
Regular Physical Exercise Modulates Iron Homeostasis in the 5xFAD Mouse Model of Alzheimer’s Disease

 , , ,
, , , 
 , and
, and
Abstract
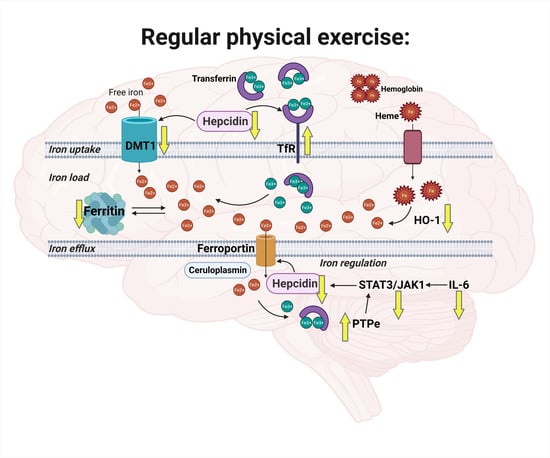
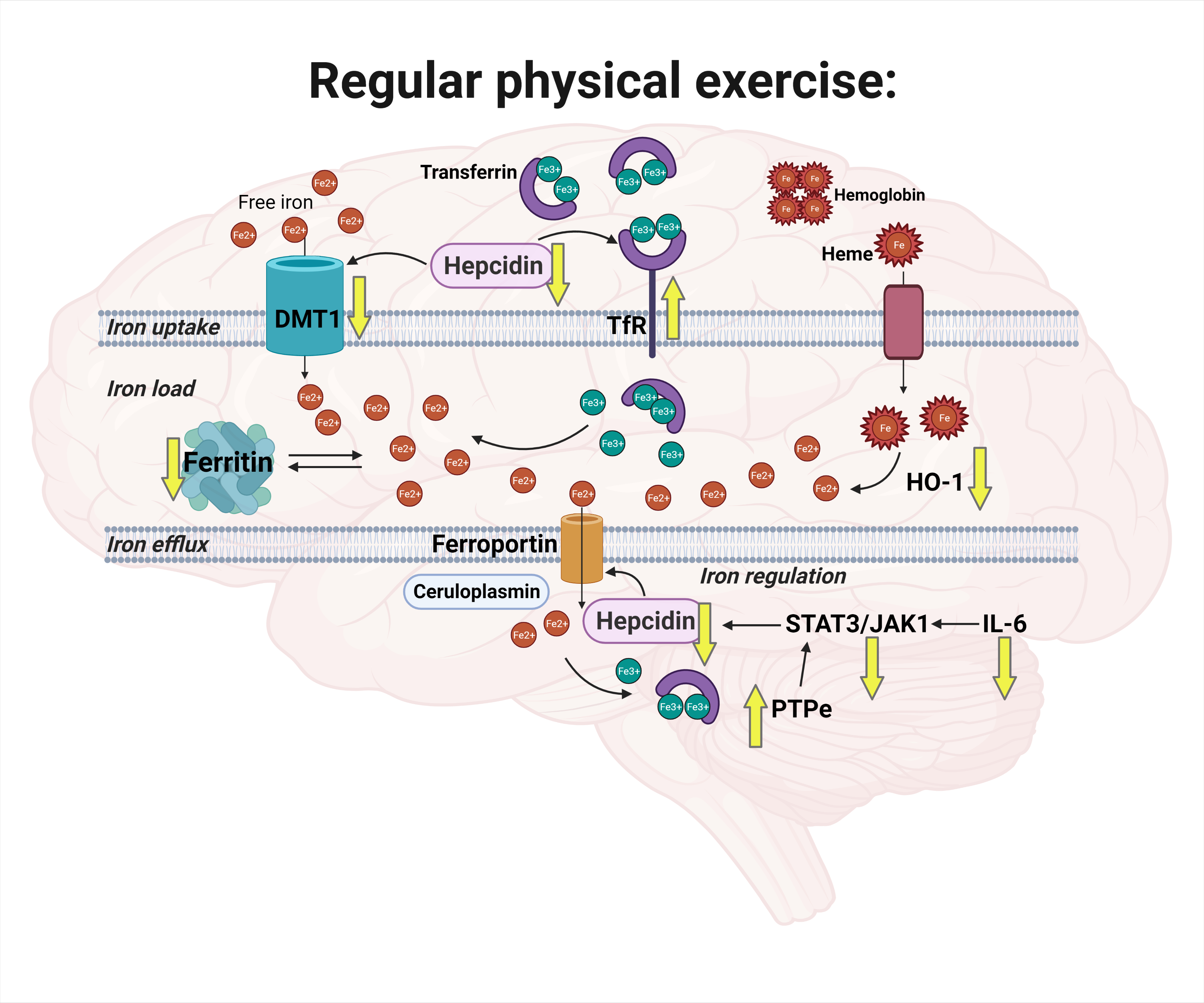
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
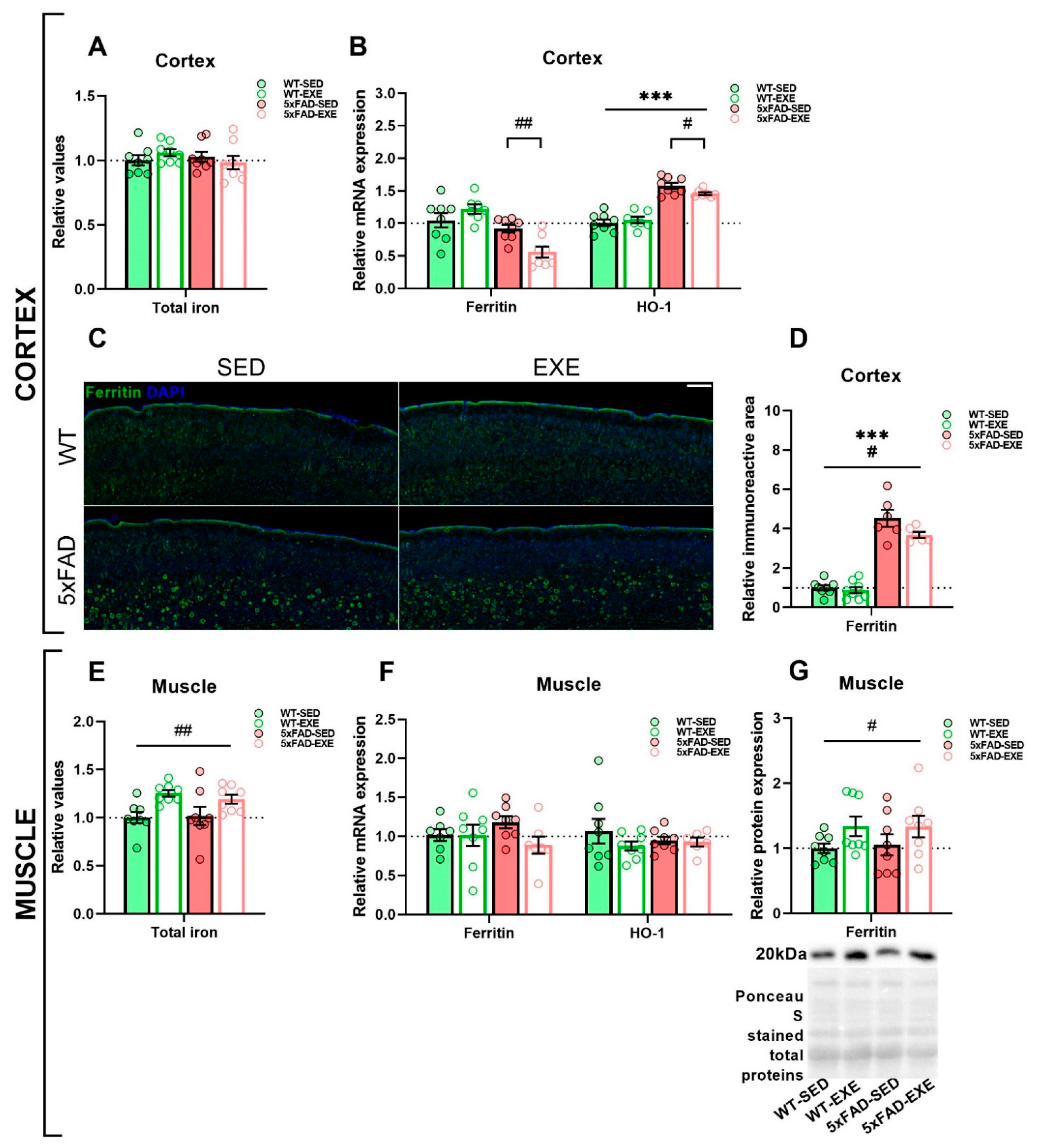
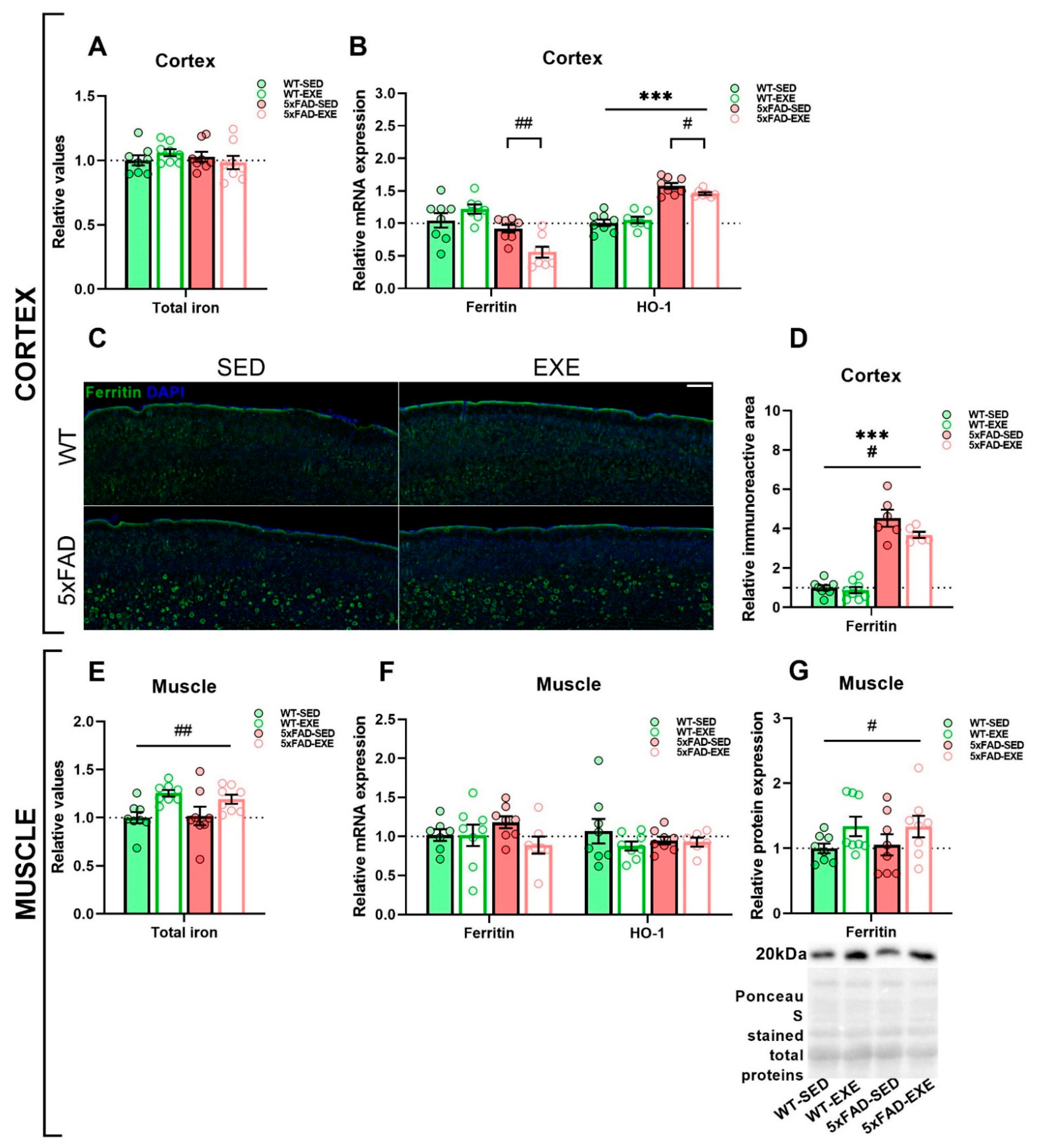
2.1. Exercise Effects on Iron Load in Muscle and Cortex
2.2. Exercise Effects on Iron Trafficking in Muscle and Cortex
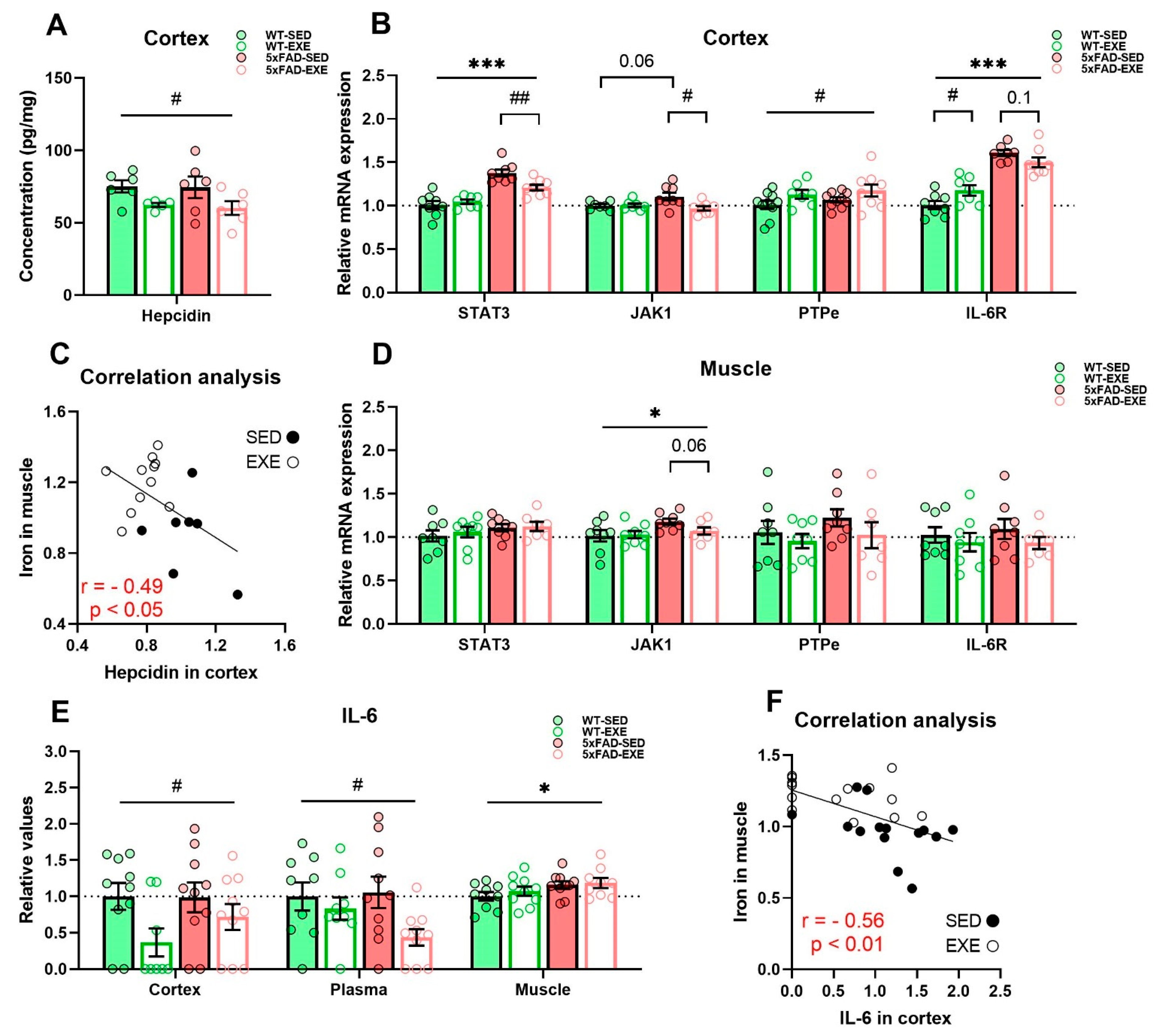
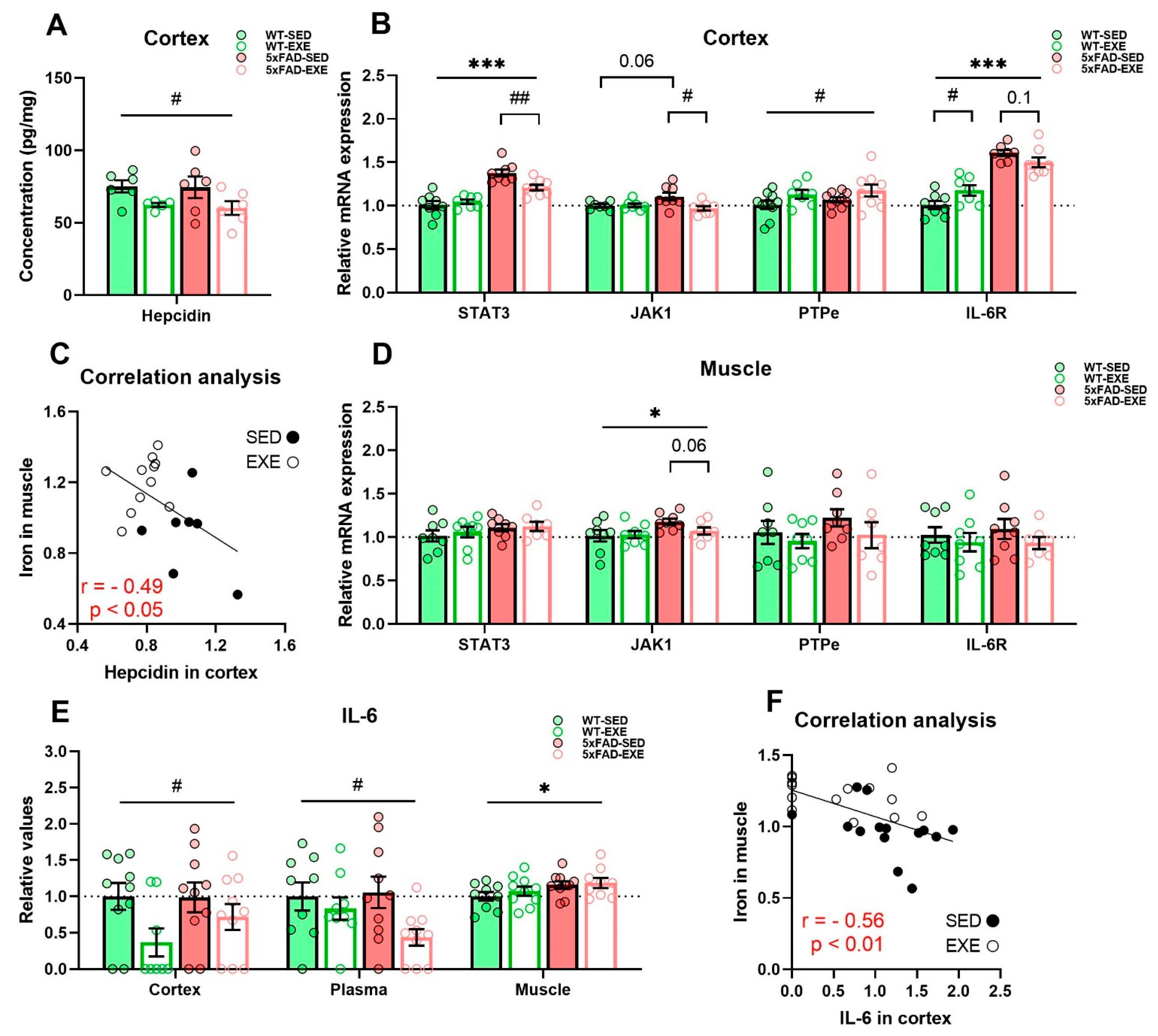
2.3. Exercise Effects on Regulation of Iron Homeostasis in Muscle and Cortex
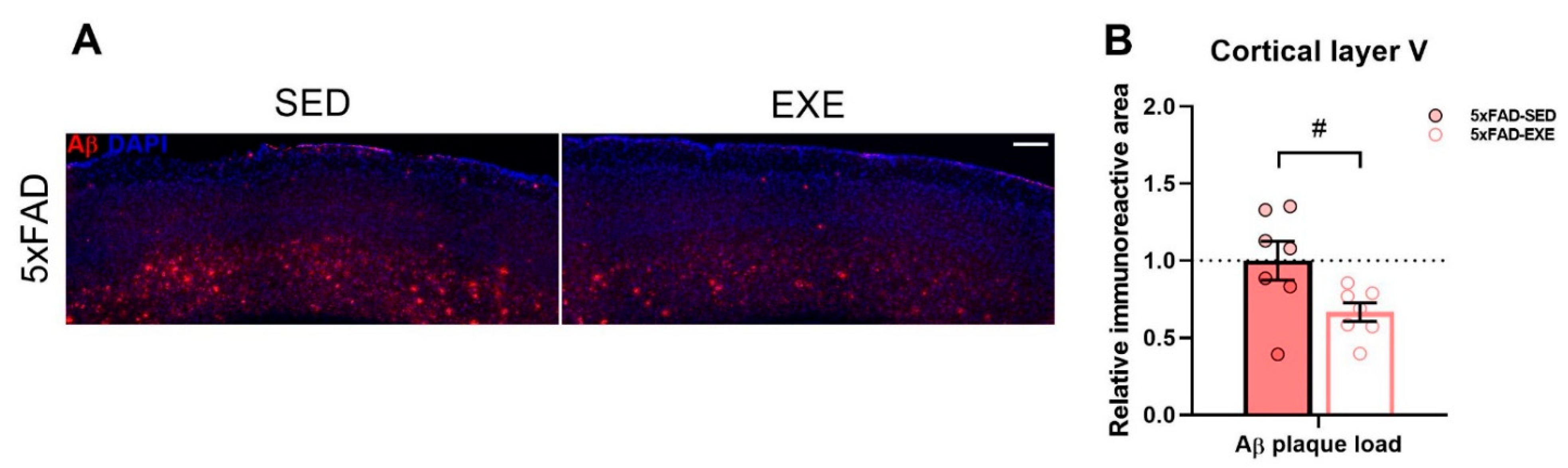
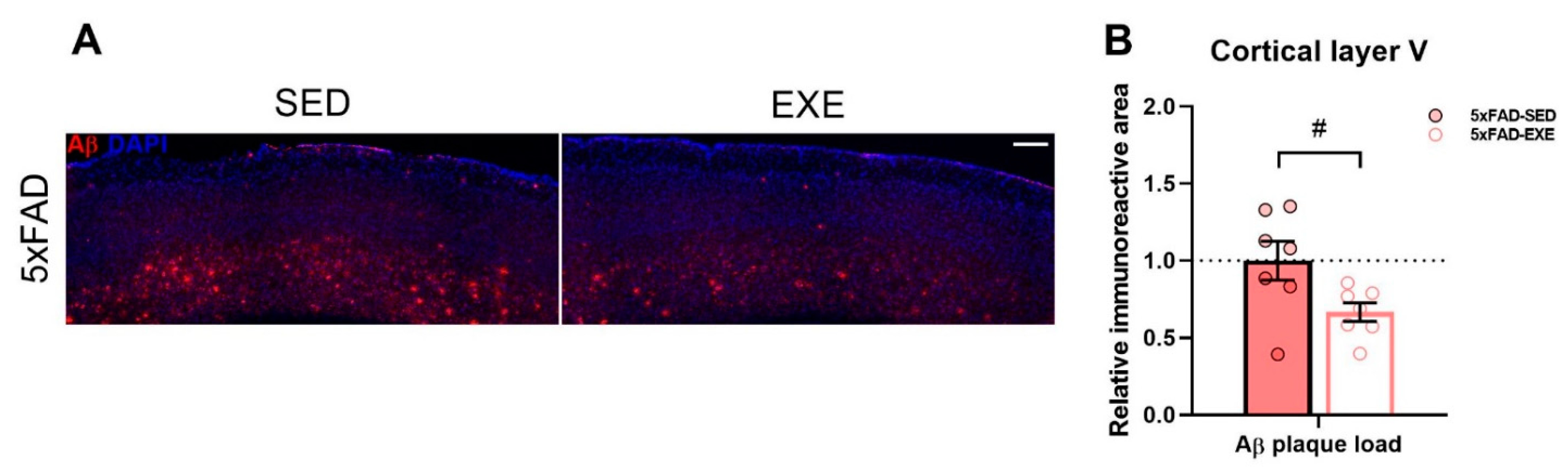
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. Tissue Collection
4.3. Iron Quantitation Via ICP-MS
4.4. Protein and RNA Extraction
4.5. Quantitative Real-Time PCR (qPCR)
4.6. Western Blot
4.7. Enzyme-Linked Immunosorbent Assay (ELISA)
4.8. Cytokine Bead Array (CBA)
4.9. Immunohistochemistry (IHC)
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nelson, P.T.; Braak, H.; Markesbery, W.R. Neuropathology and Cognitive Impairment in Alzheimer Disease: A Complex but Coherent Relationship. J. Neuropathol. Exp. Neurol. 2009, 68, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s Disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Silverman, M.N.; Deuster, P.A. Biological Mechanisms Underlying the Role of Physical Fitness in Health and Resilience. Interface Focus 2014, 4, 20140040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleeson, M.; Bishop, N.C.; Stensel, D.J.; Lindley, M.R.; Mastana, S.S.; Nimmo, M.A. The Anti-Inflammatory Effects of Exercise: Mechanisms and Implications for the Prevention and Treatment of Disease. Nat. Rev. Immunol. 2011, 11, 607–615. [Google Scholar] [CrossRef]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C.; et al. Combined Adult Neurogenesis and BDNF Mimic Exercise Effects on Cognition in an Alzheimer’s Mouse Model. Science 2018, 361, eaan8821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Praag, H.; Shubert, T.; Zhao, C.; Gage, F.H. Exercise Enhances Learning and Hippocampal Neurogenesis in Aged Mice. J. Neurosci. 2005, 25, 8680–8685. [Google Scholar] [CrossRef]
- Ubaida-Mohien, C.; Gonzalez-Freire, M.; Lyashkov, A.; Moaddel, R.; Chia, C.W.; Simonsick, E.M.; Sen, R.; Ferrucci, L. Physical Activity Associated Proteomics of Skeletal Muscle: Being Physically Active in Daily Life May Protect Skeletal Muscle from Aging. Front. Physiol. 2019, 10, 312. [Google Scholar] [CrossRef]
- Deldicque, L. Endoplasmic Reticulum Stress in Human Skeletal Muscle: Any Contribution to Sarcopenia? Front. Physiol. 2013, 4, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ayton, S.; Portbury, S.; Kalinowski, P.; Agarwal, P.; Diouf, I.; Schneider, J.A.; Morris, M.C.; Bush, A.I. Regional Brain Iron Associated with Deterioration in Alzheimer’s Disease: A Large Cohort Study and Theoretical Significance. Alzheimer’s Dement. 2021, 1–13. [Google Scholar]
- Zecca, L.; Youdim, M.B.H.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, Brain Ageing and Neurodegenerative Disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- Ang, E.T.; Tai, Y.K.; Lo, S.Q.; Seet, R.; Soong, T.W. Neurodegenerative Diseases: Exercising toward Neurogenesis and Neuroregeneration. Front. Aging Neurosci. 2010, 2, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, N.C. Iron Homeostasis: Insights from Genetics and Animal Models. Nat. Rev. Genet. 2000, 1, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The Role of Iron in Brain Ageing and Neurodegenerative Disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- Ziolkowski, W.; Ziemann, E.; Hermann-Antosiewicz, A.; Borkowska, A.; Laskowski, R.; Antosiewicz, J. Are the Health Effects of Exercise Related to Changes in Iron Metabolism? Med. J. Nutr. Metab. 2014, 7, 33–43. [Google Scholar] [CrossRef]
- Pedersen, B.K. Physical Activity and Muscle–brain Crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Pedersen, M.; Krabbe, K.S.; Bruunsgaard, H.; Matthews, V.B.; Febbraio, M.A. Role of Exercise-Induced Brain-Derived Neurotrophic Factor Production in the Regulation of Energy Homeostasis in Mammals. Exp. Physiol. 2009, 94, 1153–1160. [Google Scholar] [CrossRef]
- Marsland, A.L.; Gianaros, P.J.; Kuan, D.C.H.; Sheu, L.K.; Krajina, K.; Manuck, S.B. Brain Morphology Links Systemic Inflammation to Cognitive Function in Midlife Adults. Brain Behav. Immun. 2015, 48, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an Endocrine Organ: Focus on Muscle-Derived Interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.H.; Moon, K.M.; Min, K.-W. Exercise-Induced Myokines Can Explain the Importance of Physical Activity in the Elderly: An Overview. Healthcare 2020, 8, 378. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Chennaoui, M.; Gomez-Merino, D.; Drogou, C.; Geoffroy, H.; Dispersyn, G.; Langrume, C.; Ciret, S.; Gallopin, T.; Sauvet, F. Effects of Exercise on Brain and Peripheral Inflammatory Biomarkers Induced by Total Sleep Deprivation in Rats. J. Inflamm. 2015, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Hashiguchi, D.; Campos, H.C.; Wuo-Silva, R.; Faber, J.; Gomes Da Silva, S.; Coppi, A.A.; Arida, R.M.; Longo, B.M. Resistance Exercise Decreases Amyloid Load and Modulates Inflammatory Responses in the APP/PS1 Mouse Model for Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 73, 1525–1539. [Google Scholar] [CrossRef]
- Vela, D. Hepcidin, an Emerging and Important Player in Brain Iron Homeostasis. J. Transl. Med. 2018, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Haertel, C.; Maelicke, A.; Montag, D. Galantamine Slows Down Plaque Formation and Behavioral Decline in the 5XFAD Mouse Model of Alzheimer’s Disease. PLoS ONE 2014, 9, e89454. [Google Scholar]
- Belaya, I.; Ivanova, M.; Sorvari, A.; Ilicic, M.; Loppi, S.; Koivisto, H.; Varricchio, A.; Tikkanen, H.; Walker, F.R.; Atalay, M.; et al. Astrocyte Remodeling in the Beneficial Effects of Long-Term Voluntary Exercise in Alzheimer’s Disease. J. Neuroinflamm. 2020, 17, 271. [Google Scholar] [CrossRef] [PubMed]
- Moos, T.; Nielsen, T.R.; Skjørringe, T.; Morgan, E.H. Iron Trafficking inside the Brain. J. Neurochem. 2007, 103, 1730–1740. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.D.; Donovan, A.; Ward, D.M.V.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kezele, T.G.; Ćurko-Cofek, B. Age-Related Changes and Sex-Related Differences in Brain Iron Metabolism. Nutrients 2020, 12, 2601. [Google Scholar] [CrossRef] [PubMed]
- You, L.H.; Yan, C.Z.; Zheng, B.J.; Ci, Y.Z.; Chang, S.Y.; Yu, P.; Gao, G.F.; Li, H.Y.; Dong, T.Y.; Chang, Y.Z. Astrocyte Hepcidin Is a Key Factor in Lps-Induced Neuronal Apoptosis. Cell Death Dis. 2017, 8, e2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanuma, N.; Nakamura, K.; Shima, H.; Kikuchi, K. Protein-Tyrosine Phosphatase PTPεC Inhibits Jak-STAT Signaling and Differentiation Induced by Interleukin-6 and Leukemia Inhibitory Factor in M1 Leukemia Cells. J. Biol. Chem. 2000, 275, 28216–28221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Qu, C.K. Protein Tyrosine Phosphatases in the JAK/STAT Pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, D.G.; Connor, J.R.; Meadowcroft, M.D. The Relationship between Iron Dyshomeostasis and Amyloidogenesis in Alzheimer’s Disease: Two Sides of the Same Coin. Neurobiol. Dis. 2015, 81, 49–65. [Google Scholar] [CrossRef] [Green Version]
- Kabir, M.T.; Uddin, M.S.; Zaman, S.; Begum, Y.; Ashraf, G.M.; Bin-Jumah, M.N.; Bungau, S.G.; Mousa, S.A.; Abdel-Daim, M.M. Molecular Mechanisms of Metal Toxicity in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Buratti, P.; Gammella, E.; Rybinska, I.; Cairo, G.; Recalcati, S. Recent Advances in Iron Metabolism: Relevance for Health, Exercise, and Performance. Med. Sci. Sports Exerc. 2015, 47, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Falangola, M.F.; Lee, S.P.; Nixon, R.A.; Duff, K.; Helpern, J.A. Histological Co-Localization of Iron in Aβ Plaques of PS/APP Transgenic Mice. Neurochem. Res. 2005, 30, 201–205. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Bush, A.I. Iron Neurochemistry in Alzheimer’s Disease and Parkinson’s Disease: Targets for Therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef] [Green Version]
- Meadowcroft, M.D.; Connor, J.R.; Yang, Q.X. Cortical Iron Regulation and Inflammatory Response in Alzheimer’s Disease and APPSWE/PS1ΔE9 Mice: A Histological Perspective. Front. Neurosci. 2015, 9, 255. [Google Scholar] [CrossRef] [Green Version]
- Svobodová, H.; Kosnáč, D.; Balázsiová, Z.; Tanila, H.; Miettinen, P.O.; Sierra, A.; Vitovič, P.; Wagner, A.; Polák, S.; Kopáni, M. Elevated Age-Related Cortical Iron, Ferritin and Amyloid Plaques in APPswe/PS1ΔE9 Transgenic Mouse Model of Alzheimer’s Disease. Physiol. Res. 2019, 68, 445–451. [Google Scholar] [CrossRef]
- Choi, D.H.; Kwon, K.C.; Hwang, D.J.; Koo, J.H.; Um, H.S.; Song, H.S.; Kim, J.S.; Jang, Y.; Cho, J.Y. Treadmill Exercise Alleviates Brain Iron Dyshomeostasis Accelerating Neuronal Amyloid-β Production, Neuronal Cell Death, and Cognitive Impairment in Transgenic Mice Model of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3208–3223. [Google Scholar] [CrossRef]
- Yu, W.J.; An, S.J.; Shao, T.M.; Xu, H.J.; Chen, H.X.; Ning, J.D.; Zhou, Y.J.; Chai, X.Q. Active Compounds of Herbs Ameliorate Impaired Cognition in APP/PS1 Mouse Model of Alzheimer’s Disease. Aging Albany 2019, 11, 11186–11201. [Google Scholar] [CrossRef]
- Ashraf, A.; Clark, M.; So, P.W. The Aging of Iron Man. Front. Aging Neurosci. 2018, 10, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, R.L.; Liu, M.; Choi, D.Y.; Cass, W.A.; Bing, G. Inflammation and Age-Related Iron Accumulation in F344 Rats. Curr. Aging Sci. 2008, 1, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Gurel, B.; Cansev, M.; Sevinc, C.; Kelestemur, S.; Ocalan, B.; Cakir, A.; Aydin, S.; Kahveci, N.; Ozansoy, M.; Taskapilioglu, O.; et al. Early Stage Alterations in CA1 Extracellular Region Proteins Indicate Dysregulation of IL6 and Iron Homeostasis in the 5XFAD Alzheimer’s Disease Mouse Model. J. Alzheimer’s Dis. 2018, 61, 1399–1410. [Google Scholar] [CrossRef]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor Deficits, Neuron Loss, and Reduced Anxiety Coinciding with Axonal Degeneration and Intraneuronal Aβ Aggregation in the 5XFAD Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2012, 33, 196.e29–196.e40. [Google Scholar] [CrossRef]
- Halon-Golabek, M.; Borkowska, A.; Herman-Antosiewicz, A.; Antosiewicz, J. Iron Metabolism of the Skeletal Muscle and Neurodegeneration. Front. Neurosci. 2019, 13, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasi, U.; Abbina, S.; Gill, A.; Bhagat, V.; Kizhakkedathu, J.N. A Facile Colorimetric Method for the Quantification of Labile Iron Pool and Total Iron in Cells and Tissue Specimens. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrecht, C.; Mühlenhoff, U. The Role of Mitochondria in Cellular Iron–sulfur Protein Biogenesis and Iron Metabolism. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1491–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Zhou, Q.; Wu, D.; Chen, L. Mitochondrial Iron Metabolism and Its Role in Diseases. Clin. Chim. Acta 2021, 513, 6–12. [Google Scholar] [CrossRef]
- Egan, B.; Zierath, J.R. Exercise Metabolism and the Molecular Regulation of Skeletal Muscle Adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [Green Version]
- Ghio, A.J.; Soukup, J.M.; Ghio, C.; Gordon, C.J.; Richards, J.E.; Schladweiler, M.C.; Snow, S.J.; Kodavanti, U.P. Iron and Zinc Homeostases in Female Rats with Physically Active and Sedentary Lifestyles. BioMetals 2020, 34, 97–105. [Google Scholar] [CrossRef]
- Ward, D.M.; Kaplan, J. Ferroportin-Mediated Iron Transport: Expression and Regulation. Biochim. Biophys. Acta 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [Green Version]
- Xian-Hui, D.; Wei-Juan, G.; Tie-Mei, S.; Hong-Lin, X.; Jiang-Tao, B.; Jing-Yi, Z.; Xi-Qing, C. Age-Related Changes of Brain Iron Load Changes in the Frontal Cortex in APPswe/PS1δE9 Transgenic Mouse Model of Alzheimer’s Disease. J. Trace Elem. Med. Biol. 2015, 30, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Duan, X.; Liu, J.; Zhao, H.; Liu, Y.; Chang, Y. Nitric Oxide Contributes to the Regulation of Iron Metabolism in Skeletal Muscle in vivo and in vitro. Mol. Cell. Biochem. 2010, 342, 87–94. [Google Scholar] [CrossRef]
- Wolff, N.A.; Ghio, A.J.; Garrick, L.M.; Garrick, M.D.; Zhao, L.; Fenton, R.A.; Thévenod, F. Evidence for Mitochondrial Localization of Divalent Metal Transporter 1 (DMT1). FASEB J. 2014, 28, 2134–2145. [Google Scholar] [CrossRef]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thévenod, F. A Role for Divalent Metal Transporter (DMT1) in Mitochondrial Uptake of Iron and Manganese. Sci. Rep. 2018, 8, 1–12. [Google Scholar]
- Du, F.; Qian, C.; Ming Qian, Z.; Wu, X.M.; Xie, H.; Yung, W.H.; Ke, Y. Hepcidin Directly Inhibits Transferrin Receptor 1 Expression in Astrocytes via a Cyclic AMP-Protein Kinase a Pathway. Glia 2011, 59, 936–945. [Google Scholar] [CrossRef]
- Raha, A.A.; Vaishnav, R.A.; Friedland, R.P.; Bomford, A.; Raha-Chowdhury, R. The Systemic Iron-Regulatory Proteins Hepcidin and Ferroportin Are Reduced in the Brain in Alzheimer’s Disease. Acta Neuropathol. Commun. 2014, 2, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleszar, M.G.; Wiśniewski, J.; Berdowska, I.; Zieliński, B.; Zboch, M.; Diakowska, D.; Gamian, A.; Krzystek-Korpacka, M. Systemic Hepcidin Quantified with LC–MS/MS in Dementia in Association with Disease Pathology and Severity and with Structural Changes in the Brain. Peptides 2019, 122, 170169. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, Z.; Hu, Z.; Sternberg, D.; Waseh, S.; Quinn, J.F.; Wild, K.; Kaye, J.; Zhao, L.; Garrick, M. Serum Hepcidin Levels, Iron Dyshomeostasis and Cognitive Loss in Alzheimer’s Disease. Aging Dis. 2017, 8, 215–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kweon, O.J.; Youn, Y.C.; Lim, Y.K.; Lee, M.K.; Kim, H.R. Clinical Utility of Serum Hepcidin and Iron Profile Measurements in Alzheimer’s Disease. J. Neurol. Sci. 2019, 403, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Mohammadi, M.; Goozee, K.; Shah, T.M.; Sohrabi, H.R.; Dias, C.B.; Shen, K.; Asih, P.R.; Dave, P.; Pedrini, S.; et al. Serum Hepcidin Levels in Cognitively Normal Older Adults with High Neocortical Amyloid-β Load. J. Alzheimer’s Dis. 2020, 76, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Fu, L.J.; Duan, X.L.; Crooks, D.R.; Yu, P.; Qian, Z.M.; Di, X.J.; Li, J.; Rouault, T.A.; Chang, Y.Z. Role of Hepcidin in Murine Brain Iron Metabolism. Cell. Mol. Life Sci. 2010, 67, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Nay, K.; Smiles, W.J.; Kaiser, J.; McAloon, L.M.; Loh, K.; Galic, S.; Oakhill, J.S.; Gundlach, A.L.; Scott, J.W. Molecular Mechanisms Underlying the Beneficial Effects of Exercise on Brain Function and Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 4052. [Google Scholar] [CrossRef]
- Cavey, T.; Pierre, N.; Nay, K.; Allain, C.; Ropert, M.; Loréal, O.; Derbré, F. Simulated Microgravity Decreases Circulating Iron in Rats: Role of Inflammation-Induced Hepcidin Upregulation. Exp. Physiol. 2017, 102, 291–298. [Google Scholar] [CrossRef]
- Xu, Z.; Sun, W.; Li, Y.; Ling, S.; Zhao, C.; Zhong, G.; Zhao, D.; Song, J.; Song, H.; Li, J.; et al. The Regulation of Iron Metabolism by Hepcidin Contributes to Unloading-Induced Bone Loss. Bone 2017, 94, 152–161. [Google Scholar] [CrossRef]
- Nay, K.; Koechlin-Ramonatxo, C.; Rochdi, S.; Island, M.L.; Orfila, L.; Treffel, L.; Bareille, M.P.; Beck, A.; Gauquelin-Koch, G.; Ropert, M.; et al. Simulated Microgravity Disturbs Iron Metabolism and Distribution in Humans: Lessons from Dry Immersion, an Innovative Ground-Based Human Model. FASEB J. 2020, 34, 14920–14929. [Google Scholar] [CrossRef]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 Mediates Hypoferremia of Inflammation by Inducing the Synthesis of the Iron Regulatory Hormone Hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, S.X.; Williams, L.J.; Holloway-Kew, K.L.; Hyde, N.K.; Pasco, J.A. Skeletal Muscle Health and Cognitive Function: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 255. [Google Scholar] [CrossRef]
- Hampel, H.; Haslinger, A.; Scheloske, M.; Padberg, F.; Fischer, P.; Unger, J.; Teipel, S.J.; Neumann, M.; Rosenberg, C.; Oshida, R.; et al. Pattern of Interleukin-6 Receptor Complex Immunoreactivity between Cortical Regions of Rapid Autopsy Normal and Alzheimer’s Disease Brain. Eur. Arch. Psychiatry Clin. Neurosci. 2005, 255, 269–278. [Google Scholar] [CrossRef]
- Manji, Z.; Rojas, A.; Wang, W.; Dingledine, R.; Varvel, N.H.; Ganesh, T. 5xFAD Mice Display Sex-Dependent Inflammatory Gene Induction during the Prodromal Stage of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 70, 1259–1274. [Google Scholar] [CrossRef]
- Colbert, L.H.; Visser, M.; Simonsick, E.M.; Tracy, R.P.; Newman, A.B.; Kritchevsky, S.B.; Pahor, M.; Taaffe, D.R.; Brach, J.; Rubin, S.; et al. Physical Activity, Exercise, and Inflammatory Markers in Older Adults: Findings from the Health, Aging and Body Composition Study. J. Am. Geriatr. Soc. 2004, 52, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Sellami, M.; Bragazzi, N.L.; Aboghaba, B.; Elrayess, M.A. The Impact of Acute and Chronic Exercise on Immunoglobulins and Cytokines in Elderly: Insights from a Critical Review of the Literature. Front. Immunol. 2021, 12, 631873. [Google Scholar] [CrossRef]
- Zheng, G.; Qiu, P.; Xia, R.; Lin, H.; Ye, B.; Tao, J.; Chen, L. Effect of Aerobic Exercise on Inflammatory Markers in Healthy Middle-Aged and Older Adults: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Aging Neurosci. 2019, 11, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thielen, J.W.; Kärgel, C.; Müller, B.W.; Rasche, I.; Genius, J.; Bus, B.; Maderwald, S.; Norris, D.G.; Wiltfang, J.; Tendolkar, I. Aerobic Activity in the Healthy Elderly Is Associated with Larger Plasticity in Memory Related Brain Structures and Lower Systemic Inflammation. Front. Aging Neurosci. 2016, 8, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento, C.; Pereira, J.; Andrade, L.; Garuffi, M.; Talib, L.; Forlenza, O.; Cancela, J.; Cominetti, M.; Stella, F. Physical Exercise in MCI Elderly Promotes Reduction of Pro-Inflammatory Cytokines and Improvements on Cognition and BDNF Peripheral Levels. Curr. Alzheimer Res. 2014, 11, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Lavin, K.M.; Perkins, R.K.; Jemiolo, B.; Raue, U.; Trappe, S.W.; Trappe, T.A. Effects of Aging and Lifelong Aerobic Exercise on Basal and Exercise-Induced Inflammation. J. Appl. Physiol. 2020, 128, 87–99. [Google Scholar] [CrossRef]
- Choo, X.Y.; Liddell, J.R.; Huuskonen, M.T.; Grubman, A.; Moujalled, D.; Roberts, J.; Kysenius, K.; Patten, L.; Quek, H.; Oikari, L.E.; et al. CuII (Atsm) Attenuates Neuroinflammation. Front. Neurosci. 2018, 12, 668. [Google Scholar] [CrossRef]
- Kysenius, K.; Paul, B.; Hilton, J.B.; Liddell, J.R.; Hare, D.J.; Crouch, P.J. A Versatile Quantitative Microdroplet Elemental Imaging Method Optimised for Integration in Biochemical Workflows for Low-Volume Samples. Anal. Bioanal. Chem. 2019, 411, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Hilton, J.B.; Mercer, S.W.; Lim, N.K.H.; Faux, N.G.; Buncic, G.; Beckman, J.S.; Roberts, B.R.; Donnelly, P.S.; White, A.R.; Crouch, P.J. CuII (Atsm) Improves the Neurological Phenotype and Survival of SOD1G93A Mice and Selectively Increases Enzymatically Active SOD1 in the Spinal Cord. Sci. Rep. 2017, 7, 1–11. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belaya, I.; Kucháriková, N.; Górová, V.; Kysenius, K.; Hare, D.J.; Crouch, P.J.; Malm, T.; Atalay, M.; White, A.R.; Liddell, J.R.; et al. Regular Physical Exercise Modulates Iron Homeostasis in the 5xFAD Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 8715. https://doi.org/10.3390/ijms22168715
Belaya I, Kucháriková N, Górová V, Kysenius K, Hare DJ, Crouch PJ, Malm T, Atalay M, White AR, Liddell JR, et al. Regular Physical Exercise Modulates Iron Homeostasis in the 5xFAD Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(16):8715. https://doi.org/10.3390/ijms22168715
Chicago/Turabian StyleBelaya, Irina, Nina Kucháriková, Veronika Górová, Kai Kysenius, Dominic J. Hare, Peter J. Crouch, Tarja Malm, Mustafa Atalay, Anthony R. White, Jeffrey R. Liddell, and et al. 2021. "Regular Physical Exercise Modulates Iron Homeostasis in the 5xFAD Mouse Model of Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 16: 8715. https://doi.org/10.3390/ijms22168715
APA StyleBelaya, I., Kucháriková, N., Górová, V., Kysenius, K., Hare, D. J., Crouch, P. J., Malm, T., Atalay, M., White, A. R., Liddell, J. R., & Kanninen, K. M. (2021). Regular Physical Exercise Modulates Iron Homeostasis in the 5xFAD Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences, 22(16), 8715. https://doi.org/10.3390/ijms22168715