
FAM20C Overview: Classic and Novel Targets, Pathogenic Variants and Raine Syndrome Phenotypes

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. FAM20 Family
2.1. FAM20A
2.2. FAM20B
2.3. FAM20C
3. FAM20C Biological Function in Bone: SIBLING Proteins
3.1. OPN
3.2. DMP1
3.3. MEPE
3.4. BSP
4. FAM20C in Kidney and Parathyroid Gland
4.1. FGF23 and 1,25(OH)2D3
4.2. PTH
5. FAM20C Biological Functions in Non-Mineralized Tissues
5.1. FAM20C in Neural Tissue: Neuropeptides
5.2. FAM20C in Neural and Cardiovascular Tissues: Sortilin
5.3. FAM20C in Brain: Sortilin 1 and Sonic Hedgehog (SHH)
5.4. FAM20C in Cardiac Tissue: HRC, STIM1 and CSQ2
5.5. FAM20C in Metabolism: PCSK7 and PCSK9
5.6. FAM20C in Endoplasmic Reticulum–Redox Homeostasis: ERO1a
5.7. FAM20C in Clotting: vWF
5.8. FAM20C in Salivary Glands: BMP4
6. FAM20C Pathogenic Variants
7. Clinical Aspects of RS
7.1. Lethal Raine Syndrome
7.2. Craniofacial and Skeletal Features
7.3. Extra-Skeletal Features
7.4. Survival
8. Non-Lethal RS Phenotype
8.1. NLRS Craniofacial and Skeletal Features
8.2. Extra-Craniofacial Skeletal Features
8.3. Extra-Craniofacial and Skeletal Features
9. Differences between Lethal and Non-Lethal RS
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simpson, M.A.; Hsu, R.; Keir, L.S.; Hao, J.; Sivapalan, G.; Ernst, L.M.; Zackai, E.H.; Al-Gazali, L.I.; Hulskamp, G.; Kingston, H.M.; et al. Mutations in FAM20C Are Associated with Lethal Osteosclerotic Bone Dysplasia (Raine Syndrome), Highlighting a Crucial Molecule in Bone Development. Am. J. Hum. Genet. 2007, 81, 906–912. [Google Scholar] [CrossRef]
- Raine, A.; Allbutt, J. Factors of schizoid personality. Br. J. Clin. Psychol. 1989, 28, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Faundes, V.; Castillo-Taucher, S.; Gonzalez-Hormazabal, P.; Chandler, K.; Crosby, A.; Chioza, B. Raine syndrome: An overview. Eur. J. Med. Genet. 2014, 57, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Tada, K.; Takeuchi, A.; Nakamura, M.; Marunaka, H.; Washio, Y.; Tanaka, H.; Miya, F.; Okamoto, N.; Kageyama, M. Fetal ultrasonographic findings including cerebral hyperechogenicity in a patient with non-lethal form of Raine syndrome. Am. J. Med. Genet. Part A 2018, 176, 682–686. [Google Scholar] [CrossRef]
- Rolvien, T.; Kornak, U.; Schinke, T.; Amling, M.; Oheim, R. A novel FAM20C mutation causing hypophosphatemic osteomalacia with osteosclerosis (mild Raine syndrome) in an elderly man with spontaneous osteonecrosis of the knee. Osteoporos. Int. 2019, 30, 685–689. [Google Scholar] [CrossRef]
- Sheth, J.; Bhavsar, R.; Gandhi, A.; Sheth, F.; Pancholi, D. A case of Raine syndrome presenting with facial dysmorphy and review of literature. BMC Med. Genet. 2018, 19, 76. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.J.; Kolawole, T.M.; Al-Mofada, S.; Malabarey, T.M.; Hulailah, A. Osteopetrosis: Brain ultrasound and computed tomography findings. Eur. J. Pediatr. 1992, 151, 827–828. [Google Scholar] [CrossRef]
- Rickert, C.H.; Rieder, H.; Rehder, H.; Hülskamp, G.; Hörnig-Franz, I.; Louwen, F.; Paulus, W. Neuropathology of Raine syndrome. Acta Neuropathol. 2002, 103, 281–287. [Google Scholar] [CrossRef]
- Chitayat, D.; Shannon, P.; Keating, S.; Toi, A.; Blaser, S.; Friedberg, T.; Superti-Furga, A.; Chong, K.; Unger, S. Raine syndrome: A rare lethal osteosclerotic bone dysplasia. Prenatal diagnosis, autopsy, and neuropathological findings. Am. J. Med. Genet. 2007, 143, 3280–3285. [Google Scholar] [CrossRef]
- Michael, K.; Nelson, D.M.; Ortmeier, A. Raine syndrome. J. Diagn. Med. Sonogr. 2011, 27, 167–170. [Google Scholar] [CrossRef]
- Koob, M.; Doray, B.; Fradin, M.; Astruc, D.; Dietemann, J.L. Raine syndrome: Expanding the radiological spectrum. Pediatr. Radiol. 2011, 41, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.O.; Xu, A.; Ogura, E.; Manning, G.; Irvine, K.D. The raine syndrome protein FAM20C is a golgi kinase that phosphorylates bio-mineralization proteins. PLoS ONE 2012, 7, e42988. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Wiley, S.E.; Guo, X.; Kinch, L.N.; Durrant, E.; Wen, J.; Xiao, J.; Cui, J.; Nguyen, K.B.; Engel, J.L.; et al. A Single Kinase Generates the Majority of the Secreted Phosphoproteome. Cell 2015, 161, 1619–1632. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yan, W.; Tian, Y.; Ma, P.; Opperman, L.A.; Wang, X. Family with sequence similarity member 20C is the primary but not the only kinase for the small-integrin-binding ligand N-linked glycoproteins in bone. FASEB J. 2016, 30, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Sreelatha, A.; Kinch, L.N.; Tagliabracci, V.S. The secretory pathway kinases. Biochim. Biophys. Acta. Proteins Proteom. 2015, 1854, 1687–1693. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xia, Y.; Chen, B.-Y.; Sun, X.-L.; Duan, L.; Gao, G.-D.; Wang, J.-J.; Kam-Lin Yung, K.; Chen, L.-W. Presence of proNGF-Sortilin Signaling Complex in Nigral Dopamine Neurons and Its Variation in Relation to Aging, Lactacystin and 6-OHDA Insults. Int. J. Mol. Sci. 2013, 14, 14085–14104. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, H.; Jani, P.; Wang, X.; Lu, Y.; Li, N.; Xiao, J.; Qin, C. FAM20C regulates osteoblast behaviors and intracellular signaling pathways in a cell-autonomous manner. J. Cell. Physiol. 2018, 233, 3476–3486. [Google Scholar] [CrossRef]
- Nalbant, D.; Youn, H.; Nalbant, S.I.; Sharma, S.; Cobos, E.; Beale, E.G.; Du, Y.; Williams, S.C. FAM20: An evolutionarily conserved family of secreted proteins expressed in hematopoietic cells. BMC Genom. 2005, 6, 11. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Kaewgahya, M.; Khemaleelakul, U.; Dejkhamron, P.; Sutthimethakorn, S.; Thongboonkerd, V.; Iamaroon, A. Enamel-renal-gingival syndrome and FAM20A mutations. Am. J. Med. Genet. Part A 2014, 164, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Izumikawa, T.; Tamura, J.I.; Kitagawa, H. FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem. J. 2009, 421, 157–162. [Google Scholar] [CrossRef]
- Wen, J.; Xiao, J.; Rahdar, M.; Choudhury, B.P.; Cui, J.; Taylor, G.S.; Esko, J.D.; Dixon, J.E. Xylose phosphorylation functions as a molecular switch to regulate proteoglycan biosynthesis. Proc. Natl. Acad. Sci. USA 2014, 111, 15723–15728. [Google Scholar] [CrossRef] [PubMed]
- Paganini, C.; Costantini, R.; Superti-Furga, A.; Rossi, A. Bone and connective tissue disorders caused by defects in glycosaminoglycan biosynthesis: A panoramic view. FEBS J. 2019, 286, 3008–3032. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Murakami, H.; Enomoto, Y.; Tsurusaki, Y.; Takahashi, K.; Mitsuzuka, K.; Ishimoto, H.; Nishimura, G.; Kurosawa, K. A novel gene (FAM20B encoding glycosaminoglycan xylosylkinase) for neonatal short limb dysplasia resembling Desbuquois dysplasia. Clin. Genet. 2019, 95, 713–717. [Google Scholar] [CrossRef]
- Xiao, J.; Tagliabracci, V.S.; Wen, J.; Kim, S.A.; Dixon, J.E. Crystal structure of the Golgi casein kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 10574–10579. [Google Scholar] [CrossRef] [PubMed]
- Tagliabracci, V.S.; Engel, J.L.; Wen, J.; Wiley, S.E.; Worby, C.A.; Kinch, L.N.; Xiao, J.; Grishin, N.V.; Dixon, J.E. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science 2012, 336, 1150–1153. [Google Scholar] [CrossRef]
- Cozza, G.; Pinna, L.A. Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2016, 20, 319–340. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Pinna, L.A.; Dixon, J.E. Secreted protein kinases. Trends Biochem. Sci. 2013, 38, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Ma, S.; Zhang, H.; Liu, C.; Lu, Y.; Chen, L.; Qin, C. Specific ablation of mouse Fam20C in cells expressing type i collagen leads to skeletal defects and hypophosphatemia. Sci. Rep. 2017, 7, 3590. [Google Scholar] [CrossRef]
- Wilson, C.; Naves, T.; Saada, S.; Pinet, S.; Vincent, F.; Lalloue, F.; Jauberteau, M.-O. The Implications of Sortilin/Vps10p Domain Receptors in Neurological and Human Diseases. CNS Neurol. Disord. Drug Targets 2014, 13, 1354–1365. [Google Scholar] [CrossRef]
- Boskey, A.L.; Maresca, M.; Ullrich, W.; Doty, S.B.; Butler, W.T.; Prince, C.W. Osteopontin-hydroxyapatite interactions in vitro: Inhibition of hydroxyapatite formation and growth in a gelatin-gel. Bone Miner. 1993, 22, 147–159. [Google Scholar] [CrossRef]
- Poundarik, A.A.; Boskey, A.; Gundberg, C.; Vashishth, D. Biomolecular Regulation, Composition and Nanoarchitecture of Bone Mineral. Sci. Rep. 2018, 8, 1191. [Google Scholar] [CrossRef]
- Gericke, A.; Qin, C.; Spevak, L.; Fujimoto, Y.; Butler, W.T.; Sørensen, E.S.; Boskey, A.L. Importance of phosphorylation for osteopontin regulation of biomineralization. Calcif. Tissue Int. 2005, 77, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; David, V.; Laurence, J.S.; Schwarz, P.M.; Lafer, E.M.; Hedge, A.M.; Rowe, P.S.N. Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-Peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology 2008, 149, 1757–1772. [Google Scholar] [CrossRef] [PubMed]
- Holm, E.; Gleberzon, J.S.; Liao, Y.; Sørensen, E.S.; Beier, F.; Hunter, G.K.; Goldberg, H.A. Osteopontin mediates mineralization and not osteogenic cell development in vitro. Biochem. J. 2014, 464, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24. [Google Scholar] [CrossRef]
- Rittling, S.R.; Matsumoto, H.N.; Mckee, M.D.; Nanci, A.; An, X.R.; Novick, K.E.; Kowalski, A.J.; Noda, M.; Denhardt, D.T. Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J. Bone Miner. Res. 1998, 13, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Tibaldi, E.; Brocca, A.; Sticca, A.; Gola, E.; Pizzi, M.; Bordin, L.; Pagano, M.A.; Mazzorana, M.; Donà, G.; Violi, P.; et al. Fam20C-mediated phosphorylation of osteopontin is critical for its secretion but dispensable for its action as a cytokine in the activation of hepatic stellate cells in liver fibrogenesis. FASEB J. 2020, 34, 1122–1135. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, K.; Rittling, S.R.; Tsuji, K.; Kurosawa, H.; Nifuji, A.; Denhardt, D.T.; Noda, M. Osteopontin Deficiency Induces Parathyroid Hormone Enhancement of Cortical Bone Formation. Endocrinology 2003, 144, 2132–2140. [Google Scholar] [CrossRef][Green Version]
- Gericke, A.; Qin, C.; Sun, Y.; Redfern, R.; Redfern, D.; Fujimoto, Y.; Taleb, H.; Butler, W.T.; Boskey, A.L. Different forms of DMP1 play distinct roles in mineralization. J. Dent. Res. 2010, 89, 355–359. [Google Scholar] [CrossRef]
- Suzuki, S.; Haruyama, N.; Nishimura, F.; Kulkarni, A.B. Dentin sialophosphoprotein and dentin matrix protein-1: Two highly phosphorylated proteins in mineralized tissues. Arch. Oral Biol. 2012, 57, 1165–1175. [Google Scholar] [CrossRef]
- Qin, C.; Brunn, J.C.; Cook, R.G.; Orkiszewski, R.S.; Malone, J.P.; Veis, A.; Butler, W.T. Evidence for the proteolytic processing of dentin matrix protein 1: Identification and characterization of processed fragments and cleavage sites. J. Biol. Chem. 2003, 278, 34700–34708. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.; Xie, Y.; Yuan, B.; Yu, X.; Rauch, F.; Davis, S.I.; Zhang, S.; et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315. [Google Scholar] [CrossRef]
- Lu, Y.; Yuan, B.; Qin, C.; Cao, Z.; Xie, Y.; Dallas, S.L.; McKee, M.D.; Drezner, M.K.; Bonewald, L.F.; Feng, J.Q. The biological function of DMP-1 in osteocyte maturation is mediated by its 57-kDa c-terminal fragment. J. Bone Miner. Res. 2011, 26, 331–340. [Google Scholar] [CrossRef]
- Farrow, E.G.; Davis, S.I.; Ward, L.M.; Summers, L.J.; Bubbear, J.S.; Keen, R.; Stamp, T.C.B.; Baker, L.R.I.; Bonewald, L.F.; White, K.E. Molecular analysis of DMP1 mutants causing autosomal recessive hypophosphatemic rickets. Bone 2009, 44, 287–294. [Google Scholar] [CrossRef]
- Ye, L.; Mishina, Y.; Chen, D.; Huang, H.; Dallas, S.L.; Dallas, M.R.; Sivakumar, P.; Kunieda, T.; Tsutsui, T.W.; Boskey, A.; et al. Dmp1-deficient mice display severe defects in cartilage formation responsible for a chondrodysplasia-like phenotype. J. Biol. Chem. 2005, 280, 6197–6203. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, J.; Yuan, B.; Lu, Y.; Feng, J.Q.; Qin, C. Overexpression of Dmp1 fails to rescue the bone and dentin defects in Fam20C knockout mice. Connect. Tissue Res. 2014, 55, 299–303. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nishino, J.; Yamazaki, M.; Kawai, M.; Tachikawa, K.; Yamamoto, K.; Miyagawa, K.; Kogo, M.; Ozono, K.; Michigami, T. Extracellular Phosphate Induces the Expression of Dentin Matrix Protein 1 Through the FGF Receptor in Osteoblasts. J. Cell. Biochem. 2017, 118, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Oya, K.; Ishida, K.; Nishida, T.; Sato, S.; Kishino, M.; Hirose, K.; Ogawa, Y.; Ikebe, K.; Takeshige, F.; Yasuda, H.; et al. Immunohistochemical analysis of dentin matrix protein 1 (Dmp1) phosphorylation by Fam20C in bone: Implications for the induction of biomineralization. Histochem. Cell Biol. 2017, 147, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhu, Q.; Wang, X.; Yu, J.; Chen, X.; Wang, J.; Wang, X.; Xiao, J.; Wang, C.; Wang, L. Secretory kinase Fam20C tunes endoplasmic reticulum redox state via phosphorylation of Ero1α. EMBO J. 2018, 37, e98699. [Google Scholar] [CrossRef]
- David, V.; Martin, A.; Hedge, A.M.; Rowe, P.S.N. Matrix extracellular phosphoglycoprotein (MEPE) is a new bone renal hormone and vascularization modulator. Endocrinology 2009, 150, 4012–4023. [Google Scholar] [CrossRef]
- Lu, C.; Huang, S.; Miclau, T.; Helms, J.A.; Colnot, C. Mepe is expressed during skeletal development and regeneration. Histochem. Cell Biol. 2004, 121, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Hayashibara, T.; Hiraga, T.; Sugita, A.; Wang, L.; Hata, K.; Ooshima, T.; Yoneda, T. Regulation of osteoclast differentiation and function by phosphate: Potential role of osteoclasts in the skeletal abnormalities in hypophosphatemic conditions. J. Bone Miner. Res. 2007, 22, 1743–1751. [Google Scholar] [CrossRef]
- Marks, J.; Churchill, L.J.; Debnam, E.S.; Unwin, R.J. Matrix extracellular phosphoglycoprotein inhibits phosphate transport. J. Am. Soc. Nephrol. 2008, 19, 2313–2320. [Google Scholar] [CrossRef]
- Gowen, L.C.; Petersen, D.N.; Mansolf, A.L.; Qi, H.; Stock, J.L.; Tkalcevic, G.T.; Simmons, H.A.; Crawford, D.T.; Chidsey-Frink, K.L.; Ke, H.Z.; et al. Targeted disruption of the osteoblast/osteocyte factor 45 gene (Of45) results in increased bone formation and bone mass. J. Biol. Chem. 2003, 278, 1998–2007. [Google Scholar] [CrossRef] [PubMed]
- Kiela, P.R.; Ghishan, F.K. Recent advances in the renal-skeletal-gut axis that controls phosphate homeostasis. Lab. Investig. 2009, 89, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.; Schytte, G.N.; Scavenius, C.; Enghild, J.J.; McKee, M.D.; Sørensen, E.S. FAM20C-Mediated Phosphorylation of MEPE and Its Acidic Serine- and Aspartate-Rich Motif. JBMR Plus 2020, 4, e10378. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Rowe, P.S.N.; Liu, S.; Simpson, L.G.; Xiao, Z.S.; Darryl Quarles, L. Inhibition of MEPE cleavage by Phex. Biochem. Biophys. Res. Commun. 2002, 297, 38–45. [Google Scholar] [CrossRef]
- David, V.; Martin, A.; Hedge, A.M.; Drezner, M.K.; Rowe, P.S.N. Asarm peptides: PHEX-dependent and -independent regulation of serum phosphate. Am. J. Physiol. Ren. Physiol. 2011, 300, 783–791. [Google Scholar] [CrossRef]
- Liu, S.; Rowe, P.S.N.; Vierthaler, L.; Zhou, J.; Quarles, L.D. Phosphorylated acidic serine-aspartate-rich MEPE-associated motif peptide from matrix extracellular phosphoglycoprotein inhibits phosphate regulating gene with homologies to endopeptidases on the X-chromosome enzyme activity. J. Endocrinol. 2007, 192, 261–267. [Google Scholar] [CrossRef]
- Rowe, P.S.N. Fgf23, Mepe and Phex. Crit. Rev. Oral Biol. Med. 2004, 15, 264–281. [Google Scholar] [CrossRef]
- Addison, W.N.; Masica, D.L.; Gray, J.J.; McKee, M.D. Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage. J. Bone Miner. Res. 2010, 25, 695–705. [Google Scholar] [CrossRef]
- Rowe, P.S.N.; Garrett, I.R.; Schwarz, P.M.; Carnes, D.L.; Lafer, E.M.; Mundy, G.R.; Gutierrez, G.E. Surface plasmon resonance (SPR) confirms that MEPE binds to PHEX via the MEPE-ASARM motif: A model for impaired mineralization in X-linked rickets (HYP). Bone 2005, 36, 33–46. [Google Scholar] [CrossRef]
- Gullard, A.; Gluhak-Heinrich, J.; Papagerakis, S.; Sohn, P.; Unterbrink, A.; Chen, S.; MacDougall, M. MEPE Localization in the Craniofacial Complex and Function in Tooth Dentin Formation. J. Histochem. Cytochem. 2016, 64, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Zelenchuk, L.V.; Hedge, A.M.; Rowe, P.S.N. Age dependent regulation of bone-mass and renal function by the MEPE ASARM-MOTIF. Bone 2015, 79, 131–142. [Google Scholar] [CrossRef]
- Wang, H.; Kawashima, N.; Iwata, T.; Xu, J.; Takahashi, S.; Sugiyama, T.; Suda, H. Differentiation of odontoblasts is negatively regulated by MEPE via its C-terminal fragment. Biochem. Biophys. Res. Commun. 2010, 398, 406–412. [Google Scholar] [CrossRef]
- Schrauwen, I.; Valgaeren, H.; Tomas-Roca, L.; Sommen, M.; Altunoglu, U.; Wesdorp, M.; Beyens, M.; Fransen, E.; Nasir, A.; Vandeweyer, G.; et al. Variants affecting diverse domains of MEPE are associated with two distinct bone disorders, a craniofacial bone defect and otosclerosis. Genet. Med. 2019, 21, 1199–1208. [Google Scholar] [CrossRef]
- Kato, K.; Jeanneau, C.; Tarp, M.A.; Benet-Pagès, A.; Lorenz-Depiereux, B.; Bennett, E.P.; Mandel, U.; Strom, T.M.; Clausen, H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis: Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem. 2006, 281, 18370–18377. [Google Scholar] [CrossRef]
- Xiao, Z.; Huang, J.; Cao, L.; Liang, Y.; Han, X.; Quarles, L.D. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS ONE 2014, 9, e104154. [Google Scholar] [CrossRef] [PubMed]
- Vincent, K.; Durrant, M.C. A structural and functional model for human bone sialoprotein. J. Mol. Graph. Model. 2013, 39, 108–117. [Google Scholar] [CrossRef]
- Stubbs, J.T.; Mintz, K.P.; Eanes, E.D.; Torchia, D.A.; Fisher, L.W. Characterization of native and recombinant bone sialoprotein: Delineation of the mineral-binding and cell adhesion domains and structural analysis of the RGD domain. J. Bone Miner. Res. 1997, 12, 1210–1222. [Google Scholar] [CrossRef] [PubMed]
- Baht, G.S.; O’Young, J.; Borovina, A.; Chen, H.; Tye, C.E.; Karttunen, M.; Lajoie, G.A.; Hunter, G.K.; Goldberg, H.A. Phosphorylation of Ser136 is critical for potent bone sialoprotein-mediated nucleation of hydroxyapatite crystals. Biochem. J. 2010, 428, 385–395. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gordon, J.A.R.; Tye, C.E.; Sampaio, A.V.; Underhill, T.M.; Hunter, G.K.; Goldberg, H.A. Bone sialoprotein expression enhances osteoblast differentiation and matrix mineralization In Vitro. Bone 2007, 41, 462–473. [Google Scholar] [CrossRef]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. Part A 2019, 179, 2393–2419. [Google Scholar] [CrossRef]
- Bouet, G.; Bouleftour, W.; Juignet, L.; Linossier, M.-T.; Thomas, M.; Vanden-Bossche, A.; Aubin, J.E.; Vico, L.; Marchat, D.; Malaval, L. The Impairment of Osteogenesis in Bone Sialoprotein (BSP) Knockout Calvaria Cell Cultures Is Cell Density Dependent. PLoS ONE 2015, 10, e0117402. [Google Scholar] [CrossRef] [PubMed]
- Bouleftour, W.; Boudiffa, M.; Wade-Gueye, N.M.; Bouët, G.; Cardelli, M.; Laroche, N.; Vanden-Bossche, A.; Thomas, M.; Bonnelye, E.; Aubin, J.E.; et al. Skeletal development of mice lacking Bone Sialoprotein (BSP)—Impairment of long bone growth and progressive establishment of high trabecular bone mass. PLoS ONE 2014, 9, e95144. [Google Scholar] [CrossRef] [PubMed]
- Malaval, L.; Wade-Guéye, N.M.; Boudiffa, M.; Fei, J.; Zirngibl, R.; Chen, F.; Laroche, N.; Roux, J.P.; Burt-Pichat, B.; Duboeuf, F.; et al. Bone sialoprotein plays a functional role in bone formation and osteoclastogenesis. J. Exp. Med. 2008, 205, 1145–1153. [Google Scholar] [CrossRef]
- Foster, B.L.; Soenjaya, Y.; Nociti, F.H.; Holm, E.; Zerfas, P.M.; Wimer, H.F.; Holdsworth, D.W.; Aubin, J.E.; Hunter, G.K.; Goldberg, H.A.; et al. Deficiency in acellular cementum and periodontal attachment in Bsp null mice. J. Dent. Res. 2013, 92, 166–172. [Google Scholar] [CrossRef]
- Monfoulet, L.; Malaval, L.; Aubin, J.E.; Rittling, S.R.; Gadeau, A.P.; Fricain, J.C.; Chassande, O. Bone sialoprotein, but not osteopontin, deficiency impairs the mineralization of regenerating bone during cortical defect healing. Bone 2010, 46, 447–452. [Google Scholar] [CrossRef]
- Malaval, L.; Monfoulet, L.; Fabre, T.; Pothuaud, L.; Bareille, R.; Miraux, S.; Thiaudiere, E.; Raffard, G.; Franconi, J.M.; Lafage-Proust, M.H.; et al. Absence of bone sialoprotein (BSP) impairs cortical defect repair in mouse long bone. Bone 2009, 45, 853–861. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Engel, J.L.; Wiley, S.E.; Xiao, J.; Gonzalez, D.J.; Appaiah, H.N.; Koller, A.; Nizet, V.; White, K.E.; Dixon, J.E. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 5520–5525. [Google Scholar] [CrossRef]
- White, K.E.; Evans, W.E.; O’Riordan, J.L.H.; Speer, M.C.; Econs, M.J.; Lorenz-Depiereux, B.; Grabowski, M.; Meitinger, T.; Strom, T.M. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 2000, 26, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Bayer, J.; Schüler, C.; Zeitz, U.; Murali, S.K.; Ada, S.; Alvarez-Pez, J.M.; Smorodchenko, A.; Erben, R.G. Klotho Lacks an FGF23-Independent Role in Mineral Homeostasis. J. Bone Miner. Res. 2017, 32, 2049–2061. [Google Scholar] [CrossRef] [PubMed]
- Takeyari, S.; Yamamoto, T.; Kinoshita, Y.; Fukumoto, S.; Glorieux, F.H.; Michigami, T.; Hasegawa, K.; Kitaoka, T.; Kubota, T.; Imanishi, Y.; et al. Hypophosphatemic osteomalacia and bone sclerosis caused by a novel homozygous mutation of the FAM20C gene in an elderly man with a mild variant of raine syndrome. Bone 2014, 67, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, C.A.; Adler, F.; Nelson-Williams, C.; Iijima, J.; Li, P.; Imura, A.; Nabeshima, Y.I.; Reyes-Mugica, M.; Carpenter, T.O.; Lifton, R.P. A translocation causing increased α-Klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc. Natl. Acad. Sci. USA 2008, 105, 3455–3460. [Google Scholar] [CrossRef]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef]
- He, Q.; Shumate, L.T.; Matthias, J.; Aydin, C.; Wein, M.N.; Spatz, J.M.; Goetz, R.; Mohammadi, M.; Plagge, A.; Pajevic, P.D.; et al. A G protein-coupled, IP3/protein kinase C pathway controlling the synthesis of phosphaturic hormone FGF23. JCI Insight. 2019, 4, e125007. [Google Scholar] [CrossRef]
- Knab, V.M.; Corbin, B.; Andrukhova, O.; Hum, J.M.; Ni, P.; Rabadi, S.; Maeda, A.; White, K.E.; Erben, R.G.; Jüppner, H.; et al. C-terminal but not intact fibroblast growth factor 23 levels. Endocrinology 2017, 158, 1130–1139. [Google Scholar] [CrossRef]
- Shimada, T.; Urakawa, I.; Yamazaki, Y.; Hasegawa, H.; Hino, R.; Yoneya, T.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem. Biophys. Res. Commun. 2004, 314, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Larsson, T.; Marsell, R.; Schipani, E.; Ohlsson, C.; Ljunggren, Ö.; Tenenhouse, H.S.; Jüppner, H.; Jonsson, K.B. Transgenic mice expressing fibroblast growth factor 23 under the control of the α1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 2004, 145, 3087–3094. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.Y.; Miao, D.; Goltzman, D.; Karaplis, A.C. The autosomal dominant hypophosphatemic rickets R176Q mutation in fibroblast growth factor 23 resists proteolytic cleavage and enhances in vivo biological potency. J. Biol. Chem. 2003, 278, 9843–9849. [Google Scholar] [CrossRef]
- Bai, X.; Miao, D.; Li, J.; Goltzman, D.; Karaplis, A.C. Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology 2004, 145, 5269–5279. [Google Scholar] [CrossRef] [PubMed]
- Econs, M.J.; McEnery, P.T. Autosomal dominant hypophosphatemic rickets/osteomalacia: Clinical characterization of a novel renal phosphate-wasting disorder. J. Clin. Endocrinol. Metab. 1997, 82, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Lysaght, A.C.; Yuan, Q.; Fan, Y.; Kalwani, N.; Caruso, P.; Cunnane, M.B.; Lanske, B.; Stankovic, K.M. FGF23 deficiency leads to mixed hearing loss and middle ear malformation in mice. PLoS ONE 2014, 9, 107681. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Liu, S.; David, V.; Li, H.; Karydis, A.; Feng, J.Q.; Quarles, L.D. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB J. 2011, 25, 2551–2562. [Google Scholar] [CrossRef]
- Yuan, Q.; Jiang, Y.; Zhao, X.; Sato, T.; Densmore, M.; Schüler, C.; Erben, R.G.; McKee, M.D.; Lanske, B. Increased osteopontin contributes to inhibition of bone mineralization in FGF23-deficient mice. J. Bone Miner. Res. 2014, 29, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Gerard-O’Riley, R.L.; Acton, D.; McQueen, A.K.; Strobel, I.E.; Witcher, P.C.; Feng, J.Q.; Econs, M.J. A mutation in the Dmp1 gene alters phosphate responsiveness in mice. Endocrinology 2017, 158, 470–476. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.; Li, C.; Gao, T.; Liu, Y.; Rangiani, A.; Sun, Y.; Hao, J.; George, A.; Lu, Y.; et al. Inactivation of a novel FGF23 regulator, FAM20C, leads to hypophosphatemic rickets in mice. PLoS Genet. 2012, 8, e1002708. [Google Scholar] [CrossRef]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.C.; Moe, O.W.; et al. Regulation of fibroblast growth factor-23 signaling by Klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef]
- Qin, Z.; Wang, P.; Li, X.; Zhang, S.; Tian, M.; Dai, Y.; Fu, L. Systematic network-based discovery of a Fam20C inhibitor (FL-1607) with apoptosis modulation in triple-negative breast cancer. Mol. Biosyst. 2016, 12, 2108–2118. [Google Scholar] [CrossRef]
- Yalak, G.; Shiu, J.Y.; Schoen, I.; Mitsi, M.; Vogel, V. Phosphorylated fibronectin enhances cell attachment and upregulates mechanical cell functions. PLoS ONE 2019, 14, e0218893. [Google Scholar] [CrossRef]
- Vogel, P.; Hansen, G.M.; Read, R.W.; Vance, R.B.; Thiel, M.; Liu, J.; Wronski, T.J.; Smith, D.D.; Jeter-Jones, S.; Brommage, R. Amelogenesis imperfecta and other biomineralization defects in Fam20a and Fam20c null mice. Vet. Pathol. 2012, 49, 998–1017. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, T.; Liu, Y.; Wang, X.; Zhang, J.; Wang, X.; Shi, G.; Lou, J.; Wang, L.; Wang, C.; et al. Phosphorylation switches protein disulfide isomerase activity to maintain proteostasis and attenuate ER stress. EMBO J. 2020, 39, e103841. [Google Scholar] [CrossRef]
- Da, Q.; Han, H.; Valladolid, C.; Fernández, M.; Khatlani, T.; Pradhan, S.; Nolasco, J.; Matsunami, R.K.; Engler, D.A.; Cruz, M.A.; et al. In vitro phosphorylation of von Willebrand factor by FAM20c enhances its ability to support platelet adhesion. J. Thromb. Haemost. 2019, 17, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Ben Djoudi Ouadda, A.; Gauthier, M.S.; Susan-Resiga, D.; Girard, E.; Essalmani, R.; Black, M.; Marcinkiewicz, J.; Forget, D.; Hamelin, J.; Evagelidis, A.; et al. Ser-phosphorylation of PCSK9 (Proprotein Convertase Subtilisin-Kexin 9) by Fam20C (Family with Sequence Similarity 20, Member C) kinase enhances its ability to degrade the LDLR (Low-Density Lipoprotein Receptor). Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1996–2013. [Google Scholar] [CrossRef]
- Ashraf, Y.; Duval, S.; Sachan, V.; Essalmani, R.; Susan-Resiga, D.; Roubtsova, A.; Hamelin, J.; Gerhardy, S.; Kirchhofer, D.; Tagliabracci, V.S.; et al. Proprotein convertase 7 (PCSK7) reduces apoA-V levels. FEBS J. 2020, 287, 3565–3578. [Google Scholar] [CrossRef]
- Goettsch, C.; Hutcheson, J.D.; Aikawa, M.; Iwata, H.; Pham, T.; Nykjaer, A.; Kjolby, M.; Rogers, M.; Michel, T.; Shibasaki, M.; et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J. Clin. Investig. 2016, 126, 1323–1336. [Google Scholar] [CrossRef]
- Pollak, A.J.; Haghighi, K.; Kunduri, S.; Arvanitis, D.A.; Bidwell, P.A.; Liu, G.-S.; Singh, V.P.; Gonzalez, D.J.; Sanoudou, D.; Wiley, S.E.; et al. Phosphorylation of serine96 of histidine-rich calcium-binding protein by the Fam20C kinase functions to prevent cardiac arrhythmia. Proc. Natl. Sci. USA 2017, 114, 9098–9103. [Google Scholar] [CrossRef]
- Sathe, G.; Na, C.H.; Renuse, S.; Madugundu, A.; Albert, M.; Moghekar, A.; Pandey, A. Phosphotyrosine profiling of human cerebrospinal fluid. Clin. Proteom. 2018, 15, 29. [Google Scholar] [CrossRef]
- Salvi, M.; Cesaro, L.; Tibaldi, E.; Pinna, L.A. Motif analysis of phosphosites discloses a potential prominent role of the golgi casein kinase (GCK) in the generation of human plasma phospho-proteome. J. Proteome Res. 2010, 9, 3335–3338. [Google Scholar] [CrossRef]
- Lietz, C.B.; Toneff, T.; Mosier, C.; Podvin, S.; O’Donoghue, A.J.; Hook, V. Phosphopeptidomics Reveals Differential Phosphorylation States and Novel SxE Phosphosite Motifs of Neuropeptides in Dense Core Secretory Vesicles. J. Am. Soc. Mass Spectrom. 2018, 29, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Secher, A.; Kelstrup, C.D.; Conde-Frieboes, K.W.; Pyke, C.; Raun, K.; Wulff, B.S.; Olsen, J.V. Analytic framework for peptidomics applied to large-scale neuropeptide identification. Nat. Commun. 2016, 7, 11436. [Google Scholar] [CrossRef]
- Xu, S.Y.; Zhang, Q.L.; Zhang, Q.; Wan, L.; Jiang, J.; Tu, T.; Manavis, J.; Pan, A.; Cai, Y.; Yan, X.X. Regional and cellular mapping of sortilin immunoreactivity in adult human brain. Front. Neuroanat. 2019, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Rigbolt, K.T.G.; Prokhorova, T.A.; Akimov, V.; Henningsen, J.; Johansen, P.T.; Kratchmarova, I.; Kassem, M.; Mann, M.; Olsen, J.V.; Blagoev, B. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal. 2011, 4, rs3. [Google Scholar] [CrossRef]
- Wade-Gueye, N.M.; Boudiffa, M.; Vanden-Bossche, A.; Laroche, N.; Aubin, J.E.; Vico, L.; Lafage-Proust, M.H.; Malaval, L. Absence of bone sialoprotein (BSP) impairs primary bone formation and resorption: The marrow ablation model under PTH challenge. Bone 2012, 50, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Al Mane, K.; Al-Dayel, F.; McDonald, P. Intracranial calcification in Raine syndrome: Radiological pathological correlation. Pediatr. Radiol. 1998, 28, 820–823. [Google Scholar] [CrossRef]
- Speer, M.Y.; McKee, M.D.; Guldberg, R.E.; Liaw, L.; Yang, H.Y.; Tung, E.; Karsenty, G.; Giachelli, C.M. Inactivation of the osteopontin gene enhances vascular calcification of matrix Gla protein-deficient mice: Evidence for osteopontin as an inducible inhibitor of vascular calcification in vivo. J. Exp. Med. 2002, 196, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Bellahcène, A.; Castronovo, V.; Ogbureke, K.U.E.; Fisher, L.W.; Fedarko, N.S. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): Multifunctional proteins in cancer. Nat. Rev. Cancer 2008, 8, 212–226. [Google Scholar] [CrossRef]
- Vidavsky, N.; Kunitake, J.A.M.R.; Estroff, L.A. Multiple Pathways for Pathological Calcification in the Human Body. Adv. Healthc. Mater. 2021, 10, 2001271. [Google Scholar] [CrossRef] [PubMed]
- Willnow, T.E.; Petersen, C.M.; Nykjaer, A. VPS10P-domain receptors—Regulators of neuronal viability and function. Nat. Rev. Neurosci. 2008, 9, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Finan, G.M.; Okada, H.; Kim, T.W. BACE1 retrograde trafficking is uniquely regulated by the cytoplasmic domain of sortilin. J. Biol. Chem. 2011, 286, 12602–12616. [Google Scholar] [CrossRef]
- Hu, X.; Hu, Z.L.; Li, Z.; Ruan, C.S.; Qiu, W.Y.; Pan, A.; Li, C.Q.; Cai, Y.; Shen, L.; Chu, Y.; et al. Sortilin fragments deposit at senile plaques in human cerebrum. Front. Neuroanat. 2017, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; Beug, S.; Nickerson, P.E.B.; Peng, J.; Mazerolle, C.; Bassett, E.A.; Ringuette, R.; Jama, F.A.; Morales, C.; Christ, A.; et al. Sortilin regulates sorting and secretion of Sonic hedgehog. J. Cell Sci. 2016, 129, 3832–3844. [Google Scholar]
- Zhang, H.; Lu, Y.; Qin, C. FAM20C is Essential for Maintaining Brain Homeostasis. FASEB J. 2019, 33, 335.2. [Google Scholar]
- Arvanitis, D.A.; Sanoudou, D.; Kolokathis, F.; Vafiadaki, E.; Papalouka, V.; Kontrogianni-Konstantopoulos, A.; Theodorakis, G.N.; Paraskevaidis, I.A.; Adamopoulos, S.; Dorn, G.W.; et al. The Ser96Ala variant in histidine-rich calcium-binding protein is associated with life-threatening ventricular arrhythmias in idiopathic dilated cardiomyopathy. Eur. Heart J. 2008, 29, 2514–2525. [Google Scholar] [CrossRef]
- Amioka, M.I.; Nakano, Y.I.; Ochi, H.; Onohara, Y.; Sairaku, A.; Tokuyama, T.; Motoda, C.; Matsumura, H.; Tomomori, S.; Hironobe, N.; et al. Ser96Ala genetic variant of the human histidine-rich calcium-binding protein is a genetic predictor of recurrence after catheter ablation in patients with paroxysmal atrial fibrillation. PLoS ONE 2019, 14, e0213208. [Google Scholar] [CrossRef]
- De Meeus, A.; Stephan, E.; Debrus, S.; Jean, M.K.; Loiselet, J.; Weissenbach, J.; Demaille, J.; Bouvagnet, P. An isolated cardiac conduction disease maps to chromosome 19q. Circ. Res. 1995, 77, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Brink, P.A.; Ferreira, A.; Moolman, J.C.; Weymar, H.W.; Van Der Merwe, P.L.; Corrield, V.A. Gene for progressive familial heart block type I maps to chromosome 19q13. Circulation 1995, 91, 1633–1640. [Google Scholar] [CrossRef]
- Perisic, L.; Hedin, E.; Razuvaev, A.; Lengquist, M.; Osterholm, C.; Folkersen, L.; Gillgren, P.; Paulsson-Berne, G.; Ponten, F.; Odeberg, J.; et al. Profiling of atherosclerotic lesions by gene and tissue microarrays reveals pcsk6 as a novel protease in unstable carotid atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2432–2443. [Google Scholar] [CrossRef] [PubMed]
- Turpeinen, H.; Oksanen, A.; Kivinen, V.; Kukkurainen, S.; Uusimäki, A.; Rämet, M.; Parikka, M.; Hytönen, V.P.; Nykter, M.; Pesu, M. Proprotein Convertase Subtilisin/Kexin Type 7 (PCSK7) Is Essential for the Zebrafish Development and Bioavailability of Transforming Growth Factor β1a (TGFβ1a). J. Biol. Chem. 2013, 288, 36610–36623. [Google Scholar] [CrossRef] [PubMed]
- Peloso, G.M.; Auer, P.L.; Bis, J.C.; Voorman, A.; Morrison, A.C.; Stitziel, N.O.; Brody, J.A.; Khetarpal, S.A.; Crosby, J.R.; Fornage, M.; et al. Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. Am. J. Hum. Genet. 2014, 94, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Prat, A. The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabès, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Miao, N.; Zhan, Y.; Xu, Y.; Yuan, H.; Qin, C.; Lin, F.; Xie, X.; Mu, S.; Yuan, M.; Mu, H.; et al. Loss of Fam20c causes defects in the acinar and duct structure of salivary glands in mice. Int. J. Mol. Med. 2019, 43, 2103–2117. [Google Scholar] [CrossRef]
- Whyte, M.P.; McAlister, W.H.; Fallon, M.D.; Pierpont, M.E.; Bijanki, V.N.; Duan, S.; Otaify, G.A.; Sly, W.S.; Mumm, S. Raine Syndrome (OMIM #259775), Caused By FAM20C Mutation, Is Congenital Sclerosing Osteomalacia with Cerebral Calcification (OMIM 259660). J. Bone Miner. Res. 2017, 32, 757–769. [Google Scholar]
- Simpson, M.; Scheuerle, A.; Hurst, J.; Patton, M.; Stewart, H.; Crosby, A. Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin. Genet. 2009, 75, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Ababneh, F.K.; Alswaid, A.; Youssef, T.; Al Azzawi, M.; Crosby, A.; Albalwi, M.A. Hereditary deletion of the entire FAM20C gene in a patient with Raine syndrome. Am. J. Med. Genet. Part A 2013, 161, 3155–3160. [Google Scholar] [CrossRef]
- El-Dessouky, S.H.; Abdel-Hamid, M.S.; Abdel-Ghafar, S.F.; Aboulghar, M.M.; Gaafar, H.M.; Fouad, M.; Ahmed, A.H.; Abdel-Salam, G.M.H. Raine syndrome: Prenatal diagnosis based on recognizable fetal facial features and characteristic intracranial calcification. Prenat. Diagn. 2020, 40, 1578–1597. [Google Scholar] [CrossRef]
- Hernández-Zavala, A.; Cortés-Camacho, F.; Palma-Lara, I.; Godínez-Aguilar, R.; Espinosa, A.M.; Pérez-Durán, J.; Villanueva-Ocampo, P.; Ugarte-Briones, C.; Serrano-Bello, C.A.; Sánchez-Santiago, P.J.; et al. Two novel FAM20C variants in a family with raine syndrome. Genes 2020, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Mameli, C.; Zichichi, G.; Mahmood, N.; Elalaoui, S.C.; Mirza, A.; Dharmaraj, P.; Burrone, M.; Cattaneo, E.; Sheth, J.; Gandhi, A.; et al. Natural history of non-lethal Raine syndrome during childhood. Orphanet J. Rare Dis. 2020, 15, 93. [Google Scholar] [CrossRef]
- Acevedo, A.C.; Poulter, J.A.; Alves, P.G.; de Lima, C.L.; Castro, L.C.; Yamaguti, P.M.; Paula, L.M.; Parry, D.A.; Logan, C.V.; Smith, C.E.L.; et al. Variability of systemic and oro-dental phenotype in two families with non-lethal Raine syndrome with FAM20C mutations. BMC Med. Genet. 2015, 16, 8. [Google Scholar] [CrossRef]
- Elalaoui, S.C.; Al-Sheqaih, N.; Ratbi, I.; Urquhart, J.E.; O’Sullivan, J.; Bhaskar, S.; Williams, S.S.; Elalloussi, M.; Lyahyai, J.; Sbihi, L.; et al. Non lethal Raine syndrome and differential diagnosis. Eur. J. Med. Genet. 2016, 59, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Rafaelsen, S.H.; Ræder, H.; Fagerheim, A.K.; Knappskog, P.; Carpenter, T.O.; Johansson, S.; Bjerknes, R. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J. Bone Miner. Res. 2013, 28, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Fradin, M.; Stoetzel, C.; Muller, J.; Koob, M.; Christmann, D.; Debry, C.; Kohler, M.; Isnard, M.; Astruc, D.; Desprez, P.; et al. Osteosclerotic bone dysplasia in siblings with a Fam20C mutation. Clin. Genet. 2011, 80, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Boissel, S.; Fallet-Bianco, C.; Chitayat, D.; Kremer, V.; Nassif, C.; Rypens, F.; Delrue, M.A.; Dal Soglio, D.; Oligny, L.L.; Patey, N.; et al. Genomic study of severe fetal anomalies and discovery of GREB1L mutations in renal agenesis. Genet. Med. 2018, 20, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.Y.; Rodriguez, M.; Roberts, A.; Bauer, M.; Mihalek, I.; Bodamer, O. A novel FAM20C mutation causes a rare form of neonatal lethal Raine syndrome. Am. J. Med. Genet. Part A 2019, 179, 1866–1871. [Google Scholar] [CrossRef]
- Mamedova, E.; Dimitrova, D.; Przhiyalkovskaya, E.; Buryakina, S.; Vasilyev, E.; Tiulpakov, A.; Belaya, Z. Non-lethal Raine Syndrome in a Middle-Aged Woman Caused by a Novel FAM20C Mutation. Calcif. Tissue Int. 2019, 105, 567–572. [Google Scholar] [CrossRef]
- Mahmood, N.; Donne, A.; Weber, A.; Dharmraj, P. Raine syndrome: A review and a report of metabolic bone disease as a new link. Research 2014, 1, 1. [Google Scholar] [CrossRef]
- Seidahmed, M.Z.; Alazami, A.M.; Abdelbasit, O.B.; Al Hussein, K.; Miqdad, A.M.; Abu-Sa’da, O.; Mustafa, T.; Bahjat, S.; Alkuraya, F.S. Report of a case of Raine syndrome and literature review. Am. J. Med. Genet. Part A 2015, 167, 2394–2398. [Google Scholar] [CrossRef]
- Eras, N.; Celik, Y. Nonlethal Raine Syndrome in a Newborn Boy Caused by a Novel FAM20C Variant. Mol. Syndromol. 2021, 12, 169–173. [Google Scholar] [CrossRef]
- Eltan, M.; Alavanda, C.; Yavas Abali, Z.; Ergenekon, P.; Yalındag Ozturk, N.; Sakar, M.; Dagcinar, A.; Kirkgoz, T.; Kaygusuz, S.B.; Gokdemir, Y.; et al. A Rare Cause of Hypophosphatemia: Raine Syndrome Changing Clinical Features with Age. Calcif. Tissue Int. 2020, 107, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Kochar, G.S.; Choudhary, A.; Gadodia, A.; Gupta, N.; Simpson, M.A.; Crosby, A.H.; Kabra, M. Raine syndrome: A clinical, radiographic and genetic investigation of a case from the Indian subcontinent. Clin. Dysmorphol. 2010, 19, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Lulla, C.; Bapai, A. Raine Syndrome, a Rare Lethal Osteosclerotic Bone Dysplasia: Prenatal Diagnosis with 3-Dimensional Ultrasound and a Postnatal Clinical Exome Evaluation. J. Ultrasound Med. 2021, 40, 641–643. [Google Scholar] [CrossRef]
- Bajaj, S.; Nabi, F.; Shah, J.; Sheth, H. Recurrent variant c.1680C>A in FAM20C gene and genotype-phenotype correlation in a patient with Raine syndrome: A case report. BMC Pediatr. 2021, 21, 113. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [PubMed]
- Raine, J.; Winter, R.M.; Davey, A.; Tucker, S.M. Unknown syndrome: Microcephaly, hypoplastic nose, exophthalmos, gum hyperplasia, cleft palate, low set ears, and osteosclerosis. J. Med. Genet. 1989, 26, 786–788. [Google Scholar] [CrossRef]
- Kingston, H.M.; Freeman, J.S.; Hall, C.M. A new lethal sclerosing bone dysplasia. Skeletal Radiol. 1991, 20, 117–119. [Google Scholar] [CrossRef]
- Kan, A.E.; Kozlowski, K. New distinct lethal osteosclerotic bone dysplasia (Raine syndrome). Am. J. Med. Genet. 1992, 43, 860–864. [Google Scholar] [CrossRef]
- Al Mane, K.; Coates, R.K.; McDonald, P. Intracranial calcification in Raine syndrome. Pediatr. Radiol. 1996, 26, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Rejjal, A. Raine syndrome. Am. J. Med. Genet. 1998, 78, 382–385. [Google Scholar] [CrossRef]
- Shalev, S.A.; Shalev, E.; Reich, D.; Borochowitz, Z.U. Osteosclerosis, hypoplastic nose, and proptosis (Raine syndrome): Further delineation. Am. J. Med. Genet. 1999, 86, 274–277. [Google Scholar] [CrossRef]
- Acosta, A.X.; Peres, L.C.; Chimelli, L.C.; Pina-Neto, J.M. Raine dysplasia: A Brazilian case with a mild radiological involvement. Clin. Dysmorphol. 2000, 9, 99–101. [Google Scholar] [CrossRef]
- Mahafza, T.; El-Shanti, H.; Omari, H. Raine syndrome: Report of a case with hand and foot anomalies. Clin. Dysmorphol. 2001, 10, 227–229. [Google Scholar] [CrossRef]
- Hülskamp, G.; Wieczorek, D.; Rieder, H.; Louwen, F.; Hörnig-Franz, I.; Rickert, C.H.; Horst, J.; Harms, E.; Rehder, H. Raine syndrome: Report of a family with three affected sibs and further delineation of the syndrome. Clin. Dysmorphol. 2003, 12, 153–160. [Google Scholar] [CrossRef]
- Al-Gazali, L.I.; Jehier, K.; Nazih, B.; Abtin, F.; Haas, D.; Sadagahatian, R. Further delineation of Raine syndrome. Clin. Dysmorphol. 2003, 12, 89–93. [Google Scholar] [CrossRef]
- Gaigi, S.S.; Dorra, Z.; Aida, M.; Ahmed, C.; Sami, J.; Naima, K. Raine Syndrome. Tunis Med. 2011, 89, 723–725. [Google Scholar]
- Abu, A.J.; Bystricka, A.; Qadir, M.; Nikolay, M.K.J. Raine Syndrome: Clinical and Radiological Features of a Case from the United Arab Emirates; Swiss Society of Neonatology: Sankt Gallen, Switzerland, 2014. [Google Scholar]
- Holm, E.; Aubin, J.E.; Hunter, G.K.; Beier, F.; Goldberg, H.A. Loss of bone sialoprotein leads to impaired endochondral bone development and mineralization. Bone 2015, 71, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Beniash, E.; Deshpande, A.S.; Fang, P.A.; Lieb, N.S.; Zhang, X.; Sfeir, C.S. Possible role of DMP1 in dentin mineralization. J. Struct. Biol. 2011, 174, 100–106. [Google Scholar] [CrossRef]
- Wang, X.; Jung, J.; Liu, Y.; Yuan, B.; Lu, Y.; Feng, J.Q.; Qin, C. The specific role of FAM20C in amelogenesis. J. Dent. Res. 2013, 92, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Vishwanath, B.; Srinivasa, K.; Veera Shankar, M. Raine syndrome. Indian J. Hum. Genet. 2014, 20, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Das, D.S. Raine Syndrome: A rare case presentation. Int. J. Med. Sci. Clin. Invent. 2017, 4, 3304–3306. [Google Scholar] [CrossRef]
- Güneş, T.; Kurtoǧlu, S.; Çetin, N.; Öztürk, M.A.; Topaloǧlu, N. Raine syndrome associated with cytomegalovirus infection. Turk. J. Pediatr. 2005, 47, 89–91. [Google Scholar] [PubMed]
- Hirst, L.; Abou-Ameira, G.; Critchlow, S. Hypophosphataemic Rickets Secondary to Raine Syndrome: A Review of the Literature and Case Reports of Three Paediatric Patients’ Dental Management. Case Rep. Pediatr. 2021, 2021, 6637180. [Google Scholar] [PubMed]
- Qin, C.; D’Souza, R.; Feng, J.Q. Dentin Matrix Protein 1 (DMP1): New and important roles for biomineralization and phosphate homeostasis. J. Dent. Res. 2007, 86, 1134–1141. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| TARGET | General Function | Expression Organ/Site | FAM20C Phosphosites | FAM20C Phosphorylation Effect | Associated Diseases | Ref. |
|---|---|---|---|---|---|---|
| Fibronectin | Major component of the extracellular matrix. | Extracellular matrix. | Ser757, Ser2341, Ser2384 | [100,101] | ||
| Fibrinogen | Major component of the extracellular matrix. | Liver production and delivered to circulation. | Ser45, Ser56, Ser364, Ser524, S560, Ser609 (alpha chain) Ser68 (gamma chain) | Bleeding and thrombotic disorders. | [13] | |
| Ero1α | Thiol disulfide oxirreductase. Oxidative protein folding, ER proteostasis. ER redox homeostasis. Protection against ER stress. | Majority of cells. | Ser145 | Enhances oxidase activity and promotes oxidative protein folding. | [49,102] | |
| wVW | Coagulation. Platelet adhesion and contribution to hemostasis at sites of vascular injury as well as to arterial thrombosis. | Platelets | Ser74, Ser80, Tyr83, Ser93, Ser1517, Ser1613 | Enhances platelet adhesion. | von Willebrand disease. | [103] |
| PCSK9 | Cholesterol metabolism. It binds to LDL receptor for its internalization and degradation in endosomes and lysosomes, favoring LDL cholesterol increments. | Liver | Ser47, Ser666, Ser668, Ser688 | Enhances LDL receptor degradation, favoring lower LDL cholesterol clearance and its circulatory increase. | GoF variants: Familial Hypercholesterolemia. | [104] |
| PCSK7 | Lipid metabolism. It binds to ApoA-V (an activator of triglycerides clearance) to enhance its degradation in ER and lysosomes, indirectly lowering lipoprotein lipase activity for triglyceride clearance. | Ubiquos, but primary in liver. | Ser505 (Arg504His) | Lower degradation of apoA-V with an increase in circulation, resulting in lower blood levels and higher triglyceride uptake into adipocytes. | LoF variants: lower triglyceride levels. GoF variants: higher levels of PC7 protein and triglyceride levels. | [105] |
| Sortilin | Protein trafficking in the exocytic and endocytic pathways involved in neuronal viability and glucose homeostasis. | Nervous system, heart and other tissues. SHH regulation. | Ser825 | Vascular calcification promotion through higher TNAP activity. Intracellular sorting receptor for apoB and other proteins (Ser825). | Cardiovascular disease. | [106] |
| HRC | Regulation of calcium sequestration or release in the SR (storage, uptake and release), through interaction with SERCA2a, triadin and RyR2. | Heart, striated and arteriolar smooth muscle. | Ser96 | Calcium regulation. | Malignant arrhythmia. | [107] |
| Calsequestrin-2 | Regulation of calcium. | Heart and other tissues. | Ser385 | ER/SR calcium homeostasis and cardiac pathophysiology. | Arrhythmia. | [99] |
| STIM1 | Regulation of calcium. | Heart and other tissues. | Ser88 | ER/SR calcium homeostasis and cardiac pathophysiology. | Immunodeficiency 10 (MIM 612783) Myopathy, tubular aggregate 1 (MIM160565) Stormorken syndrome (MIM 185070). | [99] |
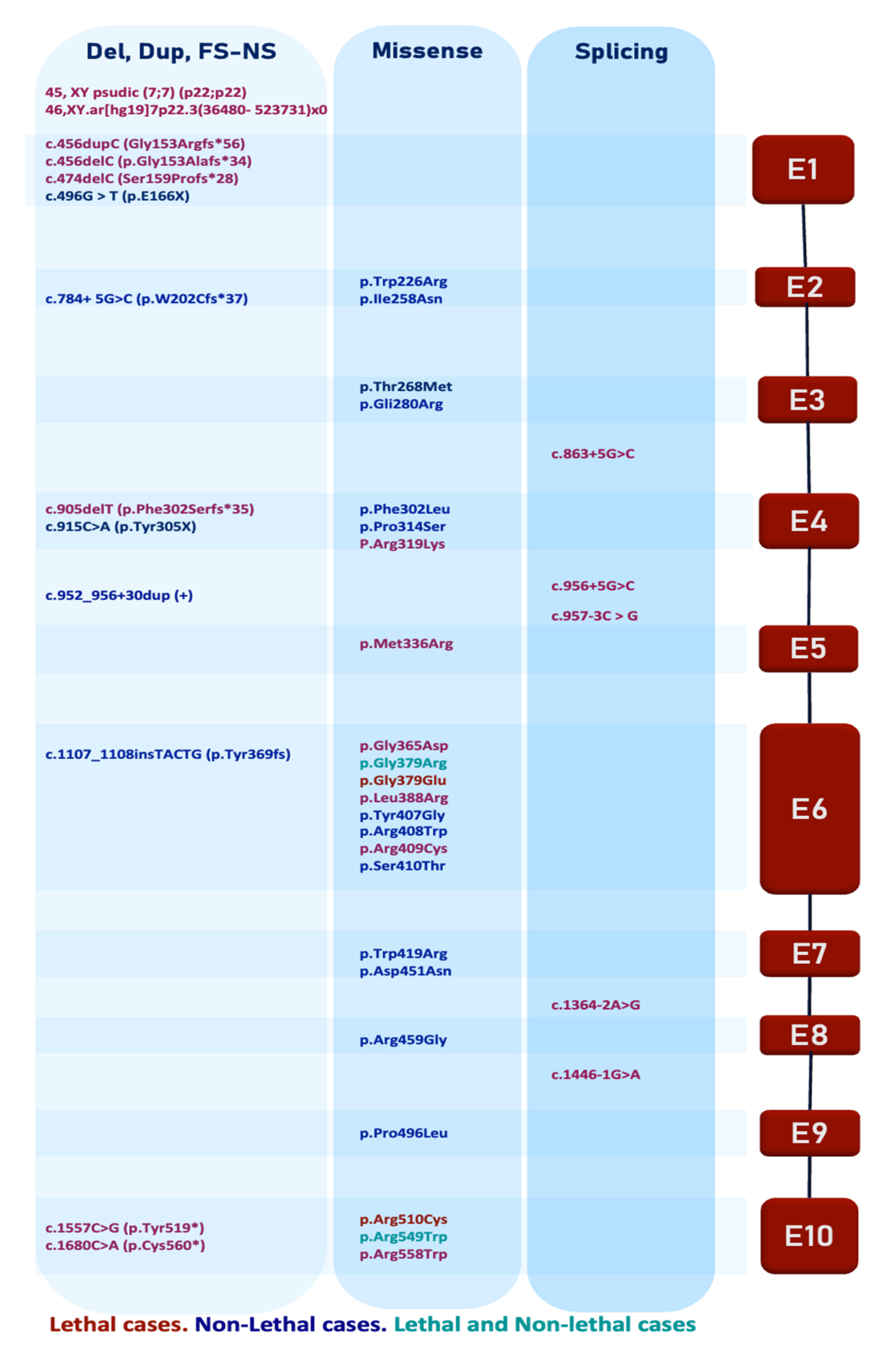
| Number | Localization | c.Description | p.Description | DNA Type | Protein Type | KD Effect | KD LOCALIZATION | Lethality | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 7pter | 45, XY psudic (7;7) (p22;p22) | - | GR | A | + | + | Simpson et al., 2007 [1] | |
| 2 | 48Kb | 46,XY.ar [hg19] 7p22.3 (36480-523731)x0 | - | GR | A | + | + | Ababneh et al., 2013 [137] | |
| 3 | E1 | c.456delC | p.Gly153Alafs*34 | Del/FS | SP | + | + | El-Dessouky et al., 2020 [138] | |
| 4 | E1 | c.456dupC | Gly153Argfs*56 | Dup/FS | SP | + | + | Hernández-Zavala et al., 2020 [139] | |
| 5 | E1 | c.474delC | Ser159Profs*28 | Del/FS | SP | + | + | Hernández-Zavala et al., 2020 [139] | |
| 6 | E1 | c.496G>T | p.Glu166* | Su | S | + | - | Mameli et al., 2020 [140] | |
| 7 | E2 | c.784+ 5G>C | p.W202Cfs*37 | Su | S/SP | + | - | Acevedo et al., 22015 [141] | |
| 8 | E2 | c.676T>A | p.Trp226Arg | Su | M | - | - | Elalaoui et al., 2016 [142] | |
| 9 | E2 | c.773T>A | p.IIe258Asn | Su | M | - | - | Simpson et al., 2019 [136] | |
| 10 | E3 | c.803C>T | p.Thr268Met | Su | M | - | - | Rafaelsen et al., 2013 [143] | |
| 11 | E3 | c.838 G>A | p.Gly280Arg | Su | M | - | - | Simpson et al., 2009 [136] | |
| 12 | E3/I | c.863+5G>C | - | Su | S | - | + | Mehme E et al., 2020 [35] | |
| 13 | E4 | c.906C>A | p.Phe302Leu | Su | M | - | - | Rolvien et al., 2018 [5] | |
| 14 | E4 | c.905delT | p.Phe302Serfs*35 | Del/FS | SP | + | + | El-Dessouky et al., 2020 [138] | |
| 15 | E4 | c.915C>A | p.Tyr305* | Su | NS | + | - | Rafaelsen et al., 2013 [143] | |
| 16 | E4 | c.940C>T | p.Pro314Ser | Su | M | - | - | Fradin et al., 2011 [144] | |
| 17 | E4/I4 | c.952_956+30dup (+) | - | Dup | S/SP | - | - | Rolvien et al., 2018 [5] | |
| 18 | E4 | c.956G>A | p.Arg319Lys | Su | M | + | Boissel et al., 2017 [145] | ||
| 19 | E4/I4 | c.956+5G>C | - | Su | SP | - | + | Simpson et al., 2007 [1] | |
| 20 | I4/E5 | c.957-3C>G | - | Su | SP | - | + | Simpson et al., 2007 [1] | |
| 21 | E5 | c.1007T>G | p.Met336Arg | Su | M | - | + | Hung et al., 2019 [146] | |
| 22 | E6 | c.1094G>A | Gly365Asp | Su | M | + | + | + | Whyte et al., 2016 [135] |
| 23 | E6 | c.1107_1108insTACTG | p.Tyr369 fs | Ins | FS | + | + | - | Mamedova et al., 2019 [147] |
| 24 | E6 | c.1135G>A | p.Gly379Arg | Su | M | + | + | + - | Simpson et al., 2007 [1] Mahmood et al., 2014 [148] |
| 25 | E6 | c.1136G>A | p.Gly379Glu | Su | M | + | + | + | Simpson et al., 2007 [1] |
| 26 | E6 | c.1163T>G | p.Leu388Arg | Su | M | + | + | + | Simpson et al., 2007 [1] |
| 27 | E6 | c.1219T>G | p.Tyr407Gly | Su | M | + | + | - | Tamai et al., 2017 [4] |
| 28 | E6 | c.1222C>T | p.Arg408Trp | Su | M | + | + | - | Takeyari et al., 2014 [83] |
| 29 | E6 | c.1225C>T | p.Arg409Cys | Su | M | + | + | + | Seidahmed et al., 2015 [149] |
| 30 | E6 | c.1228 T>A | p.Ser410Thr | Su | M | + | + | - | Sheth et al., 2018 [6] |
| 31 | E7 | c.1255T>C | p.Trp419Arg | Su | M | + | + | - | Eras et al., 2021 [150] |
| 32 | E7 | c.1351G>A | p.Asp451Asn | Su | M | + | + | - | Simpson et al., 2009 [136] |
| 33 | E7 | c.1351G>A | p.Asp451Asn | Su | M | + | + | - | Mameli et al., 2020 [140] |
| 33 | E8 | c.1375C>G | p.Arg459Gly | Su | M | + | + | - | Mamedova et al., 2019 [147] |
| 34 | I/E8 | c.1364-2A>G | - | Su | SP | + | + | + | Simpson et al., 2007 [1] |
| 35 | I/E9 | c.1446-1G>A | - | Su | SP | + | + | + | Simpson et al., 2007 [1] |
| 36 | E9 | c.1487C>T | p.Pro496Leu | Su | M | + | + | - | Acevedo et al., 2015 [141] |
| 37 | E10 | c.1528C>T | p.Arg510Cys | Su | M | + | + | + | Boissel et al., 2017 [145] |
| 38 | E10 | c.1557C>G | p.Tyr519* | Su | NS | + | + | + | El-Dessouky et al., 2020 [138] |
| 39 | E10 | c.1645C>T | p.Arg549Trp | Su | M | + | + | + - | Simpson et al., 2007 [1] Eltan et al., 2020 [151] |
| 40 | E10 | c.1672C>T | p.Arg558Trp | Su | M | + | + | + | Kochar et al., 2010 [152] |
| 41 | E10 | c-1680C>A | p.Cys560* | Su | NS | + | + | + | Lulla et al., 2020 [153] |
| Case | Reference | Year | Lethal | Brain Calcifications | Microcephay | Brachycephaly | Wide Fontanelles | Midface Hhypoplasia | Ocular Proptosis | Hypertelorism | Depressed Nasal Bridge | Choanal Stenosis/Atresia | Hypoplastic Nose | Small/Fish-Like Mouth | Macroglosia | Abnormal Teeth | Gum Hyperplasia | Cleft Palate | High Palate | Micrognatia | Low-Set Ears |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Raine [156] | 1989 | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| 2 | Kingston et al. [157] | 1991 | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||||
| 3 | Kan et al. [158] | 1992 | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| 4 | Patel et al. [7] | 1992 | + | + | + | + | + | ||||||||||||||
| 5 | Al Mane et al. [159] | 1996 | + | + | + | + | + | + | + | + | + | + | + | ||||||||
| 6 | Al Mane et al. 2 [115] | 1998 | + | + | + | + | + | + | + | + | + | + | |||||||||
| 7 | Rejjal et al. [160] | 1998 | + | + | + | + | + | + | + | + | + | + | + | ||||||||
| 8 | Shalev et al. [161] | 1999 | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||||
| 9 | Acosta et al. [162] | 2000 | + | + | + | + | + | + | + | + | + | + | + | ||||||||
| 10 | Mahafza et al. [163] | 2001 | + | + | + | + | + | + | + | + | + | + | + | ||||||||
| 11 | Hulskamp et al. [164] Ricket et al. [8] | 2003–2002 | + | + | + | + | + | + | + | + | + | + | |||||||||
| 12 | Hulskamp et al. [164] Ricket et al. [8] | 2003–2002 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||
| 13 | Hulskamp et al. [164] Ricket et al. [8] Simpson et al. [1] | 2003–2002 | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||||
| 14 | Al-Gazali et al. [165] | 2003 | + | + | + | + | + | + | + | + | |||||||||||
| 15 | Simpson et al. (1) [1] | 2007 | + | + | + | + | + | ||||||||||||||
| 16 | Simpson et al. (3) [1] | 2007 | + | + | + | + | |||||||||||||||
| 17 | Simpson et al. (6) [1] | 2007 | + | + | + | + | + | ||||||||||||||
| 18 | Simpson et al. (7) [1] | 2007 | + | + | + | + | + | ||||||||||||||
| 19 | Chitayat et al. [9] | 2007 | + | + | + | + | + | + | + | + | + | + | + | ||||||||
| 20 | Kochar et al. [152] | 2010 | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| 21 | Michael et al. [10] | 2011 | + | + | + | + | + | + | |||||||||||||
| 22 | Gaigi et al. 1 [166] | 2011 | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| 23 | Gaigi et al. 2 [166] | 2011 | + | + | + | + | + | + | + | + | + | + | + | ||||||||
| 24 | Gaigi et al. 3 [166] | 2011 | + | + | + | + | |||||||||||||||
| 25 | Gaigi et al. 4 [166] | 2011 | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| 26 | Ababneh et al. [137] | 2013 | + | + | + | + | + | + | + | + | + | + | |||||||||
| 27 | Berger et al. [167] | 2014 | + | + | + | + | + | + | + | + | + | + | + | ||||||||
| 28 | Seidahmed M et al. [149] | 2015 | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| 29 | Whyte et al. 1 [135] | 2016 | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| 30 | Whyte et al. 2 [135] | 2016 | + | + | + | + | + | + | + | + | + | + | + | ||||||||
| 31 | Boissel et al. [145] | 2017 | + | + | + | + | + | + | |||||||||||||
| 32 | Hung et al. [146] | 2019 | + | + | + | + | + | + | + | ||||||||||||
| 33 | Hernández-Zavala et al. 1 [139] | 2020 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||
| 34 | Hernández-Zavala et al. 2 [139] | 2020 | + | + | + | + | + | + | + | + | + | + | |||||||||
| 35 | Eltan et al. [151] | 2020 | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||||
| 36 | El-Dessouky et al. 1 [138] | 2020 | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| 37 | El-Dessouky et al. 2 [138] | 2020 | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| 38 | El-Dessouky et a. 3 [138] | 2020 | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| 39 | Lulla et al. [153] | 2020 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |||
| 40 | Bajaj et al. [154] | 2021 | + | + | + | + | + | + | + | + | + | + | + | + |
| Case | Reference | Year | Lethal | Age/Survival | Death Cause | Population | Consanguinity | Gender | Gestational Weeks | Weight/Percentile | Length/Percentle | Local Osteosclerosis | Generalized Osteosclerosis | Head Circumference/Percentile Circunference/Percentile | Short Neck | Cervical Ossification Defects | Vertebral Defects | Sacral Ossification Defects | Small/Narrow Thorax | Periosteal Reaction | Poor Cortico-Medullary-Differentiation | Bone-in-Bone | Bowed Bones | Fractures | Pseudo-Fractures | Short Limbs | Rhizomelic Shortening | Long Bone Shortening | Brachydactyly | Distal Phalangeal Hypoplasia | Tapered Fingers |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Raine et al. [156] | 1989 | + | 1 | R | Au | F | T | N | <3 | + | <3 | + | ||||||||||||||||||
| 2 | Kingston et al. [157]/Simpson et al. (2) [1] | 1991/2007 | + | 1 | R | Cau | + | M | 37 | N | N | + | 3 | + | + | + | + | + | + | + | |||||||||||
| 3 | Kan et al. [158] | 1992 | + | 1 | R | F | 37 | <3 | <3 | + | <3 | + | + | + | |||||||||||||||||
| 4 | Patel et al. [7] | 1992 | + | R | SA | + | M | 36 | + | + | |||||||||||||||||||||
| 5 | Al Mane et al. [159] | 1996 | + | 4 | R | SA | + | M | 39 | N | + | + | + | + | |||||||||||||||||
| 6 | Al Mane et al. [115] | 1998 | + | SA | + | F | 37 | N | + | + | + | ||||||||||||||||||||
| 7 | Rejjal et al. [160] | 1998 | + | 2 | R | SA | + | M | N | N | + | N | + | + | |||||||||||||||||
| 8 | Shalev et al. [161] | 1999 | + | 3 | R | SA | + | F | 39 | N | N | + | N | + | + | + | + | ||||||||||||||
| 9 | Acosta et al. [162] | 2000 | + | 1 | Br | + | M | 34 | 3 | <3 | + | + | + | + | |||||||||||||||||
| 10 | Mahafza et al. [163] | 2001 | + | 3 | CR | Jo | + | M | 40 | N | N | + | N | + | + | + | + | ||||||||||||||
| 11 | Hulskamp et al. (3)/Ricket et al. (1) [8,164] | 2003/2002 | + | 1 | R | T | + | M | 33 | N | N | + | N | ||||||||||||||||||
| 12 | Hulskamp et al. (2)/Ricket et al. (2) [8,164] | 2003/2002/2007 | + | 1 | R | T | + | M | 24 | N | N | + | + | + | + | + | + | + | |||||||||||||
| 13 | Hulskamp et al. (1) Ricket et al. (3)/Simpson et al. (5) [1,8,164] | 2003–2002 | + | 2 | R | T | + | F | 32 | N | N | + | N | + | + | + | + | ||||||||||||||
| 14 | Al-Gazali et al. [1,165]/Simpson et al. (4) | 2003 | + | 4 | R | E | M | 36 | N | N | + | N | + | + | + | + | + | + | + | + | |||||||||||
| 15 | Simpson et al. (1) [1] | 2007 | + | 1 | R | M | 37 | + | + | + | + | ||||||||||||||||||||
| 16 | Simpson et al. (3) [1] | 2007 | + | + | M | 38 | + | + | + | ||||||||||||||||||||||
| 17 | Simpson et al. (6) [1] | 2007 | + | 3 | F | 38 | + | + | |||||||||||||||||||||||
| 18 | Simpson et al. [1] | 2007 | + | F | 32 | + | + | + | |||||||||||||||||||||||
| 19 | Chitayat et al. [9] | 2007 | + | 3 | Jw | M | + | ||||||||||||||||||||||||
| 20 | Kochar et al. [152] | 2010 | + | 4 | PN | I | + | M | 40 | N | N | + | <3 | + | + | + | |||||||||||||||
| 21 | Michael et al. [10] | 2011 | + | 1 | Su | + | 34 | ||||||||||||||||||||||||
| 22 | Gaigi et al. 1 [166] | 2011 | + | 1 | R | M | 39 | <3 | <3 | + | <3 | + | + | ||||||||||||||||||
| 23 | Gaigi et al. 2 [166] | 2011 | + | 1 | R | M | 42 | + | |||||||||||||||||||||||
| 24 | Gaigi et al. 3 [166] | 2011 | + | 2 | R | F | 40 | <3 | <3 | + | <3 | ||||||||||||||||||||
| 25 | Gaigi et al. 4 [166] | 2011 | + | 2 | F | ||||||||||||||||||||||||||
| 26 | Ababneh et al. [137] | 2013 | + | 4 | PN | SA | + | M | T | N | + | + | + | + | + | + | + | ||||||||||||||
| 27 | Abu et al. [167] | 2014 | + | 4 | R | P | M | 31 | N | N | + | N | + | + | |||||||||||||||||
| 28 | Seidahmed M et al. [149] | 2015 | + | 1 | R | SA | + | M | 34 | N | N | + | N | + | + | + | + | + | |||||||||||||
| 29 | Whyte et al. 1 [135] | 2016 | + | 2 | R | Cau | F | <3 | + | <3 | + | + | + | ||||||||||||||||||
| 30 | Whyte et al. 2 [135] | 2016 | + | 3 | R | Cau | F | 37 | <3 | + | + | + | |||||||||||||||||||
| 31 | Boissel et al. 2017 [145] | + | 25 | SB | F | 25 | + | + | + | ||||||||||||||||||||||
| 32 | Hung et al. [146] | 2019 | + | 1 | R | E | + | M | 37 | - | - | + | + | + | + | + | |||||||||||||||
| 33 | Hernández-Zavala et al. [139] | 2020 | + | 2 | R | M | M | 40 | N | N | + | + | N | + | + | + | + | ||||||||||||||
| 34 | Hernández-Zavala et al. [139] | 2020 | + | 1 | R | M | M | 21 | N | N | + | N | + | + | + | + | |||||||||||||||
| 35 | Eltan et al. [151] | 2020 | + | 7 | R | T | - | M | 37 | N | N | + | N | + | + | + | + | + | + | + | |||||||||||
| 36 | El-Dessouky et al. 1 [138] | 2020 | + | 1 | R | E | + | M | 26 | N | N | N | + | + | + | + | + | ||||||||||||||
| 37 | El-Dessouky et al. 2 [138] | 2020 | + | 1 | R | E | + | F | 38 | N | N | N | + | + | |||||||||||||||||
| 38 | El-Dessouky et a. 3 [138] | 2020 | + | 1 | R | E | M | 38 | N | N | N | + | + | + | + | + | |||||||||||||||
| 39 | Lulla et al. [153] | 2020 | + | I | F | + | + | + | + | ||||||||||||||||||||||
| 40 | Bajaj et al. [154] | 2021 | + | I | F | N | N | N | + | N | + | + |
| Case | Reference | Year | Non-Lethal | Brain Calcifications | Microcephaly | Brachy/Turri/Plagio-Cephaly | Wide Fontanelles | Bossed Forehead | Midface Hypoplasia | Ocular Proptosis | Hypertelorism | Visual Alterations | Low/Depressed Nasal Bridge | Short/Hypoplastic Nose | Choanal Stenosis/Atresia | Gingival Hyperplasia | High Palate | Large/Protruding Tongue | Abnormal Teeth | Micrognatia | Prognathism | Low Set Ears | Dysplastic Ears | Hypoacusia |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Simpson et al. [136] | 2009 | + | + | + | + | + | + | + | + | + | + | + | + | ||||||||||
| 2 | Simpson et al. [136] | 2009 | + | + | + | + | + | + | + | + | + | + | + | + | ||||||||||
| 3 | Fradin et al. (1) [144] | 2011 | + | + | + | + | + | + | + | + | ||||||||||||||
| 4 | Fradin et al. (2) [144] | 2011 | + | + | + | + | + | + | ||||||||||||||||
| 5 | koob et al. [11] | 2011 | + | + | + | + | + | + | + | |||||||||||||||
| 6 | Rafaelsen et al. (1) [143] | 2013 | + | + | + | + | + | + | + | + | ||||||||||||||
| 7 | Rafaelsen et al. (2) [143] | 2013 | + | + | + | + | + | + | + | |||||||||||||||
| 8 | Vishwanath et al. [171] | 2014 | + | + | + | + | + | + | + | + | + | + | + | |||||||||||
| 9 | Mahmood et al. [148] | 2014 | + | + | + | + | + | + | + | + | + | + | + | |||||||||||
| 10 | Takeyari et al. [83] | 2014 | + | + | ||||||||||||||||||||
| 11 | Acevedo et al. (1) [141] | 2015 | + | + | + | + | + | + | + | + | + | + | ||||||||||||
| 12 | Acevedo et al. (2) [141] | 2015 | + | + | + | + | + | + | + | + | + | |||||||||||||
| 13 | Acevedo et al. (3) [141] | 2015 | + | + | + | + | + | + | + | + | + | |||||||||||||
| 14 | Acevedo et al. (4) [141] | 2015 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||||||
| 15 | Acevedo et al. (5) [141] | 2015 | + | + | + | + | + | + | + | + | + | + | + | + | ||||||||||
| 16 | Elalaoui et al. (1) [142] | 2016 | + | + | + | + | + | + | + | + | + | + | ||||||||||||
| 17 | Elalaoui et al. (2) [142] | 2016 | + | + | + | + | + | + | + | + | + | + | ||||||||||||
| 18 | Das et al. [172] | 2017 | + | + | + | + | + | + | + | + | ||||||||||||||
| 19 | Tamai et al. [4] | 2017 | + | + | + | + | + | + | + | + | + | + | ||||||||||||
| 20 | Sheth et al. [6] | 2018 | + | + | + | + | ||||||||||||||||||
| 21 | Rolvien et al. [5] | 2018 | + | |||||||||||||||||||||
| 22 | Mamedova et al. [147] | 2019 | + | + | + | + | + | + | + | + | ||||||||||||||
| 23 | Mameli et al. 1 [140] | 2020 | + | + | + | + | + | + | - | + | - | + | ||||||||||||
| 24 | Mameli et al. 2 [140] | 2020 | + | + | + | + | + | + | + | |||||||||||||||
| 25 | Mameli et al. 3 [140] | 2020 | + | - | + | + | + | + | + | + | + | + | ||||||||||||
| 26 | Günes et al. [173] | 2005 | + | + | + | + | + | + | + | + | + | + | + | + | + | |||||||||
| 27 | Hirst et al. 1 [174] | 2021 | + | + | + | + | + | |||||||||||||||||
| 28 | Hirst et al. 2 [174] | 2021 | + | + | + | + | + | hyp | + | + | + | |||||||||||||
| 29 | Hirst et al. 3 [174] | 2021 | + | + | + | + | + | + | + | |||||||||||||||
| 30 | Eras et al. [150] | 2021 | + | + | + | + | + | + | + | + | ||||||||||||||
| Case | Reference | Year | Non-Lethal | Age (Years) | Population | Consanguinity | Gender | Gestational Weeks | Length (Percentile) | Local Osteosclerosis | Generalized Osteosclerosis | Head Circumference | Craniosynostosis | Small Thorax | Pectus Excavatum | Cervical Ossification Defects | Vertebral Segmentation Defects | Sacral Ossification Defects | Periosteal Reaction | Short Limbs | Brachydactyily | Fingerpads | Thick Fingers | Fractures/Pseudofractures | Poor Cortico-Medullary-Differentiation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Simpson et al., 1 [136] | 2009 | + | 8y | ND | + | M | 38 | + | ||||||||||||||||
| 2 | Simpson et al., 2 [136] | 2009 | + | 11y | ND | + | + | + | + | + | |||||||||||||||
| 3 | Fradin et al., 1 [144] | 2011 | + | 4y | ND | + | 37 | N | + | N | |||||||||||||||
| 4 | Fradin et al., 2 [144] | 2011 | + | 1y | Al | + | 38 | <3 | + | N | + | + | + | + | + | ||||||||||
| 5 | Koob et al. [11] | 2011 | + | 1y | Al | + | F | N | + | + | + | + | + | ||||||||||||
| 6 | Rafaelsen et al., 1 [143] | 2013 | + | 18y | Nw | M | 36 | N | + | N | + | ||||||||||||||
| 7 | Rafaelsen et al., 2 [143] | 2013 | + | 16y | Nw | M | 37 | N | + | N | |||||||||||||||
| 8 | Vishwanath et al. [171] | 2014 | + | 1mo | I | + | <P3 | + | <P3 | + | + | ||||||||||||||
| 9 | Mahmood et al. [148] | 2014 | + | 3y | As | + | M | 36 | N | + | + | ||||||||||||||
| 10 | Takeyari et al. [83] | 2014 | + | 61y | Jp | + | M | <3 | + | + | |||||||||||||||
| 11 | Acevedo et al., 1 [141] | 2015 | + | 27y | Br | + | M | + | |||||||||||||||||
| 12 | Acevedo et al., 2 [141] | 2015 | + | 22y | Br | + | M | + | + | ||||||||||||||||
| 13 | Acevedo et al., 3 [141] | 2015 | + | 21y | Br | + | F | + | |||||||||||||||||
| 14 | Acevedo et al., 4 [141] | 2015 | + | 5y | Br | + | M | + | |||||||||||||||||
| 15 | Acevedo et al., 5 [141] | 2015 | + | 4y | Br | + | M | + | |||||||||||||||||
| 16 | Elalaoui et al., 1 [142] | 2016 | + | 18y | Mor | + | F | 40 | N | + | + | + | |||||||||||||
| 17 | Elalaoui et al., 2 [142] | 2016 | + | 15y | Mor | + | M | 40 | N | + | |||||||||||||||
| 18 | Das et al. [172] | 2017 | + | 3mo | ND | F | + | ||||||||||||||||||
| 19 | Tamai et al. [4] | 2017 | + | 2y | Jp | 38 | <3 | + | N | ||||||||||||||||
| 20 | Sheth et al. [6] | 2018 | + | 6y | I | + | F | 40 | + | ||||||||||||||||
| 21 | Rolvien et al. [5] | 2018 | + | 72y | ND | + | |||||||||||||||||||
| 22 | Mamedova et al. [147] | 2019 | + | 39y | Ar | F | + | + | + | + | |||||||||||||||
| 23 | Mameli et al., 1 [140] | 2020 | + | 12y | P | F | + | <3 | |||||||||||||||||
| 24 | Mameli et al., 2 [140] | 2020 | + | 5mo | P | M | 37 | N | + | <3 | |||||||||||||||
| 25 | Mameli et al., 3 [140] | 2020 | + | 4mo | P | M | 37 | N | + | <3 | |||||||||||||||
| 26 | Günes et al. [173] | 2005 | + | 2hr | T | + | f | ND | N | + | <10 | + | |||||||||||||
| 27 | Hirst et al., 1 [174] | 2021 | + | 14 | So | M | + | + | |||||||||||||||||
| 28 | Hirst et al., 2 [174] | 2021 | + | 13y | So | M | + | ||||||||||||||||||
| 29 | Hirst et al., 3 [174] | 2021 | + | 11y | So | F | <3 | + | + | ||||||||||||||||
| 30 | Eras et al. [150] | 2021 | + | 1y | ND | + | M | 40 | N | + | N | + | |||||||||||||
| Feature Description | LC | NLC | References (NLC) | References (LC) |
|---|---|---|---|---|
| Developmental delay/disability, Language delay | 14 | [6,136,140,141,142,143] | ||
| Seizures | 8 | [136,141,142,148,150] | ||
| Hypoacusia | 10 | [4,136,140,141,142,150] | ||
| Hydrocephaly | 3 | [136,148] | ||
| Visual impairment | 2 | [140,141] | ||
| Dystonic movements | 1 | [148] | ||
| Hypertonia | 1 | [148] | ||
| Hypotonia | 3 | 1 | [6] | [135,137] |
| Non-visualization of pituitary gland | 1 | [172] | ||
| Mild cortical atrophy | 1 | [6] | ||
| Hypoplastic appearance of posterior part of the brain | 1 | [6] | ||
| Corpus callosal (agenesia, hypoplasia) | 2 | 1 | [6] | [139,151] |
| Mild paucity of the peritrigonal white matter surrounding the trigones of bilateral lateral ventricles | 1 | [6] | ||
| Cerebellum (conical, hypoplasia) | 3 | [9,149,158] | ||
| Cortical defects (abnormal gyral pattern, dysplasia, disorganization of the cortical layers) | 3 | [9,139,149] | ||
| Gliosis/astrogliosis | 2 | [158] | ||
| Encephalocele | 2 | [115,146] | ||
| Posterior fossa (small or hypoplastic) | 2 | [9,151] | ||
| Optic nerves and chiasm (small, atrophy) | 2 | [158,161] | ||
| Choroid plexus (bilateral cysts) | 1 | [10] | ||
| Cerebral atrophy | 1 | [151] | ||
| Hydrocephaly | 1 | [151] | ||
| Microphtalmia | 1 | [151] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palma-Lara, I.; Pérez-Ramírez, M.; García Alonso-Themann, P.; Espinosa-García, A.M.; Godinez-Aguilar, R.; Bonilla-Delgado, J.; López-Ornelas, A.; Victoria-Acosta, G.; Olguín-García, M.G.; Moreno, J.; et al. FAM20C Overview: Classic and Novel Targets, Pathogenic Variants and Raine Syndrome Phenotypes. Int. J. Mol. Sci. 2021, 22, 8039. https://doi.org/10.3390/ijms22158039
Palma-Lara I, Pérez-Ramírez M, García Alonso-Themann P, Espinosa-García AM, Godinez-Aguilar R, Bonilla-Delgado J, López-Ornelas A, Victoria-Acosta G, Olguín-García MG, Moreno J, et al. FAM20C Overview: Classic and Novel Targets, Pathogenic Variants and Raine Syndrome Phenotypes. International Journal of Molecular Sciences. 2021; 22(15):8039. https://doi.org/10.3390/ijms22158039
Chicago/Turabian StylePalma-Lara, Icela, Monserrat Pérez-Ramírez, Patricia García Alonso-Themann, Ana María Espinosa-García, Ricardo Godinez-Aguilar, José Bonilla-Delgado, Adolfo López-Ornelas, Georgina Victoria-Acosta, María Guadalupe Olguín-García, José Moreno, and et al. 2021. "FAM20C Overview: Classic and Novel Targets, Pathogenic Variants and Raine Syndrome Phenotypes" International Journal of Molecular Sciences 22, no. 15: 8039. https://doi.org/10.3390/ijms22158039
APA StylePalma-Lara, I., Pérez-Ramírez, M., García Alonso-Themann, P., Espinosa-García, A. M., Godinez-Aguilar, R., Bonilla-Delgado, J., López-Ornelas, A., Victoria-Acosta, G., Olguín-García, M. G., Moreno, J., & Palacios-Reyes, C. (2021). FAM20C Overview: Classic and Novel Targets, Pathogenic Variants and Raine Syndrome Phenotypes. International Journal of Molecular Sciences, 22(15), 8039. https://doi.org/10.3390/ijms22158039