1. Introduction
Brain functions strictly depend on precise regulation of structural and functional synaptic integrity. Among the mechanisms governing synaptic protein functions, post-translational modifications (PTM) [
1,
2] play a pivotal role. PTMs may influence synaptic protein activity and turnover, localization at the synapse, and signaling cascades [
3,
4,
5,
6].
One of the PTMs is protein S-palmitoylation (S-PALM) involving covalent attachment of palmitic acid (C16:0) to cysteine residue(s) via a thioester bond. Recent studies showed that S-palmitoylation can modulate protein localization, stability, activities, and trafficking and play an essential role in various biological processes, including synaptic plasticity [
7,
8], cell signaling, cellular differentiation [
9], and apoptosis [
10].
Unlike other fatty acid modifications, S-palmitoylation is a reversible process, tightly regulated by two groups of enzymes: palmitoyl acyltransferases (PATs, palmitoylating enzymes) and palmitoyl thioesterases (depalmitoylating enzyme). It is widely accepted that repeated cycles of palmitoylation/depalmitoylation are critically involved in regulating multiple protein functions. The molecular mechanisms that lie behind site-specific protein S-palmitoylation remain largely unknown. Several human diseases are often associated with the atypical activity of PATs together with changes in the pattern of S-palmitoylation. S-PALM has been implicated in a wide range of human disease states such as cancer [
11], Alzheimer’s disease [
12], Parkinson’s disease, cardiovascular disease, schizophrenia [
13], or major depressive disorder MDD [
14]. Therefore, identifying substrates that undergo S-PALM and specific sites of these modifications may provide candidates for targeted therapy.
Twenty-three PATs have been identified in mammalian cells, which mediate the majority of protein S-palmitoylation. One of the known PATs is a zinc finger DHHC domain-containing protein 7 (Zdhhc7, abbreviated ZDHHC7). This enzyme palmitoylates various synaptic proteins involved in the regulation of cellular polarity and proliferation [
15,
16]. Moreover, Zdhhc7 is responsible for S-palmitoylation of sex steroid receptors such as estrogen and progesterone receptors [
16,
17,
18]. Importantly,
Zdhhc7-/- mice developed symptoms characteristic of human Bartter syndrome (BS) type IV because ZDHHC7 protein may affect ClC-K-barttin channel activation [
19]. Thus, targeting ZDHHC7 activity may offer a potential therapeutic strategy in certain brain pathophysiological states. Most recently, using the mass spectrometry approach, we have identified sex-dependent differences in the S-PALM of synaptic proteins potentially involved in the regulation of membrane excitability and synaptic transmission as well as in the signaling of proteins involved in the structural plasticity of dendritic spines in the mice brain [
18]. Our data showed for the first time sex-dependent action of ZDHHC7 acyltransferase. Furthermore, we revealed that different S-PALM proteins control the same biological processes in male and female synapses [
18,
19].
Several methods have been developed for the identification of S-palmitoylation target proteins. However, site-specific identification of S-palmitoylation is less studied. Large-scale identification of S-palmitoylation sites mainly relies on mass spectrometry-based methods such as PANIMoni developed in our lab [
20] or PALMPiscs or ssABE [
21]. These methods have been successfully used to identify a large number of S-palmitoylated proteins in different species, such as rats, mice, or humans. For instance, PANIMoni has been used to describe endogenous S-palmitoylation and S-nitrosylation of proteins in the rat brain excitatory synapses at the level of specific single cysteine in a mouse model of depression [
20]. In recent years, results of large-scale proteome databases obtained with PANIMoni, PALMPiscs, or ssABE methods were used to develop tools to predict sites of specific S-palmitoylation in other biological complexes. Several machine learning-based algorithms [
22,
23,
24,
25] have been developed for predictions of S-palmitoylation sites such as; NBA-PALM [
26] and CSS-PALM [
25], but their accuracy is uncertain. Therefore, with the growing number of publicly available large-scale proteome databases of the brain and somatic tissues, there is a need for the development of reliable and accurate computational tools to process them.
Considering the growing recognition for the importance of post-translational modifications of proteins in cell physiology, this study aims to develop a computational tool for predicting S-palmitoylation sites using proteomic data obtained by the mass spectrometry-based method PANIMoni [
20]. Most recently, we have successfully used this approach to create a detailed ZDHHC subtype-specific and sex-mouse S-palmitoylome [
18,
19]. Here, we have used this protein database for validation of the computational tool described in this study.
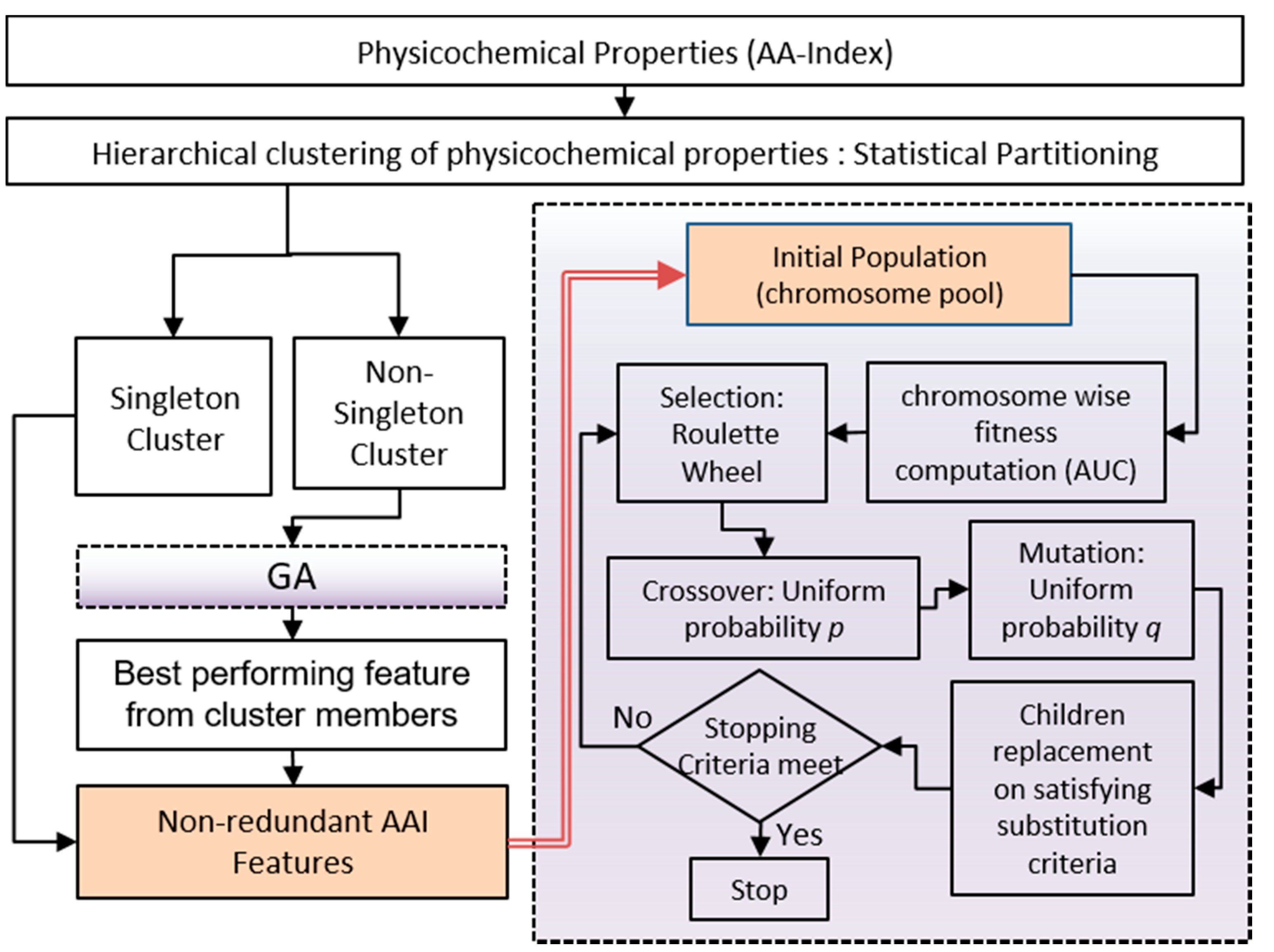
Our tool is focused on a random forest (RF) [
27] classifier-based consensus strategy, which can predict the palmitoylated cysteine sites on synaptic proteins of the male/female mouse dataset. Different heuristic selection strategies have been applied on the physicochemical features from the AAIndex feature database [
28] along with position-specific amino acid (AA) propensity information, which eventually generates three different sets of features: (a) K-Best (KB) features, (b) genetic algorithm (GA) based features [
29], and (c) a union (UN) of K-Best and GA based features. The experiment has been carried out on three categorized synaptic protein datasets originally described in our previous publications [
18,
19];
viz., male, female, and combined (male + female). In each experimental group, the weighted data is used as the training set, and the knock-out is used as the hold-out test set for performance evaluation and comparison. A novel RF-driven consensus strategy with efficient feature selection shows significant performance in predicting S-palmitoylation sites in mouse data.
2. Results
Our method, RFCM-PALM, predicts the S-palmitoylation sites from the primary sequence information of synaptic proteins. In the mouse model experiments, three categories of data,
viz., Male, Female, and Combined, and three different feature sets,
viz., KB, GA, UN, along with the RF classifier, have been used. The rationale behind the choice of the RF classifier is elaborated in the
Supplementary Section S1 and Table S1. Features are extracted from the sequence motifs of variable length, and detailed experiments are conducted to select the optimum length of such sequence motif. A summary of these experiments is discussed in
Section 4.3, and detailed results are reported in the
Supplementary Table S2. Finally, the proposed approach presents a three-star consensus model for the final classification task. The efficacy of PTM prediction depends heavily on selecting appropriate feature sets, the choice of the classifier, and the underlying evaluation strategy. In this work, GA-based features show better the area under the curve (AUC) score for male, female, and combined datasets. The UN features show promising performances for the female dataset with higher accuracy, whereas KB and GA features achieve the highest accuracy in male and combined datasets, respectively. Finally, we present a three-star consensus approach for the final classification task. The consensus model significantly improved the performance compared to individual feature-specific models. We have further compared the proposed consensus-based approach, RFCM-PALM, with two
state-of-the-art methods.
2.1. Performance Evaluation
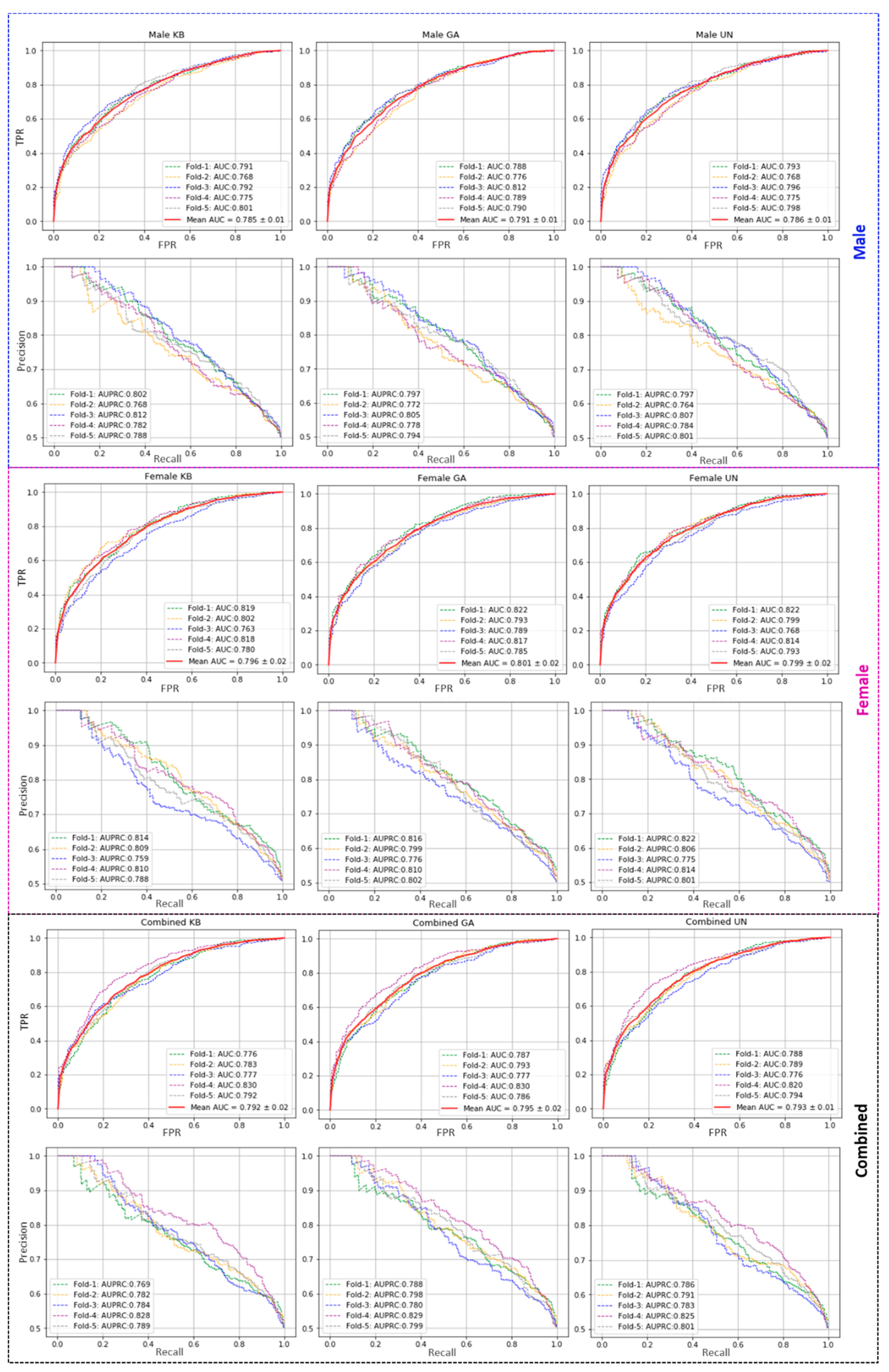
The performance of the proposed model has been evaluated with five-fold cross-validation on three different feature sets (namely KB, GA, UN) using a RF classifier. Five-fold cross-validation has been introduced to estimate the model’s strength on all three categories of datasets (Male (M), Female (F), and Combined (M + F)), and the performances are reported in
Table 1. The individual fold-wise performances on all three datasets are reported in
Supplementary Table S3. In all three datasets, the GA-based feature outperforms the rest two in AUC score. However, in our proposed method, for fold-wise testing, the GA-based feature shows a ~79% AUC score for both male and combined datasets, and 80% AUC on the female dataset, surpassing the other two features. For female data, the UN-based feature outperforms KB and GA-based features, having an accuracy score of 71.9% and F1 of 71.3% (see
Table 1). The AUC and AUPRC curves from training models are shown in
Figure 1.
The knock-out data has been used as the hold-out test set from three categories of data (Male, Female, and Combined) individually. In the knock-out hold-out test set, the GA-based feature shows better performance for all the datasets than other features with an AUC score of ~66.4% in males, 68.6% in females, and 62.5% in combined datasets (please see
Table 2). Moreover, GA has higher accuracy in all hold-out test data except the males set, where the KB-based model achieves 62% accuracy. Furthermore, we have introduced a consensus strategy for the final classification of S-PALM on the hold-out test set. Initially, the best models are extracted from the cross-validation strategy for each feature set on the three categories of data set independently.
Thus, the three best models are identified for classification from each data set (Male/Female/Combined). Finally, three consensus-based classifications are obtained for the final classification. The 1*-consensus (1*Con), 2*-consensus (2*Con), and 3*-consensus (3*Con) represent 1, 2, and 3 model confidence scores, respectively. The detailed consensus mechanism is shown in
Figure 2, and the results are depicted in
Table 2. The 2*Con (2 model confidence) has significantly improved performance compared to the corresponding individual models. Consensus-based performance with different categories of data for hold-out test sets is shown in
Table 2.
2.2. Comparison with the State-of-the-Art Approaches
To demonstrate the performance of our proposed method, we have compared our approach with existing PTM prediction models. We have identified three
state-of-the-art approaches for benchmarking purposes, CapsNet [
23], MusiteDeep [
24,
30], and ModPred [
31]. The CapsNet [
23] is a deep learning-based architecture that provides prediction models for different PTM sites. MusiteDeep [
24,
30] is a deep-learning-based system that can predict general and kinase-specific phosphorylation sites from primary sequence information. ModPred [
31] is a sequence-based PTM prediction tool developed on the structural and functional signatures of proteins. The CapsNet, provides a 10-fold cross-validation result on the benchmark dataset of animal species (metazoa), extracted from the NCBI taxonomy database [
32], which has been curated by collecting annotations from Uniprot/Swiss-Prot (August 2007 release) [
33] with less than 30% sequence similarity.
Our approach has also been trained with the similar dataset used in CapsNet [
23] for S-palmitoylated cysteine prediction for comparison purposes. When compared with all three existing approaches on similar datasets, the performance scores are directly incorporated from Wang et al. [
23]. In the proposed model, we have also presented the class-imbalanced learning by imposing a positive-negative ratio at 1:2 along with the balanced learning (1:1). The performance has been compared with the existing approaches concerning the AUC and AUPRC scores (
Table 3). Our proposed method outperforms the
state-of-the-art methods in both metrics. The AUC and AUPRC have improved by 8% in comparison with the earlier best-performing method. Additionally, the proposed approach has surpassed the prior approaches by 32% in the AUPRC score, as depicted in
Table 3. The detailed fold-wise evaluation scores are shown in the
Supplementary Table S4 (balanced) and
Table S5 (imbalanced).
To investigate the significance of our proposed model on a novel S-PALM dataset, we have evaluated and compared the performance with two web servers MusiteDeep [
30] and CSS-Palm [
25]. MusiteDeep [
24,
30] is a web resource with a deep-learning framework that can predict and visualize different post-translational modification (PTM) sites from protein sequence information. CSS-Palm [
25] is developed based on clustering and scoring strategy (CSS) algorithm and Group-based Prediction System (GPS) algorithm. CSS-Palm is evaluated with two high-performing thresholds, as stated by the authors in [
25]. The novel hold-out test data from male, female, and combined sets has been submitted in the above two web servers, and performances have been recorded for comparison purposes (see
Table 4). The proposed method has achieved a better result in more balanced metrics F1, and MCC compared to each of these web servers in S-PALM prediction depicting the efficacy of the proposed method on S-PALM prediction. In all three datasets, male, female, and combined, the proposed approach has improved the F1 score by 54%, 52%, and 48%, and MCC score by 7%, 32%, and 13%, respectively.
In this novel hold-out data set, both web servers show high precision (0.827 in MusiteDeep and 0.857 in CSS-Palm) and very low recall (0.0882 in MusiteDeep and 0.1324 in CSS-Palm). A high precision score depicts low false positivity, and low recall depicts the increase in false-negative data, which can be interpreted as a failure for predicting the positive data. This may lead to a biased classification. Low recall also results in a low F1 score, which is the harmonic mean of precision and recall. Not only the recall score, but the MCC score for both the web servers are low, which depicts the failure of the class imbalance issue [
34]. In contrast, our proposed method achieves 0.638 precision, and 0.583 recall scores on this hold-out dataset, which shows a more balanced scenario of classification outcome. In addition, our proposed method shows the highest accuracy for all three categories of the data, which outperforms the other two (accuracy improvement by 9%, 15%, and 7% in male, female, and the combined dataset).
3. Discussion
Our method, RFCM-PALM, computationally predicts the S-palmitoylation sites using the primary sequence information of the synaptic group of proteins from three categories of mouse data, designed as sex-dependent (male, female) and sex-independent (combined) mode. The computational model has been developed through a rigorous feature selection strategy and optimal model selection for predicting the S-PALM modification sites in a given subsequence window. The proposed model has been evaluated with five-fold cross-validation, and model performances have been compared with the state-of-the-art approaches using three different feature sets; KB, GA, and UN. Finally, a consensus strategy is designed based on the feature-specific best models from their cross-validated models. The performance of the consensus model improved significantly compared to state-of-the-art approaches. The significant performance improvement in predicting S-PALM modification sites portrayed the efficacy of the proposed method.
The performance of the method may further be enhanced by incorporating deep-learning models. However, the major bottleneck lies with the limitation of adequate training samples. Furthermore, due to the complex nature of the biological experiments, scalability of the experimentally validated samples may not be easy. The development of the RFCM-PALM web server is also in our plans. We also plan to extend the method for other PTM types to predict protein nitrosylation sites in the synaptic proteins.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}