
Ferroptosis Mechanisms Involved in Hippocampal-Related Diseases



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
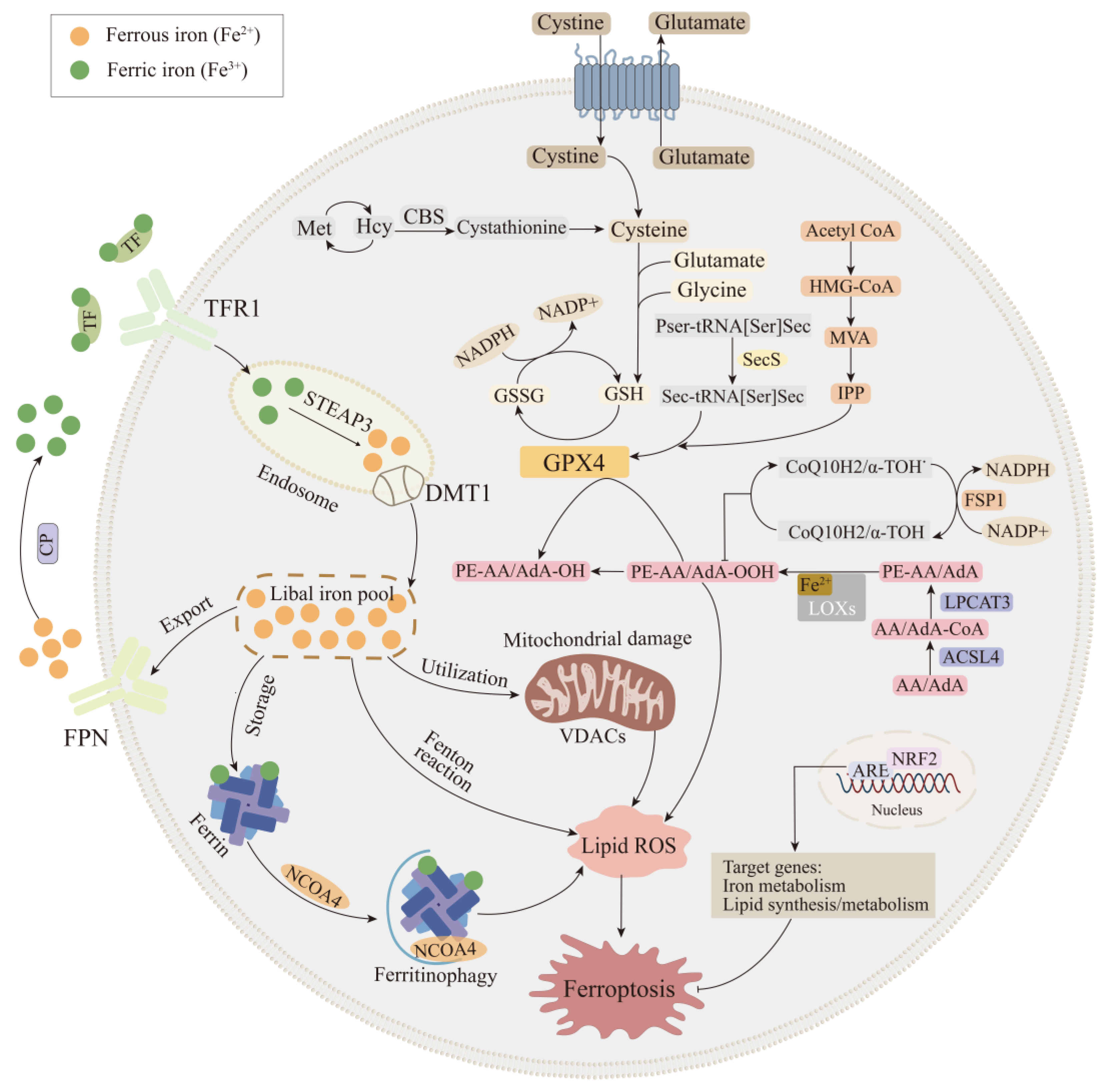
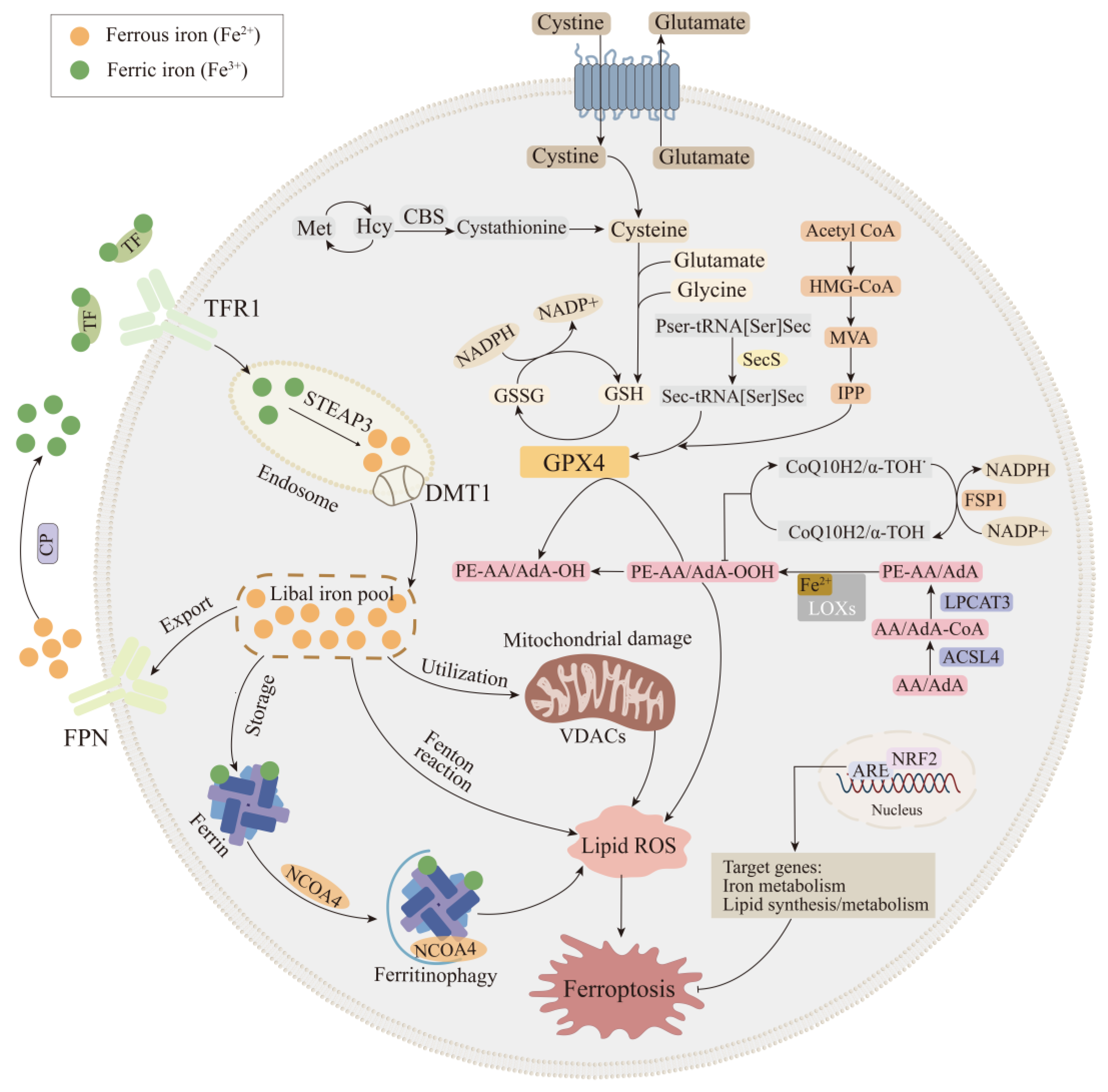
2. Key Mechanisms of the Ferroptosis Cascade
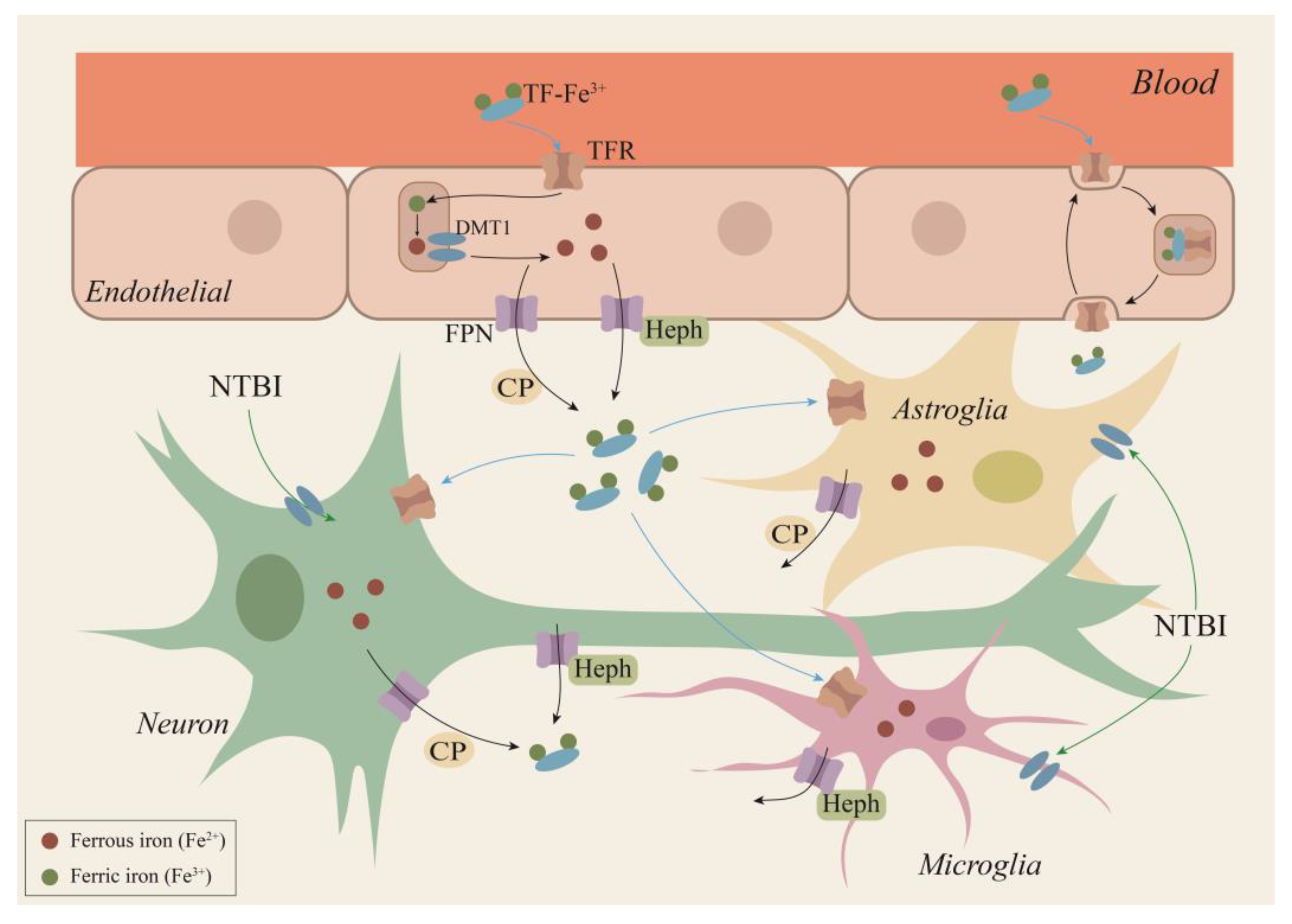
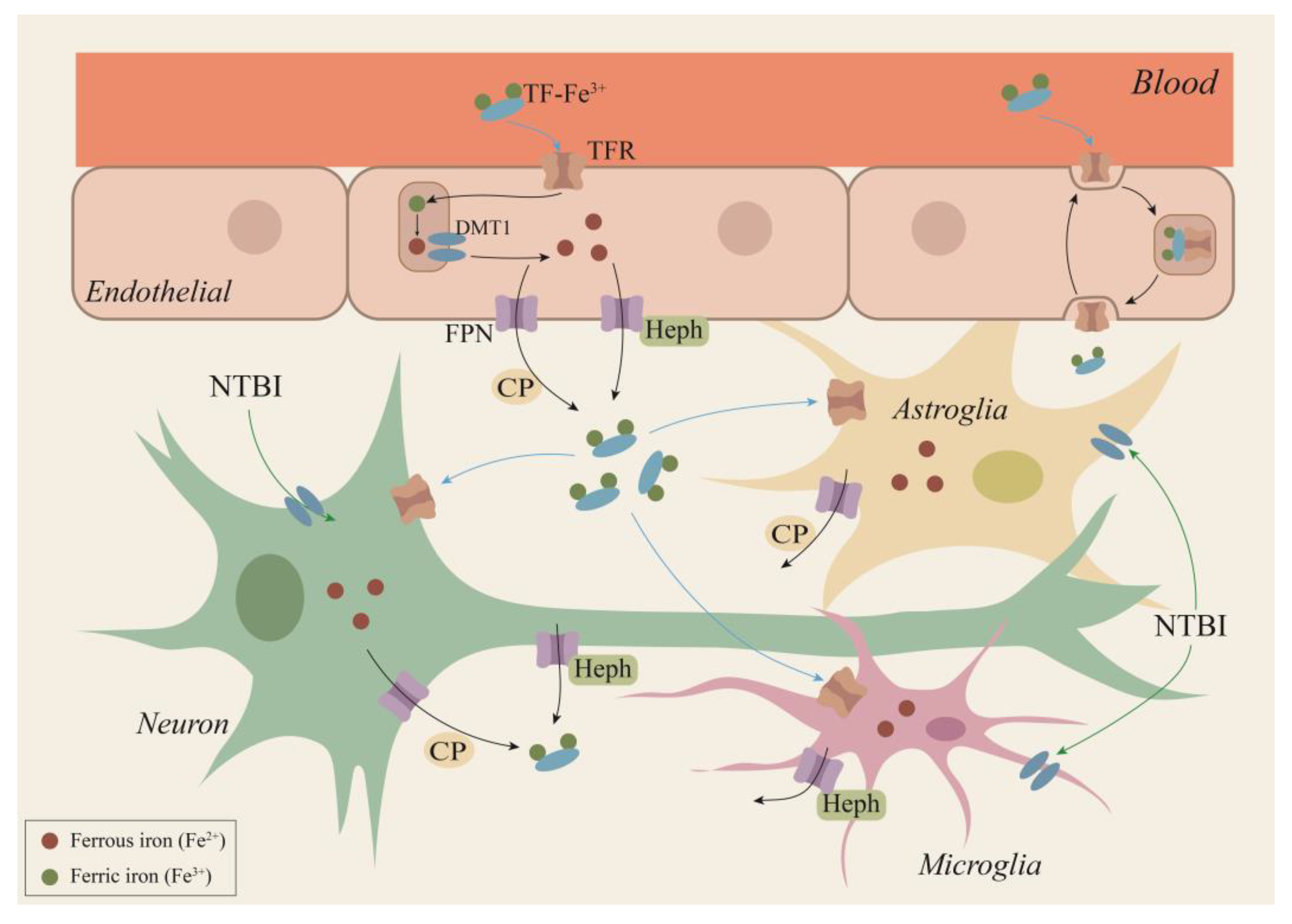
2.1. Iron Metabolic Pathway
2.2. Lipid Metabolic Pathway
2.3. Glutathione Peroxidase 4 Synthesis Pathway
2.4. FSP1-CoQ10-NAD(P)H Pathway
3. The Basic Structure of Hippocampus
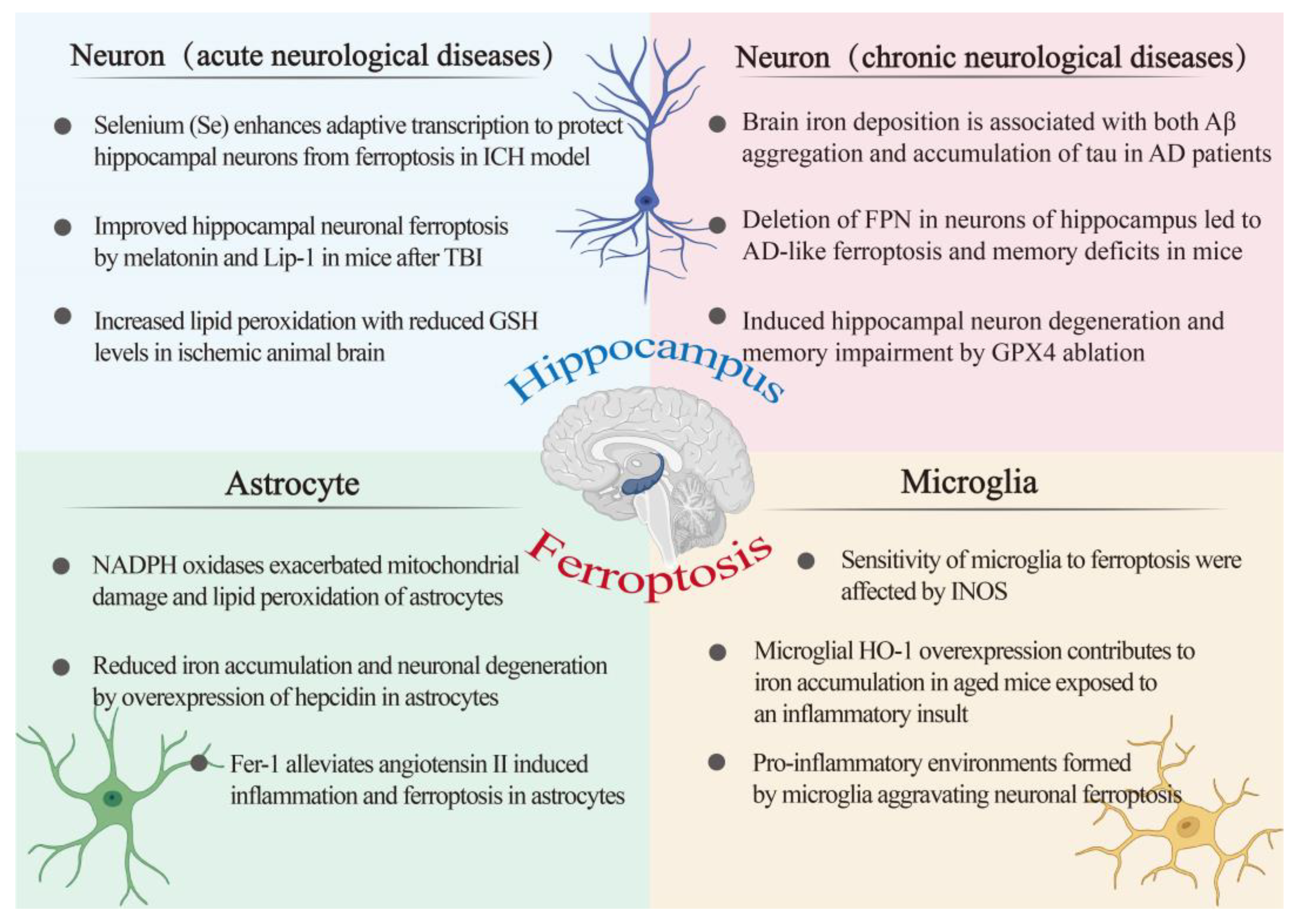
4. Ferroptosis and Hippocampus
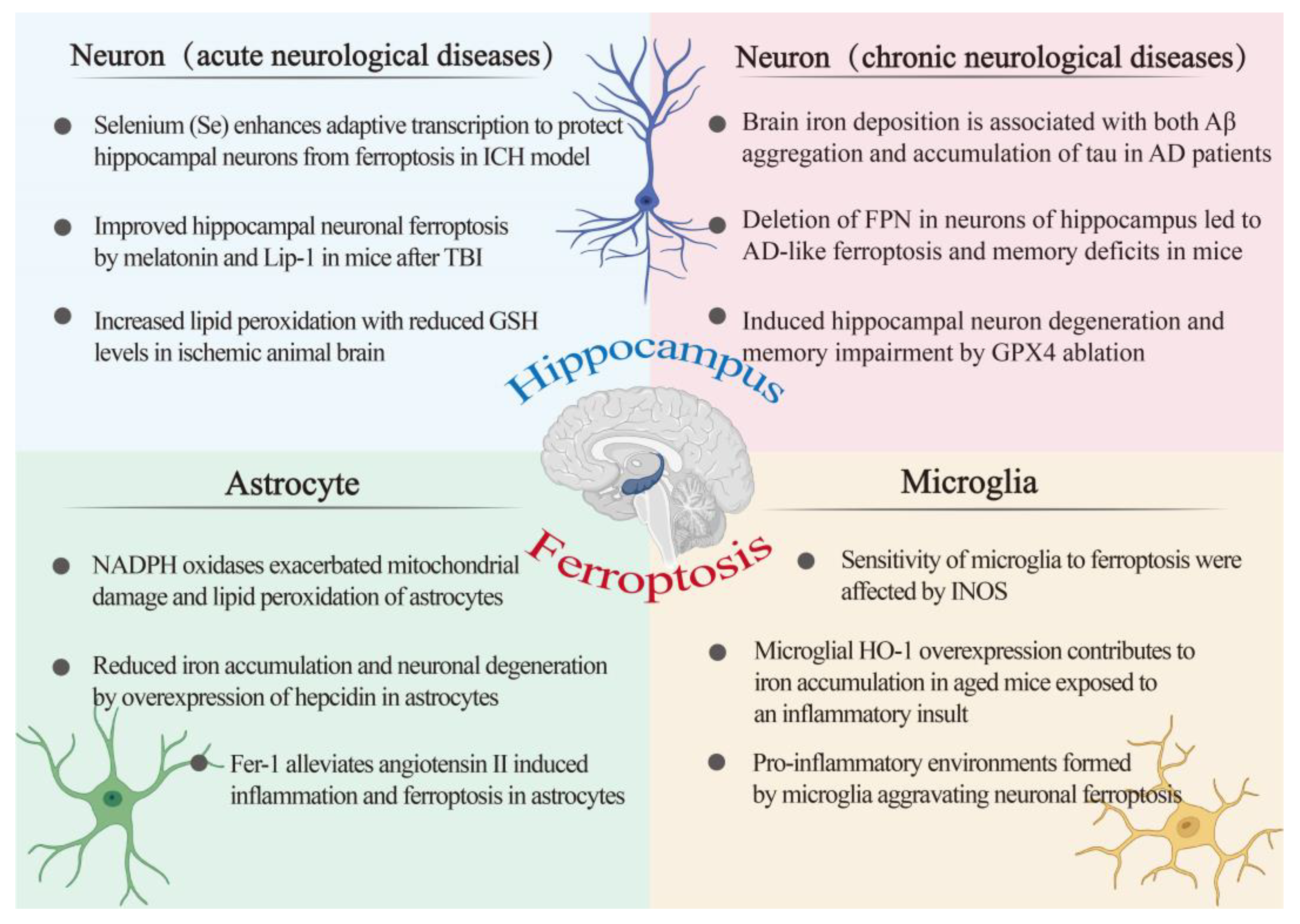
4.1. Ferroptosis and Astroglia
4.2. Ferroptosis and Microglia
4.3. Ferroptosis and Neuron
4.3.1. Ferroptosis in Acute Neurological Diseases
4.3.2. Ferroptosis in Chronic Neurological Diseases
4.4. Potential and Emerging Therapy Targeting Ferroptosis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Schweichel, J.U.; Merker, H.J. The morphology of various types of cell death in prenatal tissues. Teratology 1973, 7, 253–266. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirayama, T.; Miki, A.; Nagasawa, H. Organelle-specific analysis of labile Fe(II) during ferroptosis by using a cocktail of various colour organelle-targeted fluorescent probes. Metallomics 2019, 11, 111–117. [Google Scholar] [CrossRef]
- Hambright, W.S.; Fonseca, R.S.; Chen, L.; Na, R.; Ran, Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017, 12, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zheng, Y.F.; Wang, C.X.; Liu, Y.Z. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Magtanong, L.; Ko, P.J.; Dixon, S.J. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. 2016, 23, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, M.; Angeli, J.P.F.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; IngoId, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Ren, J.X.; Sun, X.; Yan, X.L.; Guo, Z.N.; Yang, Y. Ferroptosis in Neurological Diseases. Front. Cell Neurosci. 2020, 14, 218. [Google Scholar] [CrossRef] [PubMed]
- Vitalakumar, D.; Sharma, A.; Flora, S.J.S. Ferroptosis: A potential therapeutic target for neurodegenerative diseases. J. Biochem. Mol. Toxicol. 2021, 35, e22830. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting ferroptosis to iron out cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Kakhlon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes. Free Radic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [CrossRef]
- Knutson, M.D. Non-transferrin-bound iron transporters. Free Radic. Biol. Med. 2019, 133, 101–111. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Kosman, D.J. Iron transport across the blood-brain barrier: Development, neurovascular regulation and cerebral amyloid angiopathy. Cell. Mol. Life Sci. 2015, 72, 709–727. [Google Scholar] [CrossRef] [Green Version]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139 (Suppl. 1), 179–197. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.H.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X.J. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Gammella, E.; Recalcati, S.; Rybinska, I.; Buratti, P.; Cairo, G. Iron-induced damage in cardiomyopathy: Oxidative-dependent and independent mechanisms. Oxidative Med. Cell. Longev. 2015, 2015, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Monian, P.; Jiang, X. Metabolism and iron signaling in ferroptotic cell death. Oncotarget 2015, 6, 35145–35146. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Pierzynowska, K.; Rintz, E.; Gaffke, L.; Węgrzyn, G. Ferroptosis and Its Modulation by Autophagy in Light of the Pathogenesis of Lysosomal Storage Diseases. Cells 2021, 10, 365. [Google Scholar] [CrossRef] [PubMed]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gele, P.; Petrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Nunez, M.T.; Chana-Cuevas, P. New Perspectives in iron chelation therapy for the treatment of neurodegenerative diseases. Pharmaceuticals 2018, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Stoyanovsky, D.A.; Tyurina, Y.Y.; Shrivastava, I.; Bahar, I.; Tyurin, V.A.; Protchenko, O.; Jadhav, S.; Bolevich, S.B.; Kozlov, A.V.; Vladimirov, Y.A.; et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic. Biol. Med. 2019, 133, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Conrad, M. Iron and ferroptosis: A still Ill-defined liaison. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-Hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 31. [Google Scholar] [CrossRef]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Meng, L.; Han, L.; Jia, Y.; Zhao, Y.; Gao, H.; Kang, R.; Wang, X.; Tang, D.; Dai, E. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 2019, 508, 997–1003. [Google Scholar] [CrossRef]
- Tang, Y.; Zhou, J.; Hooi, S.C.; Jiang, Y.M.; Lu, G.D. Fatty acid activation in carcinogenesis and cancer development: Essential roles of long-chain acyl-CoA synthetases. Oncol. Lett. 2018, 16, 1390–1396. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Yang, X.F.; Liu, H.L.; Fu, N.; Ouyang, Y.; Qing, K. Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: An update. World J. Gastroenterol. 2015, 21, 3492–3498. [Google Scholar] [CrossRef] [PubMed]
- Kuch, E.M.; Vellaramkalayil, R.; Zhang, I.; Lehnen, D.; Brugger, B.; Stremmel, W.; Ehehalt, R.; Poppelreuther, M.; Fullekrug, J. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 227–239. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.W.; Qu, F.; Angeli, J.P.F.; Doll, S.; St Croix, C.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Colakoglu, M.; Tuncer, S.; Banerjee, S. Emerging cellular functions of the lipid metabolizing enzyme 15-Lipoxygenase-1. Cell Prolif. 2018, 51, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Thomas, C.; Jalil, A.; Magnani, C.; Ishibashi, M.; Queré, R.; Bourgeois, T.; Bergas, V.; Ménégaut, L.; Patoli, D.; le Guern, N.; et al. LPCAT3 deficiency in hematopoietic cells alters cholesterol and phospholipid homeostasis and promotes atherosclerosis. Atherosclerosis 2018, 275, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [Green Version]
- Shin, C.S.; Mishra, P.; Watrous, J.D.; Carelli, V.; D’Aurelio, M.; Jain, M.; Chan, D.C. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun. 2017, 8, 15074. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Freitas, F.P.; Seibt, T.; et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 2018, 172, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Roberts, B.R. Glutathione peroxidase 4: A new player in neurodegeneration? Mol. Psychiatry 2017, 22, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System Xc- cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Angeli, J.P.F.; Conrad, M. Selenium and GPX4, a vital symbiosis. Free Radic. Biol. Med. 2018, 127, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Moosmann, B.; Behl, C. Selenoproteins, cholesterol-lowering drugs, and the consequences—Revisiting of the mevalonate pathway. Trends Cardiovasc. Med. 2004, 14, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Liu, W.S.; Zhuo, Q.F.; Hu, Q.S.; Liu, M.Q.; Sun, Q.Q.; Zhang, Z.; Fan, G.X.; Xu, W.Y.; Ji, S.R.; et al. Ferroptosis: Final destination for cancer? Cell Prolif. 2020, 53, 13. [Google Scholar] [CrossRef]
- Anandhan, A.; Dodson, M.; Schmidlin, C.J.; Liu, P.F.; Zhang, D.D. Breakdown of an ironclad defense system: The critical role of NRF2 in mediating ferroptosis. Cell Chem. Biol. 2020, 27, 436–447. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L. Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion 2007, 7, S2–S7. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Zeidman, P.; Maguire, E.A. Anterior hippocampus: The anatomy of perception, imagination and episodic memory. Nat. Rev. Neurosci. 2016, 17, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.J.; Sun, A.B.; Liu, L. Epigenetic modulation during hippocampal development. Biomed. Rep. 2018, 9, 463–473. [Google Scholar] [CrossRef]
- van Strien, N.M.; Cappaert, N.L.; Witter, M.P. The anatomy of memory: An interactive overview of the parahippocampal-hippocampal network. Nat. Rev. Neurosci. 2009, 10, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Burgess, N.; Maguire, E.A.; O’Keefe, J. The human hippocampus and spatial and episodic memory. Neuron 2002, 35, 625–641. [Google Scholar] [CrossRef] [Green Version]
- Schultz, C.; Engelhardt, M. Anatomy of the hippocampal formation. Front. Neurol. Neurosci. 2014, 34, 6–17. [Google Scholar]
- Zammit, A.R.; Ezzati, A.; Katz, M.J.; Zimmerman, M.E.; Lipton, M.L.; Sliwinski, M.J.; Lipton, R.B. The association of visual memory with hippocampal volume. PLoS ONE 2017, 12, e0187851. [Google Scholar] [CrossRef] [Green Version]
- Pang, C.C.; Kiecker, C.; O’Brien, J.T.; Noble, W.; Chang, R.C. Ammon’s horn 2 (CA2) of the hippocampus: A long-known region with a new potential role in neurodegeneration. Neuroscientist 2019, 25, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Alkadhi, K.A. Cellular and molecular differences between area CA1 and the dentate gyrus of the hippocampus. Mol. Neurobiol. 2019, 56, 6566–6580. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, D.M. Interneuron networks in the hippocampus. Curr. Opin. Neurobiol. 2011, 21, 709–716. [Google Scholar] [CrossRef]
- Tatu, L.; Vuillier, F. Structure and vascularization of the human hippocampus. Front. Neurol. Neurosci. 2014, 34, 18–25. [Google Scholar]
- Ogata, K.; Kosaka, T. Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neuroscience 2002, 113, 221–233. [Google Scholar] [CrossRef]
- Bushong, E.A.; Martone, M.E.; Ellisman, M.H. Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int. J. Dev. Neurosci. 2004, 22, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Wallraff, A.; Köhling, R.; Heinemann, U.; Theis, M.; Willecke, K.; Steinhäuser, C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 5438–5447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, C.R.; Felix, L.; Zeug, A.; Dietrich, D.; Reiner, A.; Henneberger, C. Astroglial Glutamate Signaling and Uptake in the Hippocampus. Front. Mol. Neurosci. 2017, 10, 451. [Google Scholar] [CrossRef] [Green Version]
- Frost, J.L.; Schafer, D.P. Microglia: Architects of the developing nervous system. Trends Cell Biol. 2016, 26, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldskogius, H. Microglia in neuroregeneration. Microsc. Res. Tech. 2001, 54, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.; Ayton, S.; Bush, A.; Lei, P. A delicate balance: Iron metabolism and diseases of the brain. Front. Aging Neurosci. 2013, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.D.; Pang, P.; Zhou, X.T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef]
- Rui, T.; Wang, H.; Li, Q.; Cheng, Y.; Gao, Y.; Fang, X.; Ma, X.; Chen, G.; Gao, C.; Gu, Z.; et al. Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury-induced ferroptosis. J. Pineal Res. 2021, 70, e12704. [Google Scholar] [CrossRef]
- Ficiarà, E.; Munir, Z.; Boschi, S.; Caligiuri, M.E.; Guiot, C. Alteration of iron concentration in Alzheimer’s disease as a possible diagnostic biomarker unveiling ferroptosis. Int. J. Mol. Sci. 2021, 22, 4479. [Google Scholar] [CrossRef]
- Burkhart, A.; Skjørringe, T.; Johnsen, K.B.; Siupka, P.; Thomsen, L.B.; Nielsen, M.S.; Thomsen, L.L.; Moos, T. Expression of iron-related proteins at the neurovascular unit supports reduction and reoxidation of iron for transport through the blood-brain barrier. Mol. Neurobiol. 2016, 53, 7237–7253. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Kosman, D.J. Mechanisms and regulation of iron trafficking across the capillary endothelial cells of the blood-brain barrier. Front. Mol. Neurosci. 2015, 8, 31. [Google Scholar] [CrossRef]
- Bartzokis, G.; Tishler, T.A.; Lu, P.H.; Villablanca, P.; Altshuler, L.L.; Carter, M.; Huang, D.; Edwards, N.; Mintz, J. Brain ferritin iron may influence age- and gender-related risks of neurodegeneration. Neurobiol. Aging 2007, 28, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, Y.; Zhang, J.H.; Han, K.; Zhang, X.; Bai, X.; You, L.H.; Yu, P.; Shi, Z.; Chang, Y.Z.; et al. Astrocyte hepcidin ameliorates neuronal loss through attenuating brain iron deposition and oxidative stress in APP/PS1 mice. Free Radic. Biol. Med. 2020, 158, 84–95. [Google Scholar] [CrossRef]
- Park, M.W.; Cha, H.W.; Kim, J.; Kim, J.H.; Yang, H.; Yoon, S.; Boonpraman, N.; Yi, S.S.; Yoo, I.D.; Moon, J.S. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 2021, 41, 101947. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhou, C.; Zhu, Y.; Chao, Z.; Sheng, Z.; Zhang, Y.; Zhao, Y. Ferrostatin-1 alleviates angiotensin II (Ang II)- induced inflammation and ferroptosis in astrocytes. Int. Immunopharmacol. 2021, 90, 107179. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.X.; Du, F.; Jiang, L.R.; Gong, J.; Zhou, Y.F.; Luo, Q.Q.; Qian, Z.M.; Ke, Y. Effects of aspirin on expression of iron transport and storage proteins in BV-2 microglial cells. Neurochem. Int. 2015, 91, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.N.; Qian, Z.M.; Wu, K.C.; Yung, W.H.; Ke, Y. Expression of iron transporters and pathological hallmarks of Parkinson’s and Alzheimer’s diseases in the brain of young, adult, and aged rats. Mol. Neurobiol. 2017, 54, 5213–5224. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Min, L.; Chen, H.; Deng, W.; Tan, M.; Liu, H.; Hou, J. Iron overload in the motor cortex induces neuronal ferroptosis following spinal cord injury. Redox Biol. 2021, 43, 101984. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Fernández-Mendívil, C.; Luengo, E.; Trigo-Alonso, P.; García-Magro, N.; Negredo, P.; López, M.G. Protective role of microglial HO-1 blockade in aging: Implication of iron metabolism. Redox Biol. 2021, 38, 101789. [Google Scholar] [CrossRef]
- Huo, J.; Cui, Q.; Yang, W.; Guo, W. LPS induces dopamine depletion and iron accumulation in substantia nigra in rat models of Parkinson’s disease. Int. J. Clin. Exp. Pathol. 2018, 11, 4942–4949. [Google Scholar] [PubMed]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal toxicity links to Alzheimer’s disease and neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef] [PubMed]
- Puy, V.; Darwiche, W.; Trudel, S.; Gomila, C.; Lony, C.; Puy, L.; Lefebvre, T.; Vitry, S.; Boullier, A.; Karim, Z.; et al. Predominant role of microglia in brain iron retention in Sanfilippo syndrome, a pediatric neurodegenerative disease. Glia 2018, 66, 1709–1723. [Google Scholar] [CrossRef]
- Ke, Y.; Ming Qian, Z. Iron misregulation in the brain: A primary cause of neurodegenerative disorders. Lancet Neurol. 2003, 2, 246–253. [Google Scholar] [CrossRef]
- Ji, C.; Steimle, B.L.; Bailey, D.K.; Kosman, D.J. The ferroxidase hephaestin but not amyloid precursor protein is required for ferroportin-supported iron efflux in primary hippocampal neurons. Cell Mol. Neurobiol. 2018, 38, 941–954. [Google Scholar] [CrossRef]
- Alim, I.; Caulfield, J.T.; Chen, Y.; Swarup, V.; Geschwind, D.H.; Ivanova, E.; Seravalli, J.; Ai, Y.; Sansing, L.H.; Ste Marie, E.J.; et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 2019, 177, 1262–1279. [Google Scholar] [CrossRef] [Green Version]
- Tuo, Q.Z.; Lei, P.; Jackman, K.A.; Li, X.L.; Xiong, H.; Li, X.L.; Liuyang, Z.Y.; Roisman, L.; Zhang, S.T.; Ayton, S.; et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; Cao, J.; Dong, Y.; Chen, Y. Melatonin alleviates acute sleep deprivation-induced memory loss in mice by suppressing hippocampal ferroptosis. Front. Pharmacol. 2021, 12, 708645. [Google Scholar]
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Zhang, J. Iron metabolism, ferroptosis, and the links with Alzheimer’s disease. Front. Neurosci. 2019, 13, 1443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Wang, D.W.; Xu, S.F.; Zhang, S.; Fan, Y.G.; Yang, Y.Y.; Guo, S.Q.; Wang, S.; Guo, T.; Wang, Z.Y.; et al. Alpha-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 2018, 14, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Ates, G.; Goldberg, J.; Currais, A.; Maher, P. CMS121, a fatty acid synthase inhibitor, protects against excess lipid peroxidation and inflammation and alleviates cognitive loss in a transgenic mouse model of Alzheimer’s disease. Redox Biol. 2020, 36, 101648. [Google Scholar] [CrossRef]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [Green Version]
- Mazhar, M.; Din, A.U.; Ali, H.; Yang, G.; Ren, W.; Wang, L.; Fan, X.; Yang, S. Implication of ferroptosis in aging. Cell Death Discov. 2021, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Navarro Negredo, P.; Yeo, R.W.; Brunet, A. Aging and Rejuvenation of Neural Stem Cells and Their Niches. Cell Stem Cell 2020, 27, 202–223. [Google Scholar] [CrossRef] [PubMed]
- Ratan, R.R. The Chemical Biology of Ferroptosis in the Central Nervous System. Cell Chem. Biol. 2020, 27, 479–498. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Disease 2020, 11, 88. [Google Scholar] [CrossRef]
- Nirmala, J.G.; Lopus, M. Cell death mechanisms in eukaryotes. Cell Biol. Toxicol. 2020, 36, 145–164. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Wang, Z.; Cao, J.; Dong, Y.; Chen, Y. Ferroptosis Mechanisms Involved in Hippocampal-Related Diseases. Int. J. Mol. Sci. 2021, 22, 9902. https://doi.org/10.3390/ijms22189902
Wang X, Wang Z, Cao J, Dong Y, Chen Y. Ferroptosis Mechanisms Involved in Hippocampal-Related Diseases. International Journal of Molecular Sciences. 2021; 22(18):9902. https://doi.org/10.3390/ijms22189902
Chicago/Turabian StyleWang, Xintong, Zixu Wang, Jing Cao, Yulan Dong, and Yaoxing Chen. 2021. "Ferroptosis Mechanisms Involved in Hippocampal-Related Diseases" International Journal of Molecular Sciences 22, no. 18: 9902. https://doi.org/10.3390/ijms22189902
APA StyleWang, X., Wang, Z., Cao, J., Dong, Y., & Chen, Y. (2021). Ferroptosis Mechanisms Involved in Hippocampal-Related Diseases. International Journal of Molecular Sciences, 22(18), 9902. https://doi.org/10.3390/ijms22189902