-Glucopyranosyl)-4-(quinolin-2-yl)-1,2,3-triazole (L-3b)
Prepared from compound L-1b (500 mg, 0.95 mmol) according to general procedure III. The crude product was recrystallized from MeOH to yield 290 mg of white amorphous solid (85%). Rf = 0.44 (7:2 CHCl3–MeOH); [α]D = −4 (c 0.20, DMSO). 1H-NMR (400 MHz, DMSO-d6 + 1–2 drops of D2O) δ (ppm): 8.99 (1H, s, Tria-H-5), 8.51 (1H, d, J = 8.6 Hz, Qu-H-4), 8.25 (1H, d, J = 8.6 Hz, Qu-H-3), 8.06–8.01 (2H, m, Qu-H-5, Qu-H-8), 7.82 (1H, pt, J = 7.8, 7.4 Hz, Qu-H-6 or Qu-H-7), 7.63 (1H, pt, J = 7.6, 7.4 Hz, Qu-H-6 or Qu-H-7), 5.68 (1H, d, J = 9.2 Hz, H-1′), 3.90 (1H, pt, J = 9.2, 9.1 Hz, H-2′), 3.77–3.72 (1H, m, H-6′a), 3.56–3.49 (2H, m, H-5′, H-6′b), 3.46 (1H, pt, J = 9.1, 9.0 Hz, H-3′), 3.34 (1H, pt, J = 9.2, 9.0 Hz, H-4′); 13C-NMR (90 MHz, DMSO-d6) δ (ppm): 150.1, 147.5, 147.3 (Tria-C-4, Qu-C-2, Qu-C-8a), 137.3 (Qu-C-4), 130.1 (Qu-C-6 or Qu-C-7), 128.6, 128.1 (Qu-C-5, Qu-C-8), 127.3 (Qu-C-4a), 126.5 (Qu-C-6 or Qu-C-7) 123.3 (Tria-C-5), 118.3 (Qu-C-3), 87.8 (C-1′), 80.0 (C-5′), 76.8 (C-3′), 72.2 (C-2′), 69.5 (C-4′), 60.8 (C-6′). ESI-HRMS positive mode (m/z): calculated for C17H19N4O5+ [M + H]+ 359.1350; C17H18N4NaO5+ [M + Na]+ 381.1169; C34H36N8NaO10+ [2M + Na]+ 739.2447. Found: [M + H]+ 359.1349; [M + Na]+ 381.1168; [2M + Na]+ 739.2448.
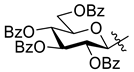
5.1.12. 2-(2′,3′,4′,6′-Tetra-O-benzoyl-β-d-glucopyranosyl)-5-(pyridin-2-yl)-1,3,4-oxadiazole (L-4)
Prepared from tetrazole [
58,
59]
2 (5.00 g, 7.71 mmol) and 2-picolinic acid (1.90 g, 14.43 mmol) according to general procedure II. Reaction time: 5 h. Purification by column chromatography (1:1 EtOAc–hexane) and crystallization from EtOH gave 1.96 g white solid (35%). R
f = 0.28 (1:1 EtOAc–hexane). [α]
D = −92 (c 0.20, CHCl
3).
1H-NMR (400 MHz, CDCl
3) δ (ppm): 8.81 (1H, d,
J = 4.8 Hz, Py-H-6), 8.21 (1H, d,
J = 7.8 Hz, Py-H-3), 8.03–7.81, 7.56–7.27 (22H, m, Ar, Py-H-4, Py-H-5), 6.10 (1H, pt,
J = 9.4, 9.4 Hz, H-3′), 6.05 (1H, pt,
J = 9.4, 9.4 Hz, H-2′), 5.86 (1H, pt,
J = 9.4, 9.4 Hz, H-4′), 5.28 (1H, d,
J = 9.4 Hz, H-1′), 4.67 (1H, dd,
J = 12.4, 2.3 Hz, H-6′a), 4.54 (1H, dd,
J = 12.4, 5.5 Hz, H-6′b), 4.37 (1H, ddd,
J = 9.4, 5.5, 2.3 Hz, H-5′);
13C-NMR (90 MHz, CDCl
3) δ (ppm): 166.2, 165.8, 165.2, 165.1, 164.9, 162.2 (4 × C=O, OD-C-2, OD-C-5), 150.5 (Py-C-6), 143.2 (Py-C-2), 137.3 (Py-C-4), 133.7, 133.6, 133.5, 133.2, 130.0–128.4 (Ar), 126.2 (Py-C-5), 123.5 (Py-C-3), 77.3 (C-5′), 73.8 (C-3′), 72.1 (C-1′), 70.6 (C-2′), 69.2 (C-4′), 63.3 (C-6′). ESI-HRMS positive mode (
m/
z): calculated for C
41H
31N
3NaO
10+ [M + Na]
+ 748.1902. Found: 748.1907.
5.1.13. 2-(2′,3′,4′,6′-Tetra-O-acetyl-β-d-glucopyranosyl)-5-(pyridin-2-yl)-1,3,4-oxadiazole (L-5)
To a solution of the 2-(β-d-glucopyranosyl)-5-(pyridin-2-yl)-1,3,4-oxadiazole (L-10, 20 mg, 0.065 mmol) in dry pyridine (0.5 mL), acetic anhydride (0.06 mL, 0.635 mmol) was added, and the mixture was stirred at 60 °C. After 1 h, the TLC (1:1 EtOAc–hexane) showed total consumption of L-10. The solvent was removed under reduced pressure, and the residue was purified by column chromatography (1:1 EtOAc–hexane). White amorphous solid, yield: 28 mg (90%). Rf = 0.21 (1:1 EtOAc–hexane); [α]D = −61 (c 0.19, CHCl3). 1H-NMR (400 MHz, CDCl3) δ (ppm): 8.82 (1H, d, J = 4.4 Hz, Py-H-6), 8.26 (1H, d, J = 7.9 Hz, Py-H-3), 7.91 (1H, dt, J = 7.9, 1.1 Hz, Py-H-4), 7.49 (1H, m, Py-H-5), 5.56 (1H, pt, J = 9.8, 9.7 Hz, H-2′), 5.40 (1H, pt, J = 9.4, 9.3 Hz, H-3′), 5.24 (1H, pt, J = 9.8, 9.7 Hz, H-4′), 4.92 (1H, d, J = 10.1 Hz, H-1′), 4.30 (1H, dd, J = 12.6, 5.1 Hz, H-6′a), 4.18 (1H, dd, J = 12.6, 2.2 Hz, H-6′b), 3.91 (1H, ddd, J = 9.7, 5.1, 2.2 Hz, H-5′), 2.09, 2.07, 2.04, 1.94 (4 × 3H, 4 s, 4 × CH3); 13C-NMR (100 MHz, CDCl3) δ (ppm): 170.7, 170.3, 169.4, 169.3 (4 × C=O), 165.1, 162.1 (OD-C-2, OD-C-5), 150.6 (Py-C-6), 143.2 (Py-C-2), 137.4 (Py-C-4), 126.3 (Py-C-5), 123.6 (Py-C-3), 76.9 (C-5′), 73.5 (C-3′), 71.6 (C-1′), 69.7 (C-2′), 68.0 (C-4′), 62.0 (C-6′), 20.8, 20.7 (2), 20.5 (4 × CH3). ESI-HRMS positive mode (m/z): calculated for C21H23N3NaO10+ [M + Na]+ 500.1276. Found: 500.1275.
5.1.14. 2-(2′,3′,4′-Tri-O-benzoyl-β-d-xylopyranosyl)-5-(pyridin-2-yl)-1,3,4-oxadiazole (L-6)
Prepared from tetrazole [
60]
3 (3.00 g, 5.83 mmol) and 2-picolinic acid (1.42 g, 11.53 mmol) according to general procedure II. Reaction time: 5 h. Purification by column chromatography (2:1 EtOAc–hexane) and crystallization from EtOH yielded 2.00 g white solid (58%). R
f = 0.35 (4:1 EtOAc–hexane); [α]
D = −123 (c 0.20, CHCl
3).
1H-NMR (360 MHz, CDCl
3) δ (ppm): 8.80 (1H, d,
J = 4.8 Hz, Py-H-6), 8.21 (1H, d, 7.9 Hz, Py-H-3), 7.99–7.83, 7.57–7.30 (17H, m, Ar, Py-H-4, Py-H-5), 6.05 (1H, pt,
J = 9.2, 9.2 Hz, H-3′), 5.96 (1H, pt,
J = 9.2, 9.2 Hz, H-2′), 5.57 (1H, ddd,
J = 10.1, 9.2, 5.3 Hz, H-4′), 5.15 (1H, d,
J = 9.2 Hz, H-1′), 4.62 (1H, dd,
J = 11.4, 5.3 Hz, H-5′eq), 3.80 (1H, pt,
J = 11.4, 10.1 Hz, H-5′ax);
13C-NMR (90 MHz, CDCl
3) δ (ppm): 165.8, 165.6, 165.1 (2), 162.5 (3 × C=O, OD-C-2, OD-C-5), 150.6 (Py-C-6), 143.3 (Py-C-2), 137.3 (Py-C-4), 133.7, 133.6, 133.5, 130.0–128.5 (Ar), 126.2 (Py-C-5), 123.5 (Py-C-3), 72.9 (C-3′), 72.5 (C-1′), 70.4 (C-2′), 69.6 (C-4′), 67.5 (C-5′). ESI-HRMS positive mode (
m/
z): calculated for C
33H
25N
3NaO
8+ [M + Na]
+ 614.1534. Found: 614.1535.
5.1.15. 2-(2′,3′,4′,6′-Tetra-O-acetyl-β-d-galactopyranosyl)-5-(pyridin-2-yl)-1,3,4-oxadiazole (L-7)
Prepared from tetrazole [
61]
4 (0.50 g, 1.25 mmol) and 2-picolinic acid (0.31 g, 2.50 mmol) according to general procedure II. Reaction time: 2 h. Purification by column chromatography (1:1 EtOAc–hexane) and crystallization from EtOH yielded 0.30 g white solid (50%). R
f = 0.17 (1:1 EtOAc–hexane); [α]
D = −41 (c 0.21, CHCl
3).
1H-NMR (360 MHz, CDCl
3) δ (ppm): 8.82 (1H, ddd,
J = 4.8, 1.8, 0.9 Hz, Py-H-6), 8.27 (1H, dd,
J = 7.9, 0.9 Hz, Py-H-3), 7.91 (1H, dt,
J = 7.9, 1.8 Hz, Py-H-4), 7.50 (1H, ddd,
J = 7.7, 4.8, 1.1 Hz, Py-H-5), 5.67 (1H, pt,
J = 10.1, 10.0 Hz, H-2′), 5.56 (1H, d,
J = 3.4 Hz, H-4′), 5.24 (1H, dd,
J = 10.1, 3.4 Hz, H-3′), 4.89 (1H, d,
J = 10.0 Hz, H-1′), 4.23–4.11 (3H, m, H-5′, H-6′a, H-6′b), 2.23, 2.06, 2.02, 1.96 (4 × 3H, 4 s, 4 × CH
3);
13C-NMR (90 MHz, CDCl
3) δ (ppm): 170.4, 170.3, 170.0, 169.4, 165.0, 162.3 (4 × C=O, OD-C-2, OD-C-5), 150.5 (Py-C-6), 143.2 (Py-C-2), 137.3 (Py-C-4), 126.2 (Py-C-5), 123.6 (Py-C-3), 75.6 (C-5′), 72.2 (C-1′), 71.4 (C-3′), 67.3 (C-4′), 66.9 (C-2′), 61.6 (C-6′), 20.8, 20.7, 20.6, 20.5 (4 × CH
3). ESI-HRMS positive mode (
m/
z): calculated for C
21H
24N
3O
10+ [M + H]
+ 478.1456; C
21H
23N
3NaO
10+ [M + Na]
+ 500.1276; C
42H
46N
6NaO
20+ [2M + Na]
+ 977.2659. Found: [M + H]
+ 478.1454; [M + Na]
+ 500.1274; [2M + Na]
+ 977.2653.
5.1.16. 2-(2′,3′,4′,6′-Tetra-O-benzoyl-β-d-galactopyranosyl)-5-(pyridin-2-yl)-1,3,4-oxadiazole (L-8)
The 2-(β-d-galactopyranosyl)-5-(pyridin-2-yl)-1,3,4-oxadiazole (L-10, 20.0 mg, 0.065 mmol) was suspended in dry pyridine (0.5 mL), and benzoyl chloride (36 µL, 0.310 mmol) was added. The reaction mixture was stirred at 60 °C until the TLC (3:2 EtOAc–hexane) showed complete disappearance of the starting material (1 h). The solvent was removed under diminished pressure. The residue was dissolved in CH2Cl2 (20 mL) and extracted with sat. aq. NaHCO3 (10 mL) and then with water (10 mL). The organic phase was dried over MgSO4, filtered, and evaporated. Purification by column chromatography (3:2 EtOAc–hexane) yielded 38 mg of white amorphous solid (81%). Rf = 0.27 (3:2 EtOAc–hexane); [α]D = +9 (c 0.20, CHCl3). 1H-NMR (400 MHz, CDCl3) δ (ppm): 8.81 (1H, d, J = 4.8 Hz, Py-H-6), 8.21 (1H, d, J = 7.9 Hz, Py-H-3), 8.17–7.23 (22H, m, Ar, Py-H-4, Py-H-5), 6.32 (1H, pt, J = 10.1, 10.0 Hz, H-2′), 6.16 (1H, d, J = 3.3 Hz, H-4′), 5.84 (1H, dd, J = 10.1, 3.3 Hz, H-3′), 5.30 (1H, d, J = 10.0 Hz, H-1′), 4.68 (1H, dd, J = 11.1, 6.4 Hz, H-6′a), 4.58 (1H, pt, J = 6.4, 5.9 Hz, H-5′), 4.49 (1H, dd, J = 11.1, 5.9 Hz, H-6′b); 13C-NMR (100 MHz, CDCl3) δ (ppm): 166.2, 165.7, 165.6, 165.1, 165.0, 162.4 (4 × C=O, OD-C-2, OD-C-5), 150.5 (Py-C-6), 143.3 (Py-C-2), 137.3 (Py-C-4), 133.8, 133.6, 133.5, 133.4, 130.2–130.0, 129.9–128.5 (Ar), 126.2 (Py-C-5), 123.6 (Py-C-3), 76.1 (C-5′), 72.5, 72.3 (C-1′, C-3′), 68.4 (C-4′), 67.9 (C-2′), 62.3 (C-6′). ESI-HRMS positive mode (m/z): calculated for C41H31N3NaO10+ [M + Na]+ 748.1902. Found: 748.1901.
5.1.17. 2-(l-Arabino-1′,2′,3′,4′-tetraacetoxybutyl)-5-(pyridin-2-yl)-1,3,4-oxadiazole (L-9)
Prepared from tetrazole [
33]
5 (3.50 g, 9.77 mmol) and 2-picolinic acid (2.41 g, 19.58 mmol) according to general procedure II. Reaction time: 5 h. Purification by column chromatography (1:1 → 2:1 → 3:1 EtOAc–hexane) yielded 0.57 g of white amorphous solid (13%). R
f = 0.27 (4:1 EtOAc–hexane); [α]
D = +6 (c 0.20, CHCl
3).
1H-NMR (400 MHz, CDCl
3) δ (ppm): 8.79 (1H, d,
J = 4.4 Hz, Py-H-6), 8.26 (1H, d,
J = 7.9 Hz, Py-H-3), 7.90 (1H, dt,
J = 7.8, 1.4 Hz, Py-H-4), 7.49 (1H, m, Py-H-5), 6.35 (1H, d,
J = 2.2 Hz, H-1′), 5.69 (1H, dd,
J = 9.4, 2.2 Hz, H-2′), 5.36 (1H, ddd,
J = 9.4, 3.9, 2.3 Hz, H-3′), 4.35 (1H, dd,
J = 12.6, 2.3 Hz, H-4′a), 4.23 (1H, dd,
J = 12.6, 3.9 Hz, H-4′b), 2.22, 2.11, 2.10, 2.06 (4 × 3H, 4 s, CH
3);
13C-NMR (100 MHz, CDCl
3) δ (ppm): 170.6, 169.7, 169.6 (2) (4 × C=O), 164.5, 162.6 (OD-C-2, OD-C-5), 150.4 (Py-C-6), 143.1 (Py-C-2), 137.4 (Py-C-4), 126.3 (Py-C-5), 123.5 (Py-C-3), 68.7 (C-2′), 67.8 (C-3′), 64.4 (C-1′), 61.5 (C-4′), 20.8, 20.7, 20.5 (2) (4 × CH
3). ESI-HRMS positive mode (
m/
z): calculated for C
19H
22N
3O
9+ [M + H]
+ 436.1351; C
19H
21N
3NaO
9+ [M + Na]
+ 458.1170. Found: [M + H]
+ 436.1349; [M + Na]
+ 458.1170.
5.1.18. 2-(β-d-Glucopyranosyl)-5-(pyridin-2-yl)-1,3,4-oxadiazole (L-10)
Prepared from compound L-4 (0.60 g, 0.83 mmol) according to general procedure III. Purified by column chromatography (9:1 CHCl3–MeOH) to give 0.22 g white amorphous solid (87%). Rf = 0.29 (8:2 CHCl3–MeOH); [α]D = +17 (c 0.20, MeOH). 1H-NMR (360 MHz, CD3OD) δ (ppm): 8.75 (1H, ddd, J = 4.9, 1.7, 0.9 Hz, Py-H-6), 8.25 (1H, dd, J = 7.9, 1.1 Hz, Py-H-3), 8.07 (1H, dt, J = 7.9, 1.7 Hz, Py-H-4), 7.64 (1H, ddd, J = 7.9, 4.9, 1.1 Hz, Py-H-5), 4.68 (1H, d, J = 9.9 Hz, H-1′), 3.90 (1H, dd, J = 12.2, 1.9 Hz, H-6′a), 3.84 (1H, dd, J = 9.9, 8.7 Hz, H-2′), 3.70 (1H, J = 12.2, 5.4 Hz, H-6′b), 3.54 (1H, pt, J = 8.9, 8.6 Hz, H-3′), 3.54–3.50 (1H, m, H-5′), 3.46 (1H, pt, J = 9.3, 8.8 Hz, H-4′); 13C-NMR (90 MHz, CD3OD) δ (ppm): 166.4, 165.7 (OD-C-2, OD-C-5), 151.4 (Py-C-6), 144.0 (Py-C-2), 139.3 (Py-C-4), 127.9 (Py-C-5), 124.5 (Py-C-3), 82.9 (C-5′), 79.1 (C-3′), 74.7 (C-1′), 73.4 (C-2′), 71.3 (C-4′), 62.8 (C-6′). ESI-HRMS positive mode (m/z): calculated for C13H15N3NaO6+ [M + Na]+ 332.0853. Found: 332.0844.
5.1.19. 2-(β-d-Xylopyranosyl)-5-(pyridin-2-yl)-1,3,4-oxadiazole (L-11)
Prepared from compound L-6 (500 mg, 0.85 mmol) according to general procedure III. Purified by column chromatography (9:1 CHCl3–MeOH) to give 82 mg white amorphous solid (35%). Rf = 0.27 (9:1 CHCl3–MeOH); [α]D = −44 (c 0.20, MeOH). 1H-NMR (360 MHz, CD3OD) δ (ppm): 8.74 (1H, ddd, J = 4.9, 1.6, 0.9 Hz, Py-H-6), 8.24 (1H, ddd, J = 7.9, 1.1, 0.9 Hz, Py-H-3), 8.06 (1H, dt, J = 7.9, 1.6 Hz, Py-H-4), 7.63 (1H, ddd, J = 7.7, 4.9, 1.1 Hz, Py-H-5), 4.59 (1H, d, J = 9.8 Hz, H-1′), 4.03 (1H, dd, J = 11.1, 5.4 Hz, H-5′eq), 3.84 (1H, pt, J = 9.8, 9.1 Hz, H-2′), 3.66 (1H, td, J = 10.1, 9.1, 5.4 Hz, H-4′), 3.47 (1H, pt, J = 9.1, 9.1 Hz, H-3′), 3.41 (1H, pt, J = 11.1, 10.1 Hz, H-5′ax); 13C-NMR (90 MHz, CD3OD) δ (ppm): 166.4, 165.7 (OD-C-2, OD-C-5), 151.4 (Py-C-6), 144.0 (Py-C-2), 139.3 (Py-C-4), 127.9 (Py-C-5), 124.5 (Py-C-3), 79.2 (C-3′), 75.4 (C-1′), 73.4 (C-2′), 71.7 (C-5′), 71.0 (C-4′). ESI-HRMS positive mode (m/z): calculated for C12H13N3NaO5+ [M + Na]+ 302.0747; C24H26N6NaO10+ [2M + Na]+ 581.1603. Found: [M + Na]+ 302.0747; [2M + Na]+ 581.1604.
5.1.20. 2-(β-d-Galactopyranosyl)-5-(pyridin-2-yl)-1,3,4-oxadiazole (L-12)
Prepared from compound L-7 (350 mg, 0.73 mmol) according to general procedure III. Purified by column chromatography (7:2 CHCl3–MeOH) to give 177 mg white amorphous solid (78%). Rf = 0.33 (7:2 CHCl3–MeOH); [α]D = +25 (c 0.21, MeOH). 1H-NMR (400 MHz, CD3OD) δ (ppm): 8.77 (1H, d, J = 4.8 Hz, Py-H-6), 8.27 (1H, d, J = 7.9 Hz, Py-H-3), 8.08 (1H, dt, J = 7.9, 1.5 Hz, Py-H-4), 7.65 (1H, m, Py-H-5), 4.63 (1H, d, J = 9.8 Hz, H-1′), 4.22 (1H, pt, J = 9.8, 9.6 Hz, H-2′), 4.01 (1H, d, J = 3.2 Hz, H-4′), 3.84–3.73 (3H, m, H-5′, H-6′a, H-6′b), 3.67 (1H, dd, J = 9.6, 3.2 Hz, H-3′); 13C-NMR (90 MHz, CD3OD) δ (ppm): 166.5, 165.7 (OD-C-2, OD-C-5), 151.4 (Py-C-6), 144.1 (Py-C-2), 139.3 (Py-C-4), 127.9 (Py-C-5), 124.5 (Py-C-3), 81.7 (C-5′), 75.8 (C-3′), 75.1 (C-1′), 70.7 (C-4′), 70.2 (C-2′), 62.8 (C-6′). ESI-HRMS positive mode (m/z): calculated for C13H16N3O6+ [M + H]+ 310.1034; C13H15N3NaO6+ [M + Na]+ 332.0853; C26H30N6NaO12+ [2M + Na]+ 641.1814. Found: [M + H]+ 310.1035; [M + Na]+ 332.0852; [2M + Na]+ 641.1815.
5.1.21. Complex Ru-1a
Prepared from complex Ru-dimer (50 mg, 0.082 mmol), compound L-1a (86 mg, 0.181 mmol, 2.2 eq.), and TlPF6 (57 mg, 0.163 mmol) according to general procedure IV. Purified by column chromatography (9:1 CHCl3–MeOH) to give 143 mg (98%) yellow powder. Rf: 0.32 (9:1 CHCl3–MeOH). Diastereomeric ratio: 1:1. 1H-NMR (400 MHz, CDCl3) δ (ppm): 9.25, 9.22 (2 × 1H, 2 d, J = 5.5 Hz in each, 2 × Py-H-6), 8.92, 8.79 (2 × 1H, 2 s, 2 × Tria-H-5), 7.96 (2H, t, J = 7.6 Hz, 2 × Py-H-4), 7.89 (2H, d, J = 7.8 Hz, 2 × Py-H-3), 7.57–7.52 (2H, m, 2 × Py-H-5), 6.00, 5.99 (2 × 1H, 2 d, J = 9.4 Hz in each, 2 × H-1′), 5.95–5.83, 5.73–5.67 (10H, m, 2 × 4 × p-cym-CHAr, 2 × H-2′), 5.47, 5.46 (2 × 1H, 2 pt, J = 9.4, 9.2 Hz in each, 2 × H-3′), 5.35, 5.33 (2 × 1H, 2 pt, J = 9.7, 9.7 Hz in each, 2 × H-4′), 4.38, 4.32 (2 × 1H, 2 dd, J = 12.8, 4.7 Hz in each, 2 × H-6′a), 4.26–4.15 (4H, m, 2 × H-5′, 2 × H-6′b), 2.79, 2.74 (2 × 1H, 2 hept, J = 6.9 Hz in each, 2 × i-Pr-CH), 2.22, 2.20 (2 × 3H, 2 s, 2 × C6H4–CH3), 2.10, 2.09, 2.05, 1.95, 1.93 (24H, singlets, 2 × 4 × COCH3), 1.17–1.12 (12H, m, 2 × 2 × i-Pr-CH3); 13C-NMR (90 MHz, CDCl3) δ (ppm): 170.8, 170.7, 170.0, 169.9, 169.6, 169.5, 169.4, 169.2 (2 × 4 × C=O), 155.5, 155.4 (2 × Py-C-6), 147.6, 147.5, 147.1, 146.7 (2 × Tria-C-4, 2 × Py-C-2), 140.3, 140.2 (2 × Py-C-4), 127.1, 126.9 (2 × Py-C-5), 125.4, 125.3 (2 × Tria-C-5), 122.9 (2) (2 × Py-C-3), 106.3, 105.5, 103.1, 101.8 (2 × 2 × p-cym-CqAr), 86.8, 86.7 (2 × C-1′), 86.4, 85.4, 85.3, 85.0, 84.8, 83.9, 83.9, 83.1 (2 × 4 × p-cym-CHAr), 75.5, 75.3 (2 × C-5′), 73.1, 73.0 (2 × C-3′), 70.2, 69.8 (2 × C-2′), 67.6, 67.5 (2 × C-4′), 61.6, 61.5 (2 × C-6′), 31.1, 31.0 (2 × i-Pr-CH), 22.5, 22.2, 22.1, 21.7 (2 × 2 × i-Pr-CH3), 20.8–20.3 (2 × 4 × COCH3), 18.7 (2) (2 × C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C31H38ClN4O9Ru+ [M − PF6]+ 747.1371. Found: 747.1370.
5.1.22. Complex Ru-1b
Prepared from complex Ru-dimer (50 mg, 0.082 mmol), compound L-1b (95 mg, 0.180 mmol, 2.2 eq.), and TlPF6 (57 mg, 0.163 mmol) according to general procedure IV. Purified by column chromatography (9:1 CHCl3–MeOH) to give 145 mg (96%) yellow powder. Rf: 0.55 (9:1 CHCl3–MeOH). Diastereomeric ratio: 2:1. 1H-NMR (400 MHz, CDCl3) δ (ppm): 9.24 (s, minor Tria-H-5), 9.03 (s, major Tria-H-5), 8.75 (d, J = 8.8 Hz, major Qu-H-8), 8.68 (d, J = 8.7 Hz, minor Qu-H-8), 8.39 (d, J = 8.6 Hz, major Qu-H-4), 8.37 (d, J = 8.9 Hz, minor Qu-H-4), 8.04–7.89, 7.75–7.70 (2 m, minor and major Qu-H-3, Qu-H-5–Qu-H-7), 6.11 (d, J = 9.4 Hz, minor H-1′), 6.08 (d, J = 9.2 Hz, major H-1′), 6.03 (pt, J = 9.2, 9.0 Hz, major H-2′), 5.97 (pt, J = 9.4, 9.0 Hz, minor H-2′), 5.96, 5.95 (2 d, J = 6.0 Hz in each, major p-cym-CHAr) 5.88, 5.84 (2 d, J = 6.0 Hz in each, minor p-cym-CHAr) 5.83–5.80 (m, minor and major p-cym-CHAr), 5.78 (d, J = 6.0 Hz, minor p-cym-CHAr), 5.67 (d, J = 5.9 Hz, major p-cym-CHAr), 5.52 (pt, J = 9.2, 9.2 Hz, major H-3′), 5.49 (pt, J = 9.5, 9.4 Hz, minor H-3′), 5.39 (pt, J = 10.2, 9.7 Hz, minor H-4′), 5.34 (pt, J = 10.0, 9.7 Hz, major H-4′), 4.44 (dd, J = 12.7, 4.7 Hz, minor H-6′a), 4.33 (dd, J = 12.7, 5.1 Hz, major H-6′a), 4.32–4.17 (m, minor and major H-5′, H-6′b), 2.57 (hept, J = 6.9 Hz, major i-Pr-CH), 2.53 (hept, J = 6.9 Hz, minor i-Pr-CH), 2.15 (s, minor C6H4–CH3), 2.13, 2.09 (singlets, COCH3), 2.07 (s, major C6H4–CH3), 2.06, 2.02, 1.96 (singlets, COCH3), 1.07, 1.04 (2 d, J = 6.9 Hz in each, 2 × major i-Pr-CH3), 1.02, 1.00 (2 d, J = 6.9 Hz in each, 2 × minor i-Pr-CH3); 13C-NMR (90 MHz, CDCl3) δ (ppm): 170.9, 170.0, 169.6, 169.4 (minor 4 × C=O), 170.8, 170.1, 169.8, 169.7 (major 4 × C=O), 149.6, 148.4, 147.5 (major Tria-C-4, Qu-C-2, Qu-C-8a), 149.2, 148.4, 148.9 (minor Tria-C-4, Qu-C-2, Qu-C-8a), 141.3 (minor Qu-C-4), 141.0 (major Qu-C-4), 133.1, 129.6, 129.2, 129.1, 129.1 (minor Qu-C-4a, Qu-C-5–Qu-C-8), 132.7, 129.5, 129.1, 129.1, 129.0 (major Qu-C-4a, Qu-C-5–Qu-C-8), 127.6 (major Tria-C-5), 127.5 (minor Tria-C-5), 119.1 (major Qu-C-3), 118.8 (minor Qu-C-3), 106.3, 102.1 (minor p-cym-CqAr), 105.4, 102.7 (major p-cym-CqAr), 88.1, 86.7, 86.5, 86.4, 85.7, 85.1, 84.6, 84.5, 84.3, 84.0 (minor and major p-cym-CHAr, minor and major C-1′), 75.6 (minor C-5′), 75.2 (major C-5′), 73.2 (minor C-3′), 73.0 (major C-3′), 70.2 (major C-2′), 69.8 (minor C-2′), 67.5 (major C-4′), 67.7 (minor C-4′), 61.6 (major C-6′), 61.5 (minor C-6′), 31.3 (minor i-Pr-CH), 31.1 (major i-Pr-CH), 22.7, 21.6 (major i-Pr-CH3), 22.4, 21.8 (minor i-Pr-CH3), 20.8, 20.7, 20.7, 20.6, 20.4 (2 × 4 × COCH3), 18.7 (minor C6H4–CH3), 18.5 (major C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C35H40ClN4O9Ru+ [M − PF6]+ 797.1528. Found: 797.1531.
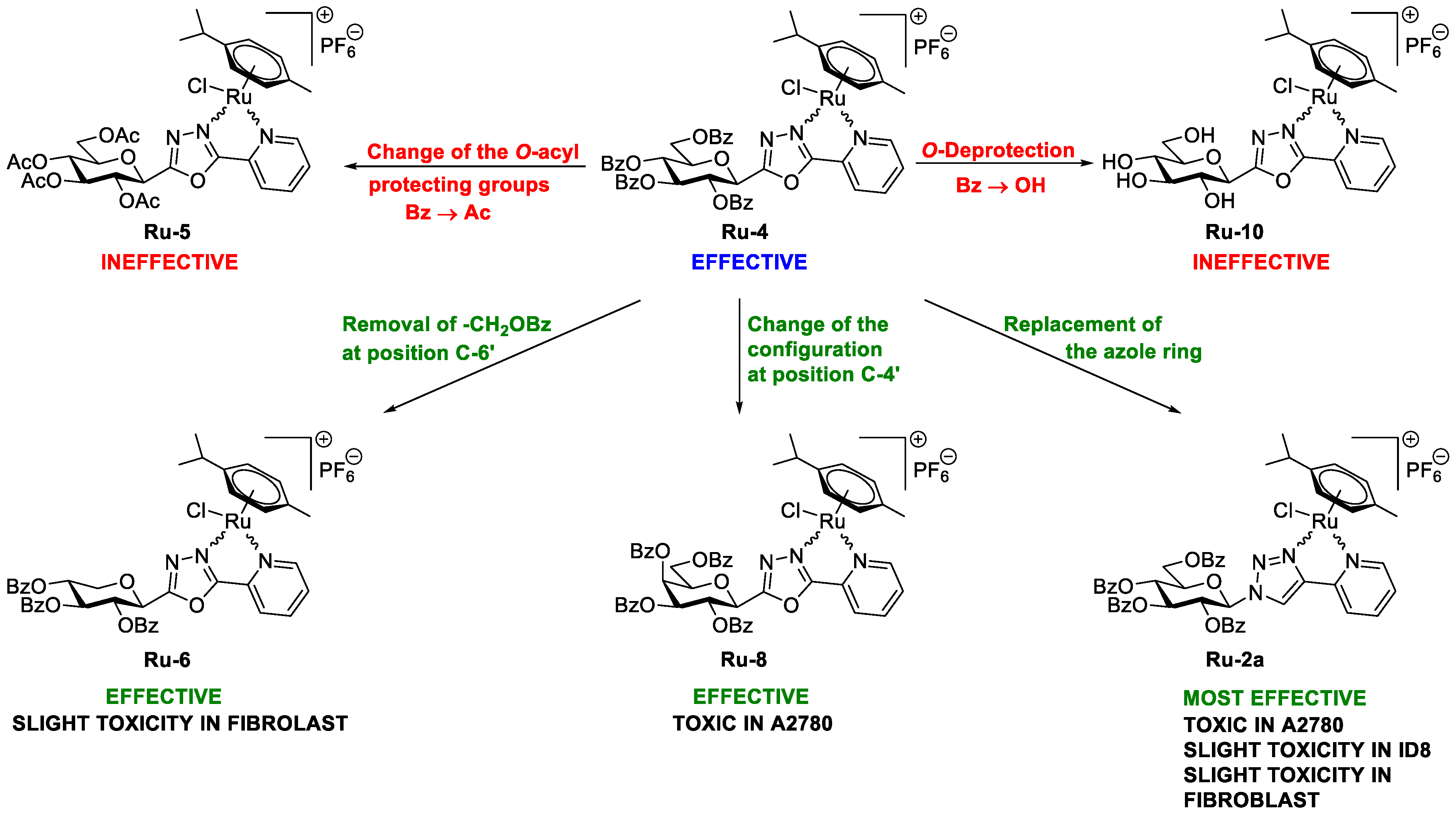
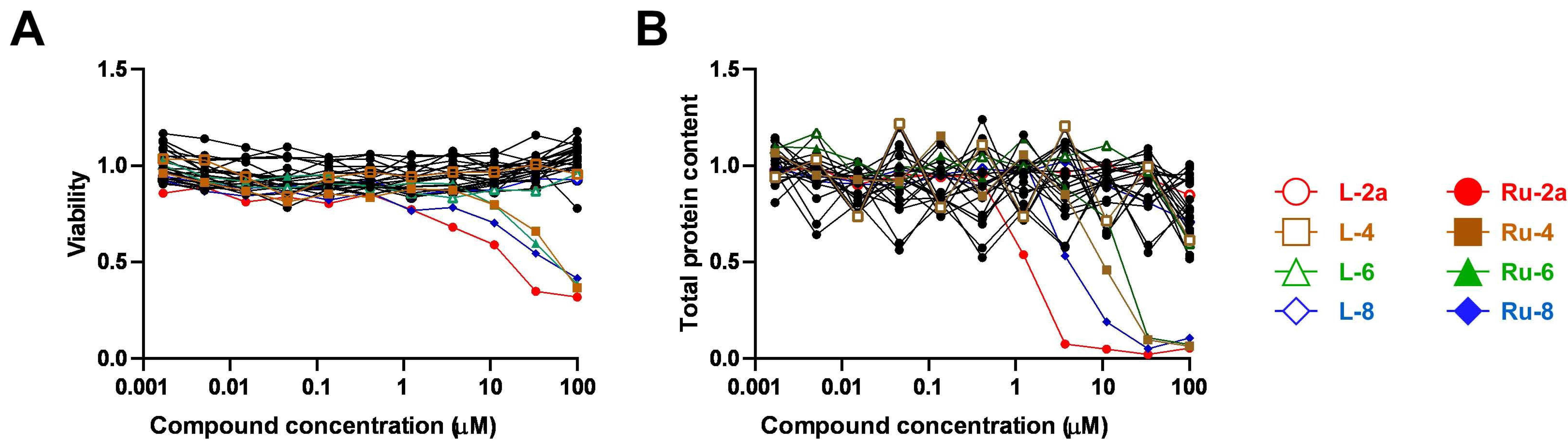
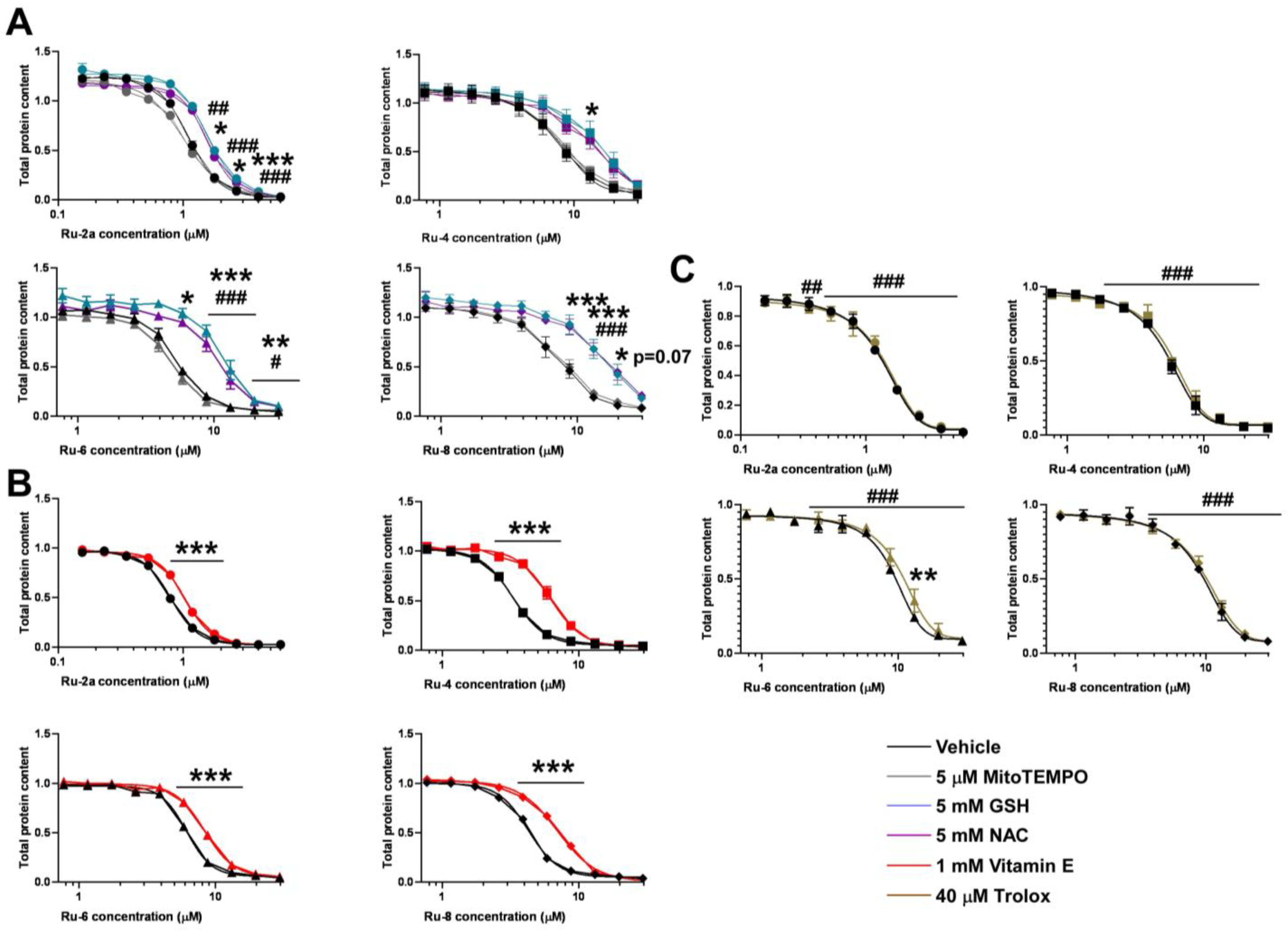
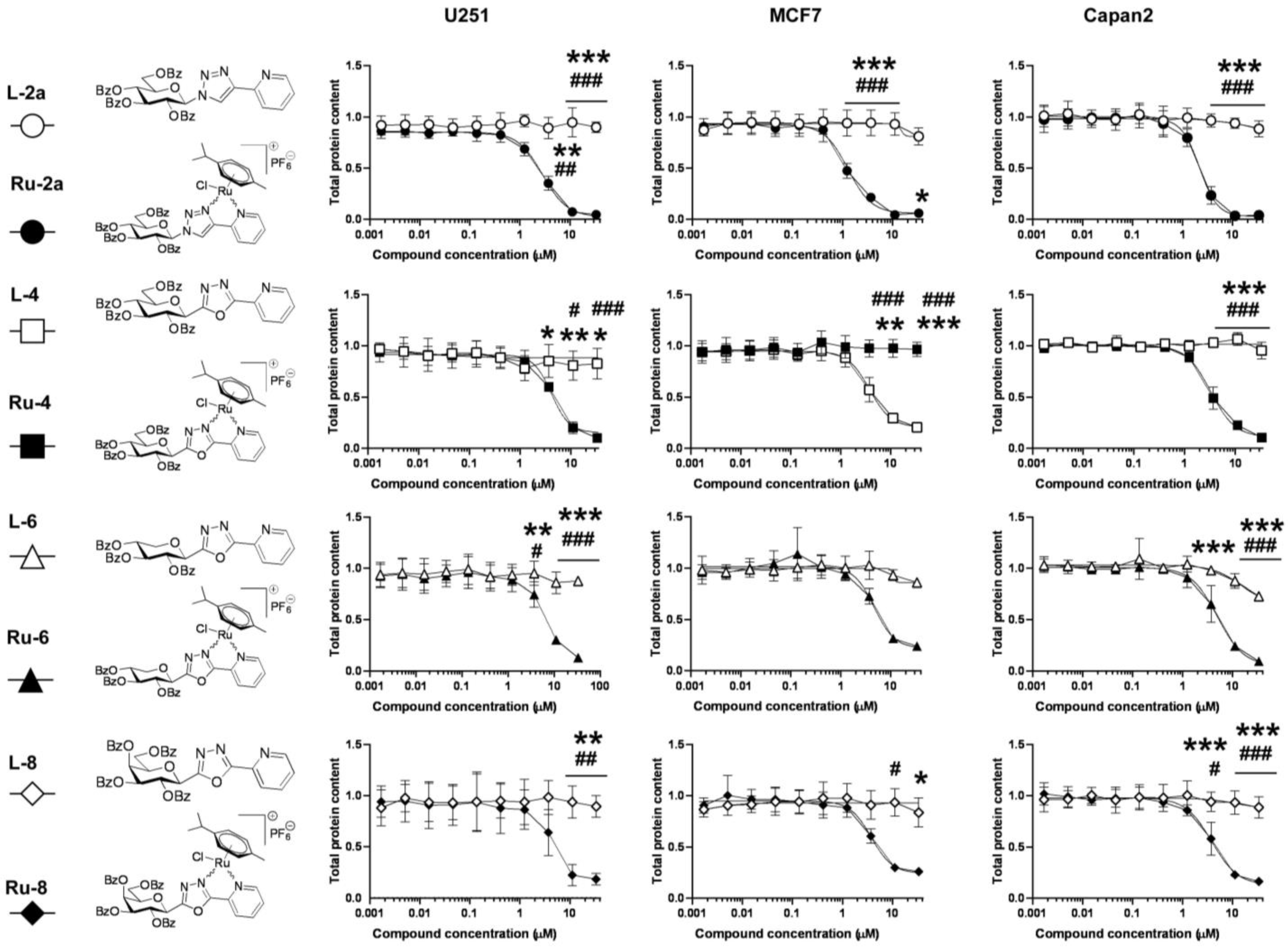
5.1.23. Complex Ru-2a
Prepared from complex Ru-dimer (10.0 mg, 0.0163 mmol), compound L-2a (24.9 mg, 0.0344 mmol, 2.1 eq.), and TlPF6 (11.4 mg, 0.0326 mmol, 2 eq.) according to general procedure IV. Purified by column chromatography (95:5 CHCl3–MeOH) to give 34.6 mg (93%) orange powder. Rf: 0.46 (95:5 CHCl3–MeOH). Diastereomeric ratio: 1:1. 1H-NMR (400 MHz, CDCl3) δ (ppm): 9.23, 9.20 (2 × 1H, 2 d, J = 5.6 Hz in each, 2 × Py-H-6), 9.02, 8.98 (2 × 1H, 2 s, 2 × Tria-H-5), 8.11–7.75, 7.62–7.24 (46H, m, 2 × 20 × Ar, 2 × Py-H-3–Py-H-5), 6.60, 6.10 (2 × 1H, 2 pt, J = 9.4, 9.3 Hz in each, 2 × H-2′), 6.50, 6.43 (2 × 1H, 2 d, J = 9.3 Hz in each, 2 × H-1′), 6.22, 6.15 (2 × 1H, 2 pt, J = 9.6, 9.4 Hz in each, 2 × H-3′), 5.97, 5.94 (2 × 1H, 2 pt, J = 9.7, 9.6 Hz in each, 2 × H-4′), 5.81, 5.65 (2), 5.57 (2), 5.53, 5.47, 5.44 (2 × 4H, 2 × 4 d, J = 6.1 Hz in each, 2 × 4 × p-cym-CHAr), 4.78–4.52 (2 × 3H, m, 2 × H-5′, 2 × H-6′a, 2 × H-6′b), 2.50, 2.36 (2 × 1H, 2 hept, J = 6.9 Hz in each, 2 × i-Pr-CH), 2.08, 2.00 (2 × 3H, 2 s, 2 × C6H4–CH3), 0.94, 0.91, 0.79, 0.74 (2 × 2 × 3H, 2 × 2 d, J = 6.9 Hz in each 2 × 2 × i-Pr-CH3); 13C-NMR (100 MHz, CDCl3) δ (ppm): 166.3, 166.2, 165.6, 165.5, 165.2 (2), 164.9, 164.8 (2 × 4 × C=O), 155.7, 155.5 (2 × Py-C-6), 147.3, 147.2, 147.0, 146.6 (2 × Tria-C-4, 2 × Py-C-2), 140.1, 140.0 (2 × Py-C-4), 134.4, 134.2, 133.9, 133.8, 133.7 (2), 133.6, 133.5, 133.3, 130.3–128.0 (Ar), 127.2, 126.9 (2 × Py-C-5), 126.2, 123.8 (2 × Tria-C-5), 122.7 (2 × Py-C-3), 105.8, 105.3, 103.9, 102.6 (2 × 2 × p-cym-CqAr), 87.2, 86.7 (2 × C-1′), 86.4, 85.8, 85.2, 85.0, 84.0, 83.9, 83.2, 82.5 (2 × 4 × p-cym-CHAr), 76.1, 75.6 (2 × C-5′), 73.6, 72.7 (2 × C-3′), 71.7, 70.4 (2 × C-2′), 68.7, 68.6 (2 × C-4′), 62.8, 62.7 (2 × C-6′), 31.0 (2) (2 × i-Pr-CH), 22.5, 22.3, 21.7, 21.4 (2 × 2 × i-Pr-CH3), 18.8, 18.6 (2 × C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C51H46ClN4O9Ru+ [M − PF6]+ 995.2002. Found: 995.1994.
5.1.24. Complex Ru-3a
Prepared from complex Ru-dimer (50.0 mg, 0.082 mmol), compound L-3a (50.4 mg, 0.163 mmol, 2 eq.), and TlPF6 (57.0 mg, 0.163 mmol) according to general procedure V. Yield: 107 mg (90%). An analytically pure sample was obtained by recrystallization from iPrOH to give 20 mg orange powder. Diastereomeric ratio: 1:1. 1H-NMR (400 MHz, CD3OD) δ (ppm): 9.42 (2H, d, J = 5.4 Hz, 2 × Py-H-6), 9.22, 9.21 (2 × 1H, 2 s, 2 × Tria-H-5), 8.19 (2H, pt, J = 7.8, 7.7 Hz, 2 × Py-H-4), 8.10 (2H, d, J = 7.8 Hz, 2 × Py-H-3), 7.69–7.66 (2H, m, 2 × Py-H-5), 6.12–6.05 (4H, m, 2 × p-cym-CHAr), 5.90–5.84 (4H, m, 2 × p-cym-CHAr), 5.88 (2H, d, 2 × H-1′), 3.95–3.89 (4H, m, 2 × H-2′, 2 × H-6′a), 3.77 (2H, dd, J = 12.1, 5.3 Hz, 2 × H-6′b), 3.69 (2H, ddd, J = 9.5, 5.3, 1.9 Hz, 2 × H-5′), 3.64 (2H, pt, J = 9.5, 8.9 Hz, 2 × H-3′), 3.56 (2H, pt, J = 9.3, 9.1 Hz, 2 × H-4′), 2.79–2.71 (2H, hept, J = 6.7 Hz, 2 × i-Pr-CH), 2.22 (6H, s, 2 × C6H4–CH3), 1.18–1.09 (12H, m, 2 × 2 × i-Pr-CH3); 13C-NMR (100 MHz, CD3OD) δ (ppm): 156.8 (2) (2 × Py-C-6), 149.6 (2), 148.2, 148.1 (2 × Py-C-2, 2 × Tria-C-4), 141.5 (2) (2 × Py-C-4), 127.6 (2) (2 × Py-C-5), 125.5, 125.2 (2 × Tria-C-5), 123.6 (2) (2 × Py-C-3), 106.8, 106.6, 104.1, 103.8 (2 × 2 × p-cym-CqAr), 91.3, 91.2 (2 × C-1′), 87.3, 87.2, 86.3, 86.1, 85.6, 85.5, 84.8, 84.6 (2 × 4 × p-cym-CHAr), 81.7, 81.6 (2 × C-5′), 78.2, 78.1 (2 × C-3′), 74.5, 74.3 (2 × C-2′), 70.8, 70.7 (2 × C-4′), 62.3, 62.2 (2 × C-6′), 32.3, 32.2 (2 × i-Pr-CH), 22.6 (2), 22.0, 21.9 (2 × 2 × i-Pr-CH3), 18.8, 18.7 (2 × C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C23H30ClN4O5Ru+ [M − PF6]+ 579.0946. Found: 579.0946.
5.1.25. Complex Ru-3b
Prepared from complex Ru-dimer (50.0 mg, 0.082 mmol), compound L-3b (58.8 mg, 0.164 mmol, 2 eq.), and TlPF6 (57 mg, 0.163 mmol) according to general procedure V. Yield: 121 mg (96%). An analytically pure sample was obtained by recrystallization from iPrOH to give 34 mg orange powder. Diastereomeric ratio: 1:1. 1H-NMR (400 MHz, CD3OD) δ (ppm): 9.42, 9.41 (2 × 1H, 2 s, 2 × Tria-H-5), 8.80 (2H, d, J = 8.8 Hz, 2 × Qu-H-8), 8.70, 8.68 (2 × 1H, 2 d, J = 8.5 Hz in each, 2 × Qu-H-4), 8.15–8.09, 7.88–7.84 (2 × 4H, m, Qu-H-3, Qu-H-5–Qu-H-7), 6.13–5.96 (2 × 4H, m, 2 × p-cym-CHAr), 5.95, 5.93 (2 × 1H, 2 d, J = 9.1 Hz in each, 2 × H-1′), 4.00, 3.99 (2 × 1H, 2 pt, J = 9.2, 9.1 Hz in each, 2 × H-2′), 3.97, 3.95 (2 × 1H, 2 dd, J = 12.1, 2.1 Hz in each, 2 × H-6′a), 3.80 (2H, dd, J = 12.1, 5.3 Hz, 2 × H-6′b), 3.74, 3.71 (2 × 1H, ddd, J = 9.3, 5.3, 2.1 Hz in each, 2 × H-5′), 3.68, 3.67 (2 × 1H, 2 pt, J = 9.0, 8.9 Hz in each, 2 × H-3′), 3.60, 3.59 (2 × 1H, 2 pt, J = 9.3, 9.2 Hz in each, 2 × H-4′), 2.48, 2.46 (2 × 1H, 2 hept, J = 6.9 Hz in each, 2 × i-Pr-CH), 2.20 (6H, s, 2 × C6H4–CH3), 1.00, 0.98, 0.92 (2) (2 × 2 × 3H, 4 d, J = 6.9 Hz in each, 2 × 2 × i-Pr-CH3); 13C NMR (90 MHz, CD3OD) δ (ppm): 151.0 (2), 149.7 (2), 149.1 (2), (2 × Tria-C-4, 2 × Qu-C-2, 2 × Qu-C-8a), 142.6 (2 × Qu-C-4), 134.0, 130.7, 130.6 (3), 130.5, 130.1 (2 × Qu-C-4a, 2 × Qu-C-5–Qu-C-8), 127.1, 126.9 (2 × Tria-C-5), 119.5, 119.4 (2 × Qu-C-3), 106.5, 106.3, 104.9, 104.7 (2 × 2 × p-cym-CqAr), 91.4, 91.2 (2 × C-1′), 88.6, 88.5, 86.5, 86.3, 86.2, 85.8, 84.6, 84.3 (2 × 4 × p-cym-CHAr), 81.8, 81.7 (2 × C-5′), 78.2, 78.1 (2 × C-3′), 74.4, 74.3 (2 × C-2′), 70.7 (2) (2 × C-4′), 62.3, 62.20 (2 × C-6′), 32.3, 32.2 (2 × i-Pr-CH), 22.7, 22.6, 21.6, 21.5 (2 × 2 × i-Pr-CH3), 18.8, 18.7 (2 × C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C27H32ClN4O5Ru+ [M − PF6]+: 629.1103. Found: 629.1103.
5.1.26. Complex Ru-4
Prepared from complex Ru-dimer (10.0 mg, 0.016 mmol), compound L-4 (24.9 mg, 0.034 mmol, 2.1 eq.), and TlPF6 (11.4 mg, 0.033 mmol) according to general procedure IV. After filtration and removal of the solvent, the residue was dissolved in CHCl3 (3 mL), and Et2O (6 mL) was added. The precipitated product was filtered off to give 28.4 mg (76%) yellow powder. The product could also be obtained by column chromatographic purification (9:1 CHCl3–MeOH), albeit in lower yield (52%). Rf: 0.38 (9:1 CHCl3–MeOH). Diastereomeric ratio: 2:1. 1H-NMR (400 MHz, CDCl3) δ (ppm): 9.50 (s, minor Py-H-6), 9.26 (s, major Py-H-6), 8.18–7.70, 7.60–7.28 (m, minor and major Ar, Py-H-3–Py-H-5), 6.19 (pt, J = 9.8, 9.7 Hz, minor H-3′), 6.16 (pt, J = 9.8, 9.5 Hz, major H-2′), 6.08 (pt, J = 9.5, 9.5 Hz, major H-3′), 5.88 (pt, J = 9.7, 9.5 Hz, major H-4′), 5.86 (pt, J = 9.7, 9.6 Hz, minor H-4′), 5.90–5.84 (m, 2 × minor p-cym-CHAr), 5.76–5.72 (m, major p-cym-CHAr, minor H-2′), 5.68–5.65 (m, 2 × minor p-cym-CHAr), 5.53–5.51 (m, major p-cym-CHAr), 5.46 (d, J = 9.8 Hz, major H-1′), 5.35 (d, J = 9.8 Hz, minor H-1′), 5.31–5.29 (m, 2 × major p-cym-CHAr), 4.74–4.68 (m, minor and major H-6′a), 4.57–4.52 (m, minor and major H-6′b), 4.50–4.42 (m, minor and major H-5′), 2.78 (hept, J = 6.8 Hz, major i-Pr-CH), 2.72 (hept, J = 6.8 Hz, minor i-Pr-CH), 2.02 (s, minor C6H4–CH3), 1.95 (s, major C6H4–CH3), 1.16, 1.15, 1.12, 1.08 (4 d, J = 6.8 Hz in each, minor and major 2 × i-Pr-CH3); 13C-NMR (90 MHz, CDCl3) δ (ppm): 166.2, 165.8, 165.7, 165.7, 165.3, 165.2, 165.2, 164.9, 164.8, 164.7 (minor and major C=O, OD-C-2, OD-C-5), 158.3 (minor Py-C-6), 156.6 (major Py-C-6), 140.3 (minor Py-C-4), 140.2 (major Py-C-2), 140.1 (major Py-C-4), 139.1 (minor Py-C-2), 134.6, 134.0, 133.9, 133.8, 133.7, 133.5, 133.4, 131.1, 130.6, 130.2–128.5, 127.8 (minor and major Ar, Py-C-5), 125.3 (minor Py-C-3), 125.2 (major Py-C-3), 107.0, 102.3 (minor p-cym-CqAr), 104.8, 102.1 (major p-cym-CqAr), 88.7, 84.6, 83.2 (2) (major p-cym-CHAr), 85.5, 85.2, 83.8, 83.6 (minor p-cym-CHAr), 77.8 (minor C-5′), 77.4 (major C-5′), 73.8, 71.5, 69.8 (major C-1′–C-3′), 72.8, 71.9, 71.4 (minor C-1′–C-3′), 68.8 (major C-4′), 68.7 (minor C-4′), 63.0 (minor C-6′), 62.6 (major C-6′), 31.3 (minor i-Pr-CH), 31.1 (major i-Pr-CH), 23.1, 21.3 (major i-Pr-CH3), 22.2, 22.1 (minor i-Pr-CH3), 18.8 (minor C6H4–CH3), 18.1 (major C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C51H45ClN3O10Ru+ [M − PF6]+ 996.1843; C53H52N3O12Ru+ [M − PF6 − Cl + OMe + MeOH]+ 1024.2604. Found: [M − PF6]+ 996.1861; [M − PF6 − Cl + OMe + MeOH]+ 1024.2598.
5.1.27. Complex Ru-5
Prepared from complex Ru-dimer (10.0 mg, 0.0163 mmol), compound L-5 (17.9 mg, 0.0375 mmol, 2.3 eq.), and TlPF6 (11.4 mg, 0.0326 mmol) according to general procedure IV. Purified by column chromatography (9:1 CHCl3–MeOH) to give 21.7 mg (74%) yellow powder. Rf: 0.54 (9:1 CHCl3–MeOH). Diastereomeric ratio: 4:3. 1H-NMR (400 MHz, CDCl3) δ (ppm): 9.45 (d, J = 3.9 Hz, minor Py-H-6), 9.30 (d, J = 4.4 Hz, major Py-H-6), 8.23–8.08 (m, minor and major Py-H-3, Py-H-4), 7.88 (m, minor Py-H-5), 7.80 (m, major Py-H-5), 6.01–5.96, 5.82–5.69 (m, minor and major p-cym-CHAr), 5.75 (pt, J = 9.7, 9.6 Hz, major H-2′), 5.44, (pt, J = 9.3, 9.2 Hz, minor H-2′), 5.40 (pt, J = 9.4, 9.0 Hz, minor and major H-3′), 5.24 (pt, J = 9.8, 9.2 Hz, minor H-4′), 5.23 (pt, J = 9.8, 9.8 Hz, major H-4′), 5.03 (d, J = 10.0 Hz, major H-1′), 5.00 (d, J = 8.7 Hz, minor H-1′), 4.30 (dd, J = 12.8, 4.8 Hz, minor and major H-6′a), 4.17 (dd, J = 12.8, 2.4 Hz, minor and major H-6′b), 3.99 (ddd, J = 10.2, 4.8, 2.4 Hz, minor and major H-5′), 3.03 (hept, J = 6.8 Hz, major i-Pr-CH), 2.91 (hept, J = 6.8 Hz, minor i-Pr-CH), 2.23, 2.17, 2.10–2.02 (singlets, minor and major C6H4–CH3, COCH3), 1.36 (d, J = 6.8 Hz, major 2 × i-Pr-CH3), 1.30, 1.25 (2 d, J = 6.8 Hz in each, minor 2 × i-Pr-CH3); 13C-NMR (90 MHz, CDCl3) δ (ppm): 170.7, 170.6, 170.1, 170.0, 169.8, 169.6, 169.4 (2 × 4 × C=O), 164.9, 164.6 (major OD-C-2, OD-C-5), 164.9, 164.7 (minor OD-C-2, OD-C-5), 157.6 (minor Py-C-6), 156.4 (major Py-C-6), 140.5 (minor Py-C-4), 140.3 (major Py-C-4), 140.3, 139.5 (minor and major Py-C-2), 130.8 (minor Py-C-5), 130.0 (major Py-C-5), 125.5 (minor Py-C-3), 125.4 (major Py-C-3), 106.4 (minor p-cym-CqAr), 105.0 (major p-cym-CqAr), 102.7 (minor p-cym-CqAr), 102.4 (major p-cym-CqAr), 88.2, 86.0, 85.6, 83.5, 83.2, 83.1 (minor and major p-cym-CHAr), 77.2 (minor C-5′), 77.0 (major C-5′), 73.7, 70.9 (major C-1′ and C-3′), 73.0, 71.2, 70.0 (minor C-1′–C-3′), 68.8 (major C-2′), 67.7 (minor C-4′), 67.6 (major C-4′), 61.9 (minor C-6′), 61.7 (major C-6′), 31.3 (minor i-Pr-CH), 31.2 (major i-Pr-CH), 22.9, 21.7 (major i-Pr-CH3), 22.4, 22.0 (minor i-Pr-CH3), 20.8, 20.7, 20.6, 20.6, 20.5 (2 × 4 × COCH3), 18.8 (minor C6H4–CH3), 18.3 (major C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C31H37ClN3O10Ru+ [M − PF6]+ 748.1211. Found: 748.1212.
5.1.28. Complex Ru-6
Prepared from complex Ru-dimer (50 mg, 0.082 mmol), compound L-6 (97 mg, 0.164 mmol, 2 eq.), and TlPF6 (57 mg, 0.163 mmol) according to general procedure IV. Purified by column chromatography (9:1 CHCl3–MeOH) to give 140 mg (85%) yellow powder. Rf: 0.67 (9:1 CHCl3–MeOH). Diastereomeric ratio: 3:2. 1H-NMR (400 MHz, CDCl3) δ (ppm): 9.46 (d, J = 4.1 Hz, minor Py-H-6), 9.26 (d, J = 4.2 Hz, major Py-H-6), 8.18–7.73, 7.60–7.32 (m, minor and major Ar, Py-H-3–Py-H-5), 6.11 (pt, J = 9.1, 9.1, major H-2′), 6.06–6.02 (m, major and minor H-3′), 5.86 (d, J = 5.4 Hz, minor p-cym-CHAr), 5.80–5.76 (m, minor and major p-cym-CHAr), 5.74 (pt, J = 9.3, 9.1 Hz, minor H-2′), 5.65, 5.63 (2 d, J = 6.1 Hz in each, minor p-cym-CHAr), 5.61–5.53 (m, major and minor H-4′), 5.53 (d, J = 5.4 Hz, major p-cym-CHAr), 5.37–5.35 (m, major p-cym-CHAr), 5.28 (d, J = 9.1 Hz, major H-1′), 5.24 (d, J = 9.1 Hz, minor H-1′), 4.64 (dd, J = 11.8, 5.1 Hz, minor H-5′eq), 4.60 (dd, J = 11.8, 5.4 Hz, major H-5′eq), 3.87 (pt, J = 10.9, 10.1, minor and major H-5′ax), 2.80 (hept, J = 6.9 Hz, major i-Pr-CH), 2.75 (hept, J = 6.9 Hz, minor i-Pr-CH), 2.04 (s, minor C6H4–CH3), 1.99 (s, major C6H4–CH3), 1.19–1.11 (m, minor and major 2 × i-Pr-CH3); 13C-NMR (90 MHz, CDCl3) δ (ppm): 165.7, 165.6 (2), 165.5, 165.4, 165.3, 165.2, 164.9, 164.7, 164.6 (minor and major C=O, OD-C-2, OD-C-5), 157.9 (minor Py-C-6), 157.0 (major Py-C-6), 140.4 (minor Py-C-4), 140.3 (major Py-C-4), 140.0 (major Py-C-2), 139.3 (minor Py-C-2), 134.4, 133.9, 133.8 (2), 133.7, 133.5, 130.8–128.0 (minor and major Ar, Py-C-5), 125.2 (minor Py-C-3), 125.1 (major Py-C-3), 106.3, 104.9, 102.4, 102.1 (minor and major p-cym-CqAr), 88.4, 86.0, 85.1, 84.3, 83.5, 83.4, 83.3, 83.1 (minor and major p-cym-CHAr), 73.1, 72.1, 71.9, 71.8, 70.5, 69.7, 69.5, 69.0 (minor and major C-1′–C-4′), 67.8, 67.4 (minor and major C-5′), 31.2, 31.0 (minor and major i-Pr-CH), 23.1, 22.4, 21.8, 21.1 (minor and major i-Pr-CH3), 18.6, 18.1 (minor and major C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C43H39ClN3O8Ru+ [M − PF6]+ 862.1473; C45H46N3O10Ru+ [M − PF6 − Cl + OMe + MeOH]+ 890.2234. Found: [M − PF6]+ 862.1470; [M − PF6 − Cl + OMe + MeOH]+ 890.2231.
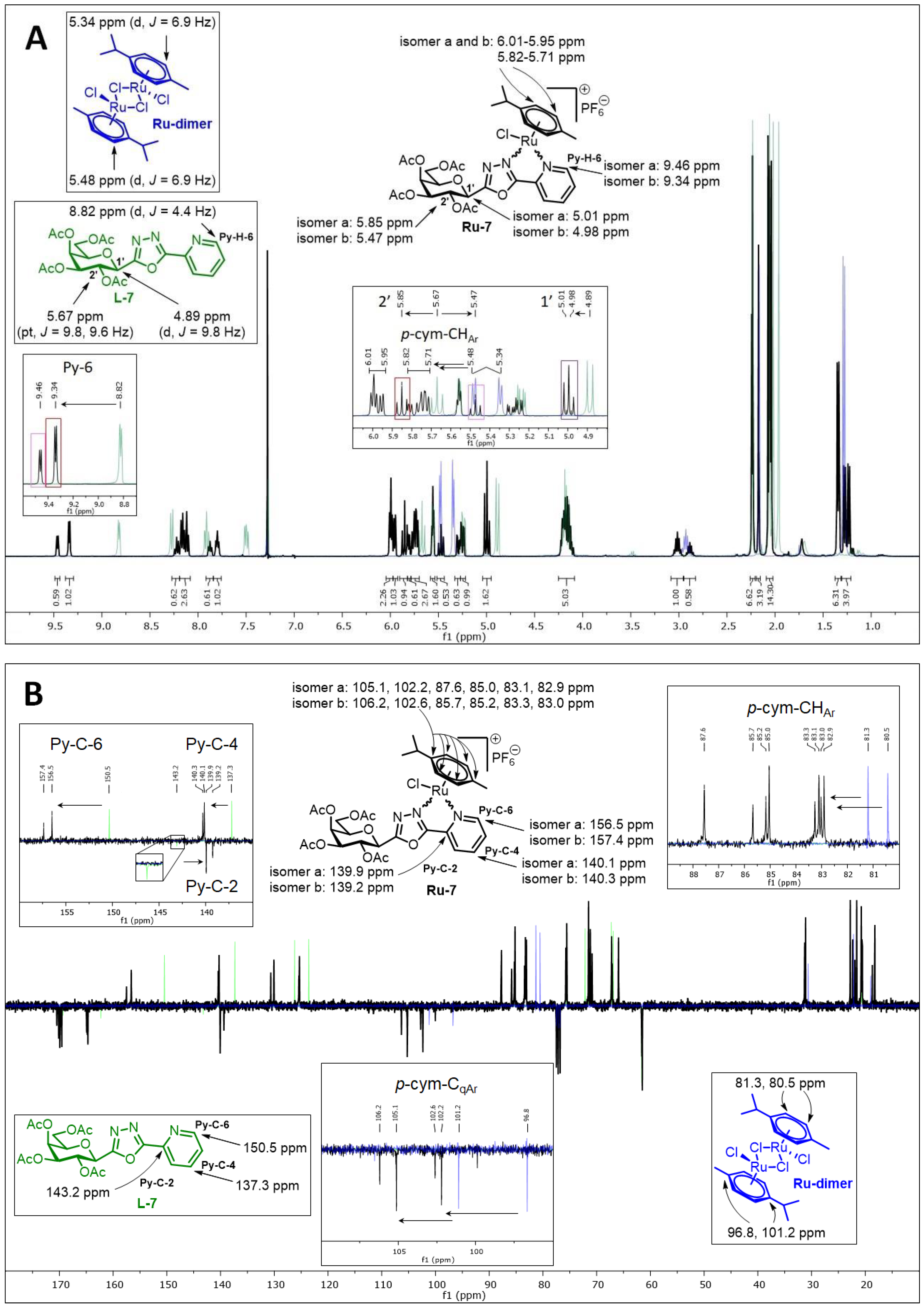
5.1.29. Complex Ru-7
Prepared from complex Ru-dimer (50 mg, 0.082 mmol), compound L-7 (78 mg, 0.163 mmol, 2 eq.), and TlPF6 (57 mg, 0.163 mmol) according to general procedure IV. Purified by column chromatography (9:1 CHCl3–MeOH) to give 108 mg (74%) yellow powder. Rf: 0.54 (9:1 CHCl3–MeOH). Diastereomeric ratio: 2:1. 1H-NMR (400 MHz, CDCl3) δ (ppm): 9.46 (d, J = 4.4 Hz, minor Py-H-6), 9.34 (d, J = 4.4 Hz, major Py-H-6), 8.24–8.10 (m, minor and major Py-H-4, Py-H-3), 7.87 (m, minor Py-H-5), 7.80 (m, major Py-H-5), 6.01–5.95 (m, minor and major p-cym-CHAr), 5.85 (pt, J = 10.2, 10.0 Hz major H-2′), 5.82–5.71 (m, minor and major p-cym-CHAr), 5.57–5.55 (m, minor and major H-4′), 5.47 (pt, J = 10.0, 10.0 Hz minor H-2′), 5.29 (dd, J = 10.0, 3.2 Hz, minor H-3′), 5.25 (dd, J = 10.0, 3.2 Hz, major H-3′), 5.01 (d, J = 10.2 Hz, major H-1′), 4.98 (d, J = 10.0 Hz, minor H-1′), 4.23–4.10 (m, minor and major H-5′, H-6′a, H-6′b), 3.01 (hept, J = 6.8 Hz, major i-Pr-CH), 2.89 (hept, J = 6.8 Hz, minor i-Pr-CH), 2.24–2.23, 2.17, 2.07–2.04 (singlets, minor and major C6H4–CH3, COCH3), 1.34 (d, J = 6.8 Hz, 2 × major i-Pr-CH3), 1.28, 1.24 (2 d, J = 6.8 Hz in each, 2 × minor i-Pr-CH3); 13C-NMR (90 MHz, CDCl3) δ (ppm): 170.3, 170.2, 170.1, 170.0, 169.8, 169.7 (2 × 4 × C=O), 164.8, 164.7, 164.5 (2) (minor and major OD-C-2, OD-C-5), 157.4 (minor Py-C-6), 156.5 (major Py-C-6), 140.3 (minor Py-C-4), 140.1 (major Py-C-4), 139.9 (major Py-C-2), 139.2 (minor Py-C-2), 130.5 (minor Py-C-5), 129.9 (major Py-C-5), 125.3 (minor Py-C-3), 125.2 (major Py-C-3), 106.2, 102.6 (minor p-cym-CqAr), 105.1, 102.2 (major p-cym-CqAr), 87.6, 85.0, 83.1, 82.9 (major p-cym-CHAr), 85.7, 85.2, 83.3, 83.0 (minor p-cym-CHAr), 75.6 (minor C-5′), 75.4 (major C-5′), 71.4, 71.0, 67.0 (major C-1′, C-3′, C-4′), 71.3, 70.7, 67.1, 66.9 (minor C-1′–C-4′), 65.8 (major C-2′), 61.4 (minor C-6′), 61.3 (major C-6′), 31.1 (minor i-Pr-CH), 31.0 (major i-Pr-CH), 22.6, 21.4 (major i-Pr-CH3), 22.2, 21.8 (minor i-Pr-CH3), 20.6, 20.6, 20.5, 20.5, 20.4, 20.4 (2 × 4 × COCH3), 18.5 (minor C6H4–CH3), 18.1 (major C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C33H44N3O12Ru+ [M − PF6 − Cl + OMe + MeOH]+ 776.1973. Found: 776.1973.
5.1.30. Complex Ru-8
Prepared from complex Ru-dimer (10.0 mg, 0.0163 mmol), compound L-8 (24.9 mg, 0.0343 mmol, 2.1 eq.), and TlPF6 (11.4 mg, 0.0326 mmol) according to general procedure IV. Purified by column chromatography (95:5 CHCl3–MeOH) to give 30.3 mg (81%) yellow powder. Rf: 0.49 (95:5 CHCl3–MeOH). Diastereomeric ratio: 5:4. 1H-NMR (400 MHz, CDCl3) δ (ppm): 9.49 (d, J = 4.7 Hz, minor Py-H-6), 9.26 (d, J = 5.1 Hz, major Py-H-6), 8.21–7.25 (m, minor and major Ar, Py-H-3–Py-H-5), 6.43 (pt, J = 10.1, 10.0 Hz, major H-2′), 6.20 (d, J = 3.3 Hz, major H-4′), 6.16 (d, J = 2.6 Hz, minor H-4′), 6.02–5.94 (m, minor H-2′ and H-3′), 5.84–5.75 (m, minor and major p-cym-CHAr, major H-3′), 5.67, 5.64 (2 d, J = 6.1 Hz in each, minor p-cym-CHAr) 5.52 (d, J = 6.1 Hz, major p-cym-CHAr), 5.47 (d, J = 10.1 Hz, major H-1′), 5.37 (d, J = 8.7 Hz, minor H-1′), 5.32, 5.27 (2 d, J = 6.1 Hz in each, major p-cym-CHAr), 4.72–4.62 (m, minor and major H-5′, H-6′a), 4.53 (dd, J = 10.3, 4.1 Hz, minor H-6′b), 4.44 (dd, J = 10.3, 4.0 Hz, major H-6′b), 2.80 (hept, J = 6.9 Hz, major i-Pr-CH), 2.73 (hept, J = 6.9 Hz, minor i-Pr-CH), 2.03 (s, minor C6H4–CH3), 1.94 (s, major C6H4–CH3), 1.17, 1.15 (2 d, J = 6.9 Hz in each, 2 × major i-Pr-CH3), 1.14, 1.09 (2 d, J = 6.9 Hz in each, 2 × minor i-Pr-CH3); 13C-NMR (100 MHz, CDCl3) δ (ppm): 166.2, 166.1, 165.9, 165.7, 165.5 (2), 165.4, 165.3, 164.9, 164.8 (2), 164.7 (2 × 4 × C=O, 2 × OD-C-2, 2 × OD-C-5), 157.9 (minor Py-C-6), 156.3 (major Py-C-6), 140.4 (major Py-C-2), 140.3 (minor Py-C-4), 140.2 (major Py-C-4), 139.3 (minor Py-C-2), 134.5, 134.2, 133.9, 133.8 (2), 133.6, 133.5 (2), 130.9–128.1 (minor and major Ar, Py-C-5), 125.3 (minor Py-C-3), 125.2 (major Py-C-3), 106.8, 104.7, 102.3, 102.0 (minor and major p-cym-CqAr), 88.8, 85.4 (2), 84.5, 83.6, 83.5, 83.0 (2) (minor and major p-cym-CHAr), 76.7 (minor C-5′), 76.3 (major C-5′), 72.5 (major C-3′), 72.2 (minor C-1′), 71.7 (major C-1′), 71.3, 68.7 (minor C-2′, C-3′), 68.3 (major C-4′), 68.2 (minor C-4′), 66.8 (major C-2′), 62.4 (minor C-6′), 62.1 (major C-6′), 31.3 (minor i-Pr-CH), 31.1 (major i-Pr-CH), 23.2, 21.3 (major i-Pr-CH3), 22.2, 22.0 (minor i-Pr-CH3), 18.7 (minor C6H4–CH3), 18.0 (major C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C51H45ClN3O10Ru+ [M − PF6]+ 996.1843. Found: 996.1842.
5.1.31. Complex Ru-9
Prepared from complex Ru-dimer (50 mg, 0.082 mmol), compound L-9 (78 mg, 0.179 mmol, 2.2 eq.), and TlPF6 (57 mg, 0.163 mmol) according to general procedure IV. Purified by column chromatography (9:1 CHCl3–MeOH) to give 130 mg (94%) yellow powder. Rf: 0.54 (9:1 CHCl3–MeOH). Diastereomeric ratio: 1:1. 1H-NMR (400 MHz, CDCl3) δ (ppm): 9.33 (2H, d, J = 4.2 Hz, 2 × Py-H-6), 8.15 (2H, t, J = 7.4 Hz, 2 × Py-H-4), 8.07 (2H, d, J = 7.4 Hz, 2 × Py-H-3), 7.79 (2H, m, 2 × Py-H-5), 6.52, 6.46 (2 × 1H, 2 d, J = 2.3 Hz in each, 2 × H-1′), 6.01–5.95 (4H, m, 2 × p-cym-CHAr), 5.84–5.71 (6H, m, 2 × p-cym-CHAr, 2 × H-2′), 5.34, 5.30 (2 × 1H, 2 ddd, J = 8.9, 4.1, 2.9 Hz in each, 2 × H-3′), 4.36, 4.35 (2 × 1H, 2 dd, J = 12.5, 2.9 Hz in each, 2 × H-4′a), 4.21, 4.20 (2 × 1H, 2 dd, J = 12.5, 4.1 in each, 2 × H-4′b), 2.98, 2.97 (2 × 1H, 2 hept, J = 6.9 Hz in each, 2 × i-Pr-CH), 2.29, 2.24, 2.21, 2.18, 2.13, 2.12, 2.11, 2.10, 2.08, 2.08 (30H, singlets, 2 × i-Pr-CH3, 2 × 4 × COCH3), 1.33–1.29 (12H, m, 2 × 2 × i-Pr-CH3); 13C-NMR (90 MHz, CDCl3) δ (ppm): 170.7, 170.6, 169.9, 169.8, 169.6, 169.5, 169.4, 169.1 (2 × 4 × C=O), 165.2, 165.1, 164.5 (2) (2 × OD-C-2, 2 × OD-C-5), 156.9, 156.8 (2 × Py-C-6), 140.4 (2) (2 × Py-C-4), 139.8, 139.7 (2 × Py-C-2), 130.3 (2) (2 × Py-C-5), 125.4, 125.3 (2 × Py-C-3), 105.9, 105.7, 102.4, 102.3 (2 × 2 × p-cym-CqAr), 87.2, 86.6, 85.4, 85.1, 83.5 (2), 83.4, 83.3 (2 × 4 × p-cym-CHAr), 68.9, 68.7 (2 × C-2′), 68.0, 67.8 (2 × C-3′), 64.3, 63.7 (2 × C-1′), 61.5 (2) (2 × C-4′), 31.3 (2) (2 × i-Pr-CH), 22.8, 22.6, 21.9, 21.8 (2 × 2 × i-Pr-CH3), 21.0, 20.9 (2), 20.8, 20.7, 20.6, 20.5 (2) (2 × 4 × COCH3), 18.5, 18.4 (2 × C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C29H35ClN3O9Ru+ [M − PF6]+ 706.1105; C31H42N3O11Ru+ [M − PF6 − Cl + OMe + MeOH]+ 734.1866. Found: [M − PF6]+ 706.1106; [M − PF6 − Cl + OMe + MeOH]+ 734.1868.
5.1.32. Complex Ru-10
Prepared from complex Ru-dimer (39 mg, 0.064 mmol), compound L-10 (39 mg, 0.126 mmol, 2 eq.), and TlPF6 (44 mg, 0.126 mmol) according to general procedure V. The crude product was triturated with Et2O to give 80 mg (87%) orange powder. Diastereomeric ratio: 1:1. 1H-NMR (400 MHz, CD3OD) δ (ppm): 9.54 (2H, d, J = 5.5 Hz, 2 × Py-H-6), 8.40–8.36 (4H, m, 2 × Py-H-3, 2 × Py-H-4), 7.98–7.93 (2H, m, 2 × Py-H-5), 6.19–6.13 (2 × 2H, m, 2 × p-cym-CHAr), 5.94–5.92 (2 × 2H, m, 2 × p-cym-CHAr), 4.85, 4.83 (2 × 1H, 2 d, J = 9.8 Hz in each, 2 × H-1′), 3.92, 3.91 (2 × 1H, 2 dd, J = 12.1, 6.1 Hz in each, 2 × H-6′a), 3.83, 3.81 (2 × 1H, 2 pt, J = 9.5, 9.3 Hz in each, 2 × H-2′), 3.71, 3.70 (2 × 1H, 2 dd, J = 12.1, <1 Hz in each, 2 × H-6′b), 3.59–3.54 (4H, m, 2 × H-3′, 2 × H-5′), 3.45, 3.45 (2 × 1H, 2 pt, J = 9.8, 9.5 Hz in each, 2 × H-4′), 2.91, 2.90 (2 × 1H, 2 hept, J = 6.9 Hz in each, 2 × i-Pr-CH), 2.23 (6H, s, 2 × C6H4–CH3), 1.28–1.23 (12H, m, 2 × 2 × i-Pr-CH3); 13C-NMR (90 MHz, CD3OD) δ (ppm): 168.5 (2), 166.0 (2) (2 × OD-C-2, 2 × OD-C-5), 158.3, 158.3 (2 × Py-C-6), 142.0 (2) (2 × Py-C-4), 141.6, 141.5 (2 × Py-C-2), 131.1 (2) (2 × Py-C-5), 126.6, 126.5 (2 × Py-C-3), 107.3, 107.2, 103.5, 103.4 (2 × 2 × p-cym-CqAr), 87.4, 87.2, 85.9, 85.8, 85.1, 85.0, 84.9, 84.8 (2 × 4 × p-cym-CHAr), 83.2, 83.1 (2 × C-5′), 78.8, 78.7 (2 × C-3′), 74.6 (2) (2 × C-1′), 73.3, 73.2 (2 × C-2′), 71.1 (2) (2 × C-4′), 62.6 (2) (2 × C-6′), 32.4 (2) (2 × i-Pr-CH), 22.8, 22.7, 22.1, 22.0 (2 × 2 × i-Pr-CH3), 18.8 (2) (2 × C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C23H29ClN3O6Ru+ [M − PF6]+ 580.0786; C25H36N3O8Ru+ [M − PF6 − Cl + OMe + MeOH]+ 608.1548. Found: [M − PF6]+ 580.0787; [M − PF6 − Cl + OMe + MeOH]+ 608.1545.
5.1.33. Complex Ru-11
Prepared from complex Ru-dimer (10.0 mg, 0.016 mmol), compound L-11 (9.1 mg, 0.033 mmol, 2 eq.), and TlPF6 (11.4 mg, 0.033 mmol) according to general procedure V. Purified by recrystallization from an iPrOH–Et2O solvent mixture (2 mL and 5 mL, respectively) to give 9.5 mg (42%) yellow powder. Diastereomeric ratio: 1:1. 1H-NMR (400 MHz, CD3OD) δ (ppm): 9.53 (2H, d, J = 5.4 Hz, 2 × Py-H-6), 8.38–8.34 (4H, m, 2 × Py-H-3, 2 × Py-H-4), 7.95–7.92 (2H, m, 2 × Py-H-5), 6.18–6.12 (2 × 2H, m, 2 × p-cym-CHAr), 5.94–5.90 (2 × 2H, m, 2 × p-cym-CHAr), 4.77, 4.76 (2 × 1H, 2 d, J = 9.6 Hz in each, 2 × H-1′), 4.07, 4.06 (2 × 1H, 2 dd, J = 11.1, 5.3 Hz in each, 2 × H-5′eq), 3.81, 3.80 (2 × 1H, 2 pt, J = 9.6, 9.0 Hz in each, 2 × H-2′), 3.68, 3.65 (2 × 1H, 2 ddd, J = 10.5, 8.9, 5.3 Hz in each, 2 × H-4′), 3.51 (2H, pt, J = 9.0, 8.9 Hz, 2 × H-3′), 3.47, 3.46 (2 × 1H, 2 pt, J = 11.1, 10.5 Hz in each, 2 × H-5′ax), 2.91 (2H, 2 hept, J = 6.8 Hz, 2 × i-Pr-CH), 2.22 (6H, s, 2 × C6H4–CH3), 1.28–1.23 (12H, m, 2 × 2 × i-Pr-CH3); 13C-NMR (100 MHz, CD3OD) δ (ppm): 168.8, 168.6, 166.1, 166.0 (2 × OD-C-2, 2 × OD-C-5), 158.2 (2) (2 × Py-C-6), 142.0 (2) (2 × Py-C-4), 141.6, 141.5 (2 × Py-C-2), 131.1 (2) (2 × Py-C-5), 126.6, 126.5 (2 × Py-C-3), 107.4 (2), 103.5, 103.4 (2 × 2 × p-cym-CqAr), 87.4, 87.3, 86.0, 85.9, 85.0 (2), 84.9, 84.8 (2 × 4 × p-cym-CHAr), 79.0, 78.9 (2 × C-3′), 75.4, 75.3 (2 × C-1′), 73.5, 73.3, (2 × C-2′), 71.8, 71.7 (2 × C-5′), 70.8 (2) (2 × C-4′), 32.4 (2) (2 × i-Pr-CH), 22.7 (2), 22.0 (2) (2 × 2 × i-Pr-CH3), 18.7 (2) (2 × C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C22H27ClN3O5Ru+ [M − PF6]+ 550.0680; C24H34N3O7Ru+ [M − PF6 − Cl + OMe + MeOH]+ 578.1442. Found: [M − PF6]+ 550.0672; [M − PF6 − Cl + OMe + MeOH]+ 578.1432.
5.1.34. Complex Ru-12
Prepared from complex Ru-dimer (10.0 mg, 0.016 mmol), compound L-12 (10.1 mg, 0.033 mmol, 2 eq.), and TlPF6 (11.4 mg, 0.033 mmol) according to general procedure V. Purified by recrystallization from an iPrOH–Et2O solvent mixture (2 mL and 5 mL, respectively) to give 11.9 mg (50%) yellow powder. Diastereomeric ratio: 1:1. 1H-NMR (360 MHz, CD3OD) δ (ppm): 9.55 (2H, d, J = 5.6 Hz, 2 × Py-H-6), 8.40–8.35 (4H, m, 2 × Py-H-3, 2 × Py-H-4), 7.96–7.93 (2H, m, 2 × Py-H-5), 6.19–6.13 (2 × 2H, m, 2 × p-cym-CHAr), 5.94–5.92 (2 × 2H, m, 2 × p-cym-CHAr), 4.78, 4.76 (2 × 1H, 2 d, J = 9.8 Hz in each, 2 × H-1′), 4.18, 4.15 (2 × 1H, 2 pt, J = 9.8, 9.4 in each, 2 × H-2′), 4.01 (2H, d, J = 3.2 Hz, 2 × H-4′), 3.84–3.73 (6H, m, 2 × H-5′, 2 × H-6′a, 2 × H-6′b), 3.69, 3.68 (2 × 1H, 2 dd, J = 9.4, 3.2 in each, 2 × H-3′), 2.91 (2H, 2 hept, J = 6.9 Hz, 2 × i-Pr-CH), 2.23 (6H, s, 2 × C6H4–CH3), 1.27–1.23 (12H, m, 2 × 2 × i-Pr-CH3); 13C-NMR (90 MHz, CD3OD) δ (ppm): 168.7, 168.6, 166.0, 165.9 (2 × OD-C-2, 2 × OD-C-5), 158.1 (2) (2 × Py-C-6), 141.9 (2) (2 × Py-C-4), 141.6, 141.5 (2 × Py-C-2), 131.1 (2) (2 × Py-C-5), 126.5, 126.4 (2 × Py-C-3), 107.4, 107.3, 103.5, 103.4 (2 × 2 × p-cym-CqAr), 87.2, 87.1, 86.0, 85.8, 85.0 (2), 84.8, 84.7 (2 × 4 × p-cym-CHAr), 82.0, 81.9 (2 × C-5′), 75.6, 75.5, 75.1, 75.0 (2 × C-1′, 2 × C-3′), 70.6, 70.5, 70.2, 70.0 (2 × C-2′, 2 × C-4′), 62.7 (2) (2 × C-6′), 32.4 (2) (2 × i-Pr-CH), 22.7, 22.6, 22.1, 22.0 (2 × 2 × i-Pr-CH3), 18.7 (2) (2 × C6H4–CH3). ESI-HRMS positive mode (m/z): calculated for C23H29ClN3O6Ru+ [M − PF6]+ 580.0786; C25H36N3O8Ru+ [M − PF6 − Cl + OMe + MeOH]+ 608.1548. Found: [M − PF6]+ 580.0792; [M − PF6 − Cl + OMe + MeOH]+ 608.1553.
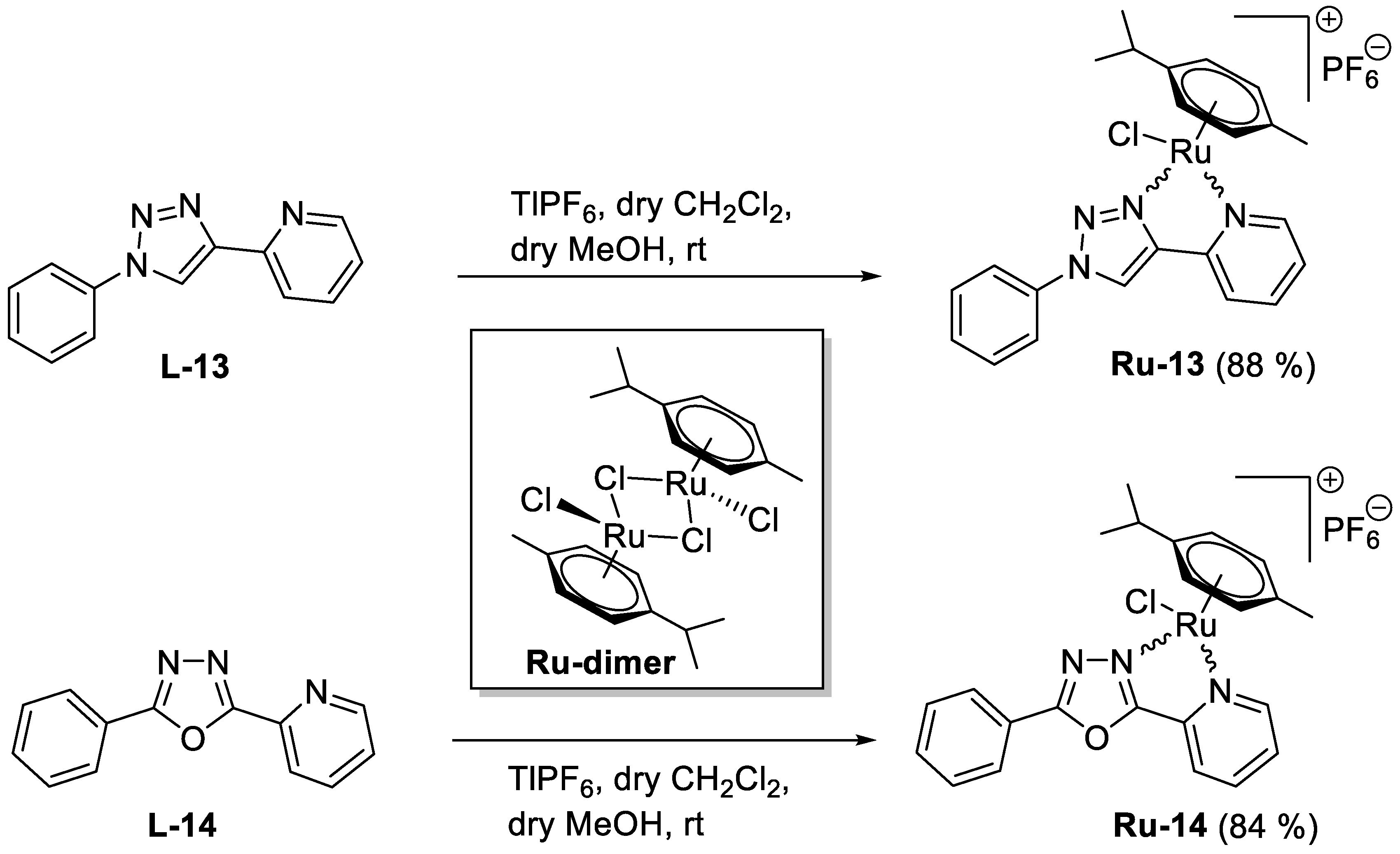
5.1.35. Complex Ru-13
Prepared from complex
Ru-dimer (20.0 mg, 0.033 mmol), 1-phenyl-4-(pyridine-2-yl)-1,2,3-triazole [
34] (
L-13, 14.5 mg, 0.065 mmol, 2 eq.), and TlPF
6 (22.8 mg, 0.065 mmol) according to general procedure V. Purified by recrystallization from a CHCl
3–Et
2O solvent mixture (3 mL and 6 mL, respectively) to give 36.8 mg (88%) yellow powder. Racemic mixture.
1H-NMR (400 MHz, acetone-
d6) δ (ppm): 9.62 (1H, s, Tria-H-5), 9.57 (1H, ddd,
J = 5.6, 1.4, 0.9 Hz, Py-H-6), 8.29 (1H, dt,
J = 7.6, 1.4 Hz, Py-H-4), 8.25 (1H, ddd,
J = 7.9, 1.9, 0.9 Hz, Py-H-3), 8.12–8.09 (2H, m, Ph), 7.79–7.68 (4H, m, Ph, Py-H-5), 6.27, 6.24, 6.04, 5.99 (4 × 1H, 4 d,
J = 6.2 Hz in each, 4 ×
p-cym-CH
Ar), 2.91 (1H, hept,
J = 6.9 Hz,
i-Pr-CH), 2.29 (3H, s, C
6H
4–C
H3), 1.23, 1.19 (2 × 3H, 2 d,
J = 6.9 Hz in each, 2 ×
i-Pr-C
H3);
13C-NMR (90 MHz, acetone-
d6) δ (ppm): 156.6 (Py-C-6), 149.2, 148.2 (Tria-C-4, Py-C-2), 141.1 (Py-C-4), 131.6, 131.2, 127.3, 124.0, 123.3, 121.9 (Ph, Py-C-3, Py-C-5, Tria-C-5), 106.2, 103.4 (2 ×
p-cym-C
qAr), 87.4, 85.8, 85.1, 84.5 (4 ×
p-cym-CH
Ar), 31.9 (
i-Pr-
CH), 22.6, 21.9 (2 ×
i-Pr-
CH
3), 18.7 (C
6H
4–
CH
3). ESI-HRMS positive mode (
m/
z): calculated for C
23H
24ClN
4Ru
+ [M − PF
6]
+: 493.0730. Found: 493.0709.
5.1.36. Complex Ru-14
Prepared from complex
Ru-dimer (20.0 mg, 0.033 mmol), 2-phenyl-5-(pyridine-2-yl)-1,3,4-oxadiazole [
35,
36] (
L-14, 14.6 mg, 0.065 mmol, 2 eq.), and TlPF
6 (22.8 mg, 0.065 mmol) according to general procedure V. Purified by recrystallization from a CHCl
3–Et
2O solvent mixture (3 mL and 6 mL, respectively) to give 34.9 mg (84%) yellow powder. Racemic mixture.
1H-NMR (360 MHz, acetone-
d6) δ (ppm): 9.69 (1H, ddd,
J = 5.6, 1.3, 0.8 Hz, Py-H-6), 8.54 (1H, ddd,
J = 7.8, 1.6, 0.8 Hz, Py-H-3), 8.48 (1H, dt,
J = 7.8, 1.3 Hz, Py-H-4), 8.33–8.29 (2H, m, Ph), 8.01 (1H, ddd,
J = 7.8, 5.6, 1.6 Hz, Py-H-5), 7.85–7.72 (3H, m, Ph), 6.30–6.27, 6.07–6.04 (2 × 2H, 2 m, 4 ×
p-cym-CH
Ar), 3.03 (1H, hept,
J = 6.9 Hz,
i-Pr-CH), 2.31 (3H, s, C
6H
4–C
H3), 1.32, 1.30 (2 × 3H, 2 d,
J = 6.9 Hz in each, 2 ×
i-Pr-C
H3);
13C-NMR (90 MHz, acetone-
d6) δ (ppm): 168.2, 164.7 (OD-C-2, OD-C-5), 157.8 (Py-C-6), 141.6 (Py-C-4), 141.5 (Py-C-2), 134.9, 130.7, 130.3, 128.5, 125.9, 122.7 (Ph, Py-C-3, Py-C-5), 106.7, 102.9 (2 ×
p-cym-C
qAr), 86.9, 85.6, 84.7, 84.5 (4 ×
p-cym-CH
Ar), 32.0 (
i-Pr-
CH), 22.7, 22.0 (2 ×
i-Pr-
CH
3), 18.8 (C
6H
4–CH
3). ESI-HRMS positive mode (
m/
z): calculated for C
23H
23ClN
3ORu
+ [M − PF
6]
+: 494.0556. Found: 494.0553.


 ,
, 

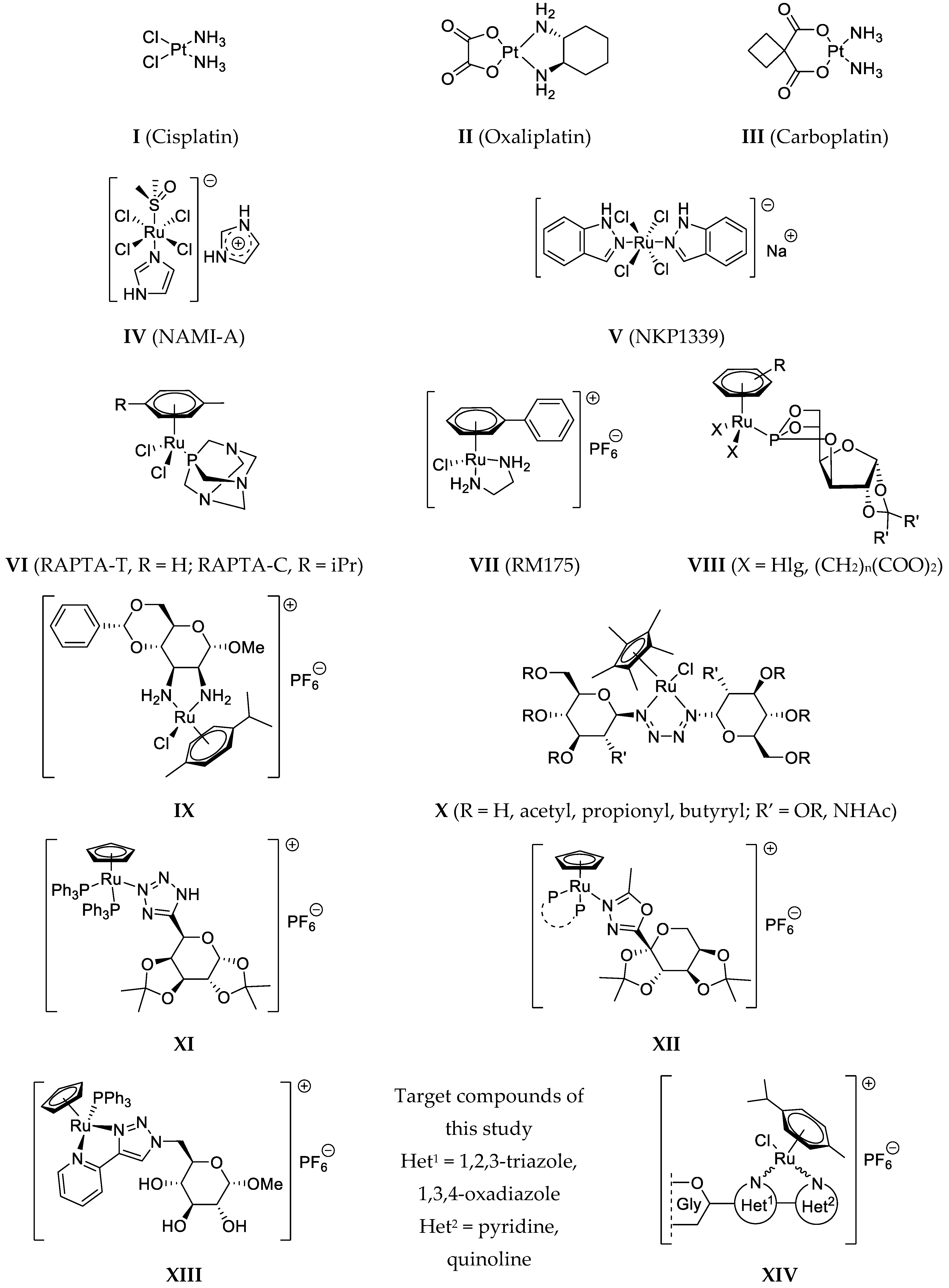



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}