
Bmi1 Augments Proliferation and Survival of Cortical Bone-Derived Stem Cells after Injury through Novel Epigenetic Signaling via Histone 3 Regulation

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results
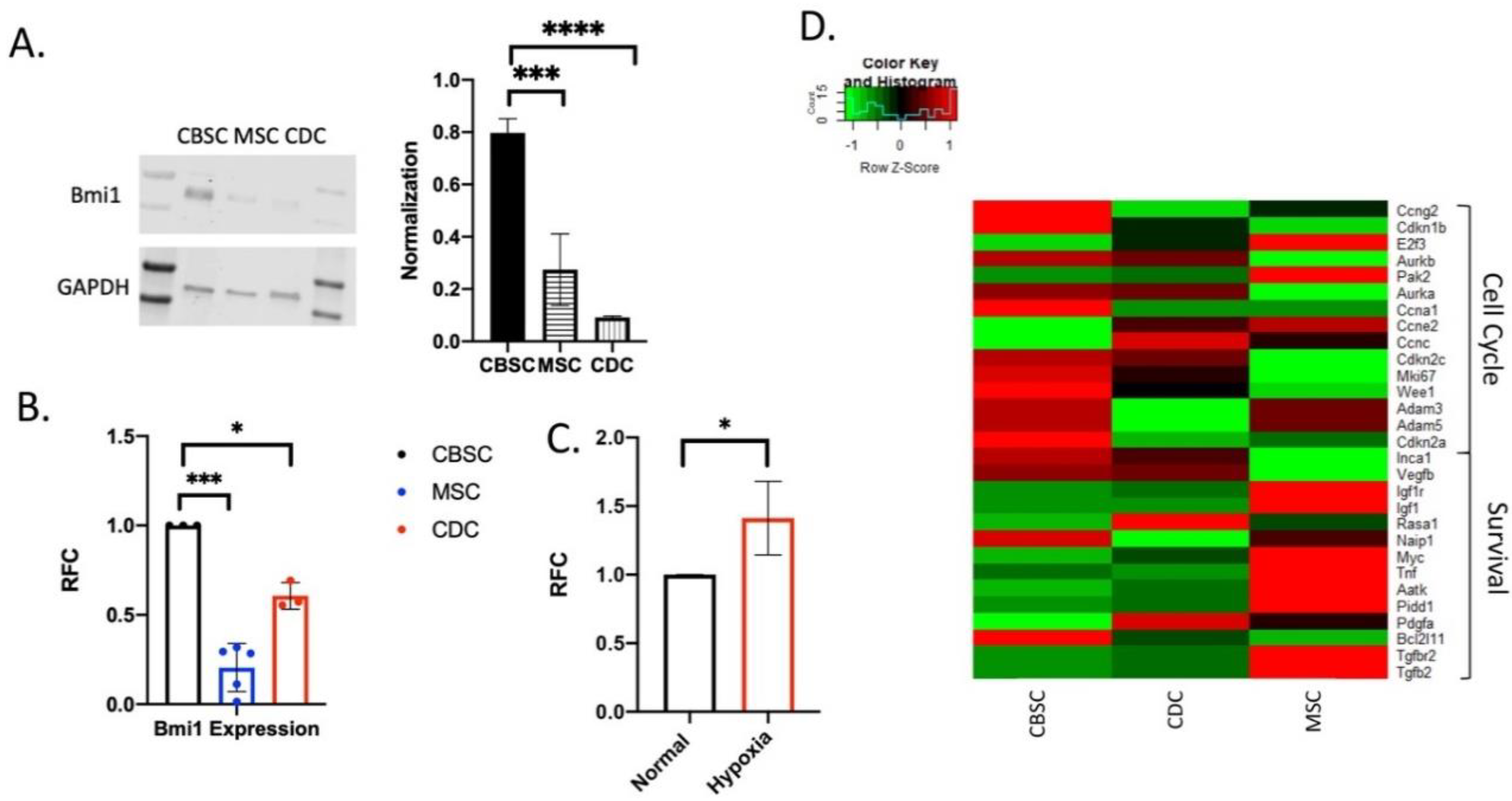
2.1. Expression of Bmi1 in CBSCs
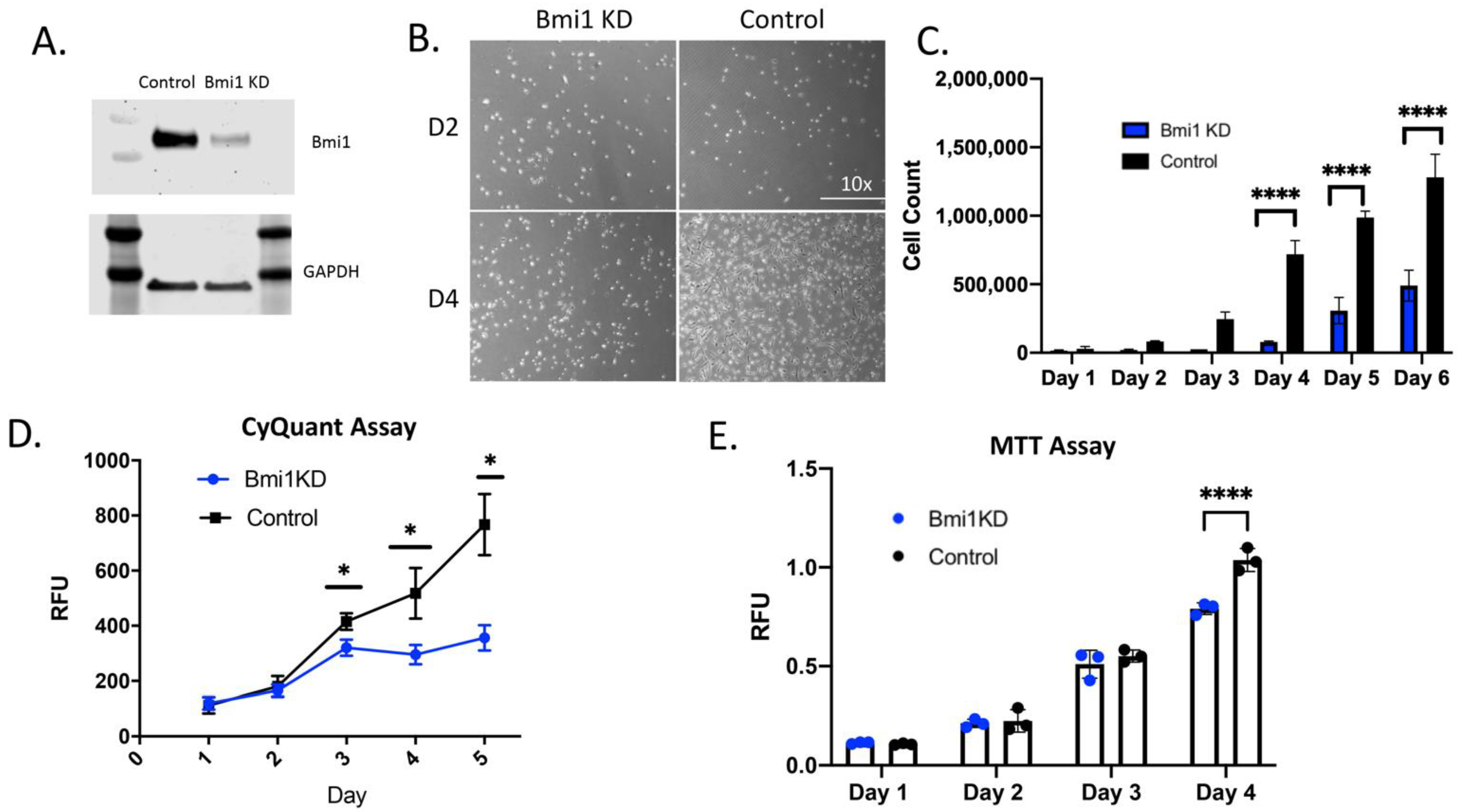
2.2. Bmi1 Regulates the Proliferative Potential of CBSCs
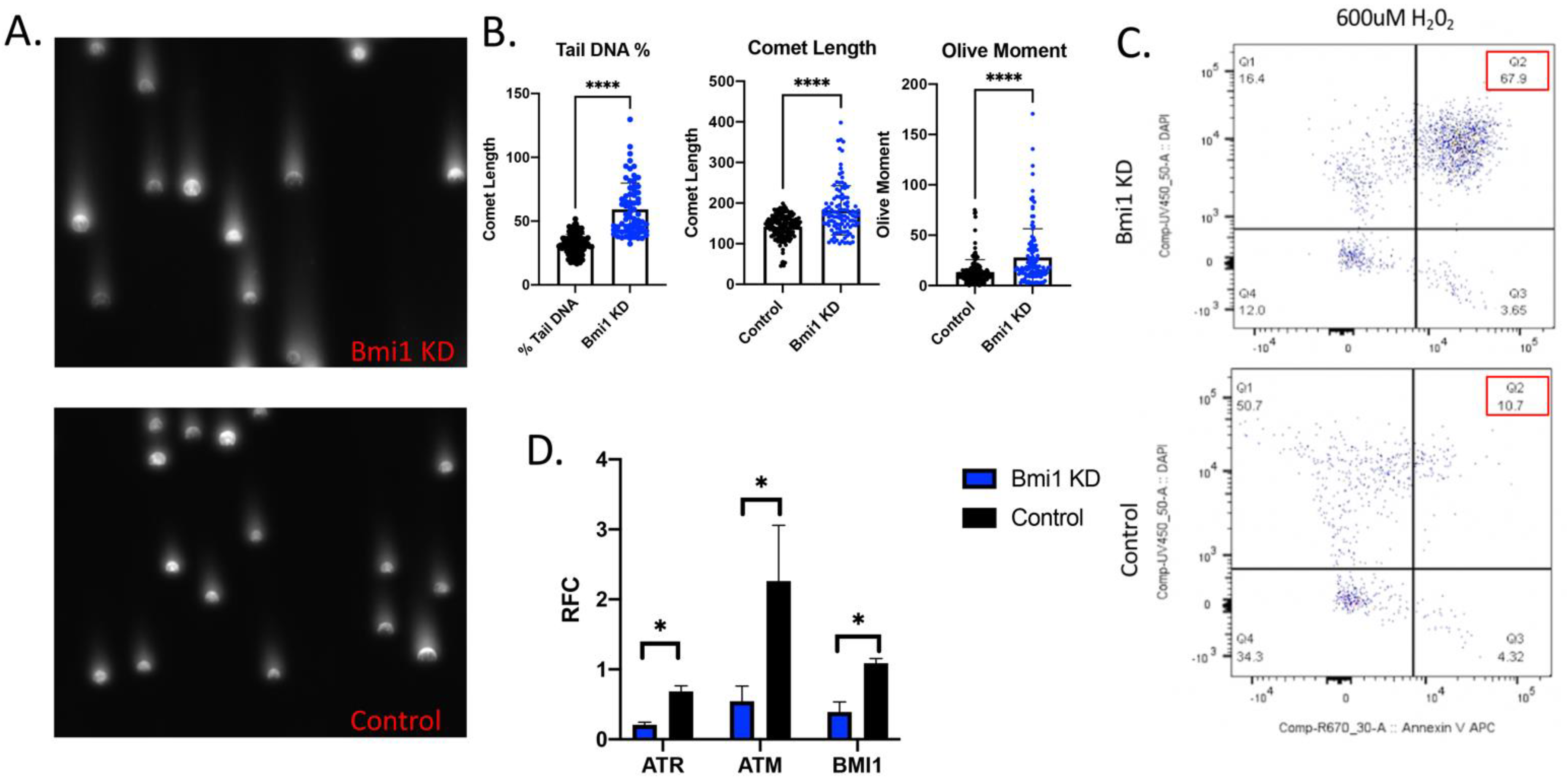
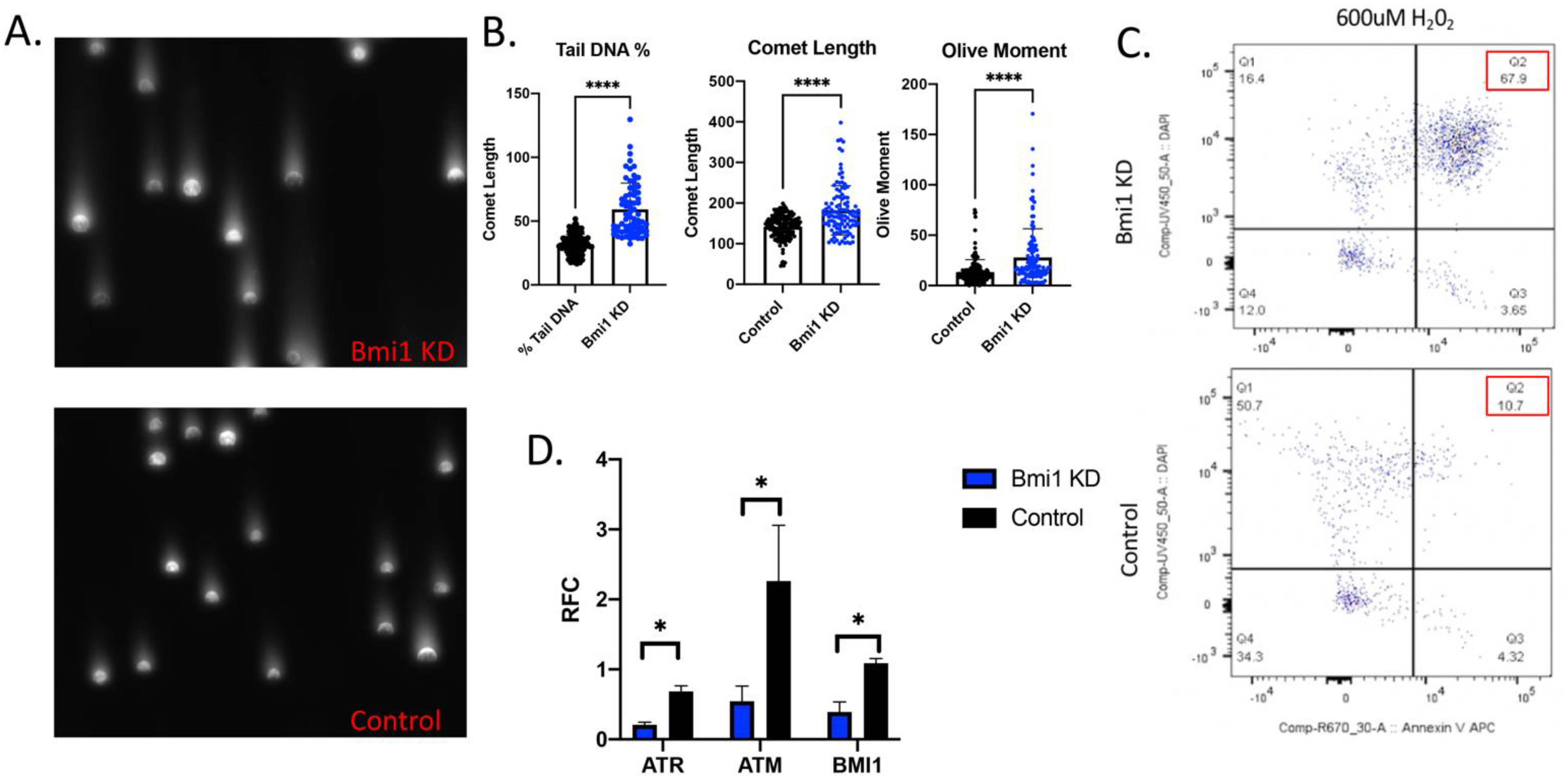
2.3. Bmi1 Knockdown Increases Apoptosis and Causes DNA Damage in CBSCs
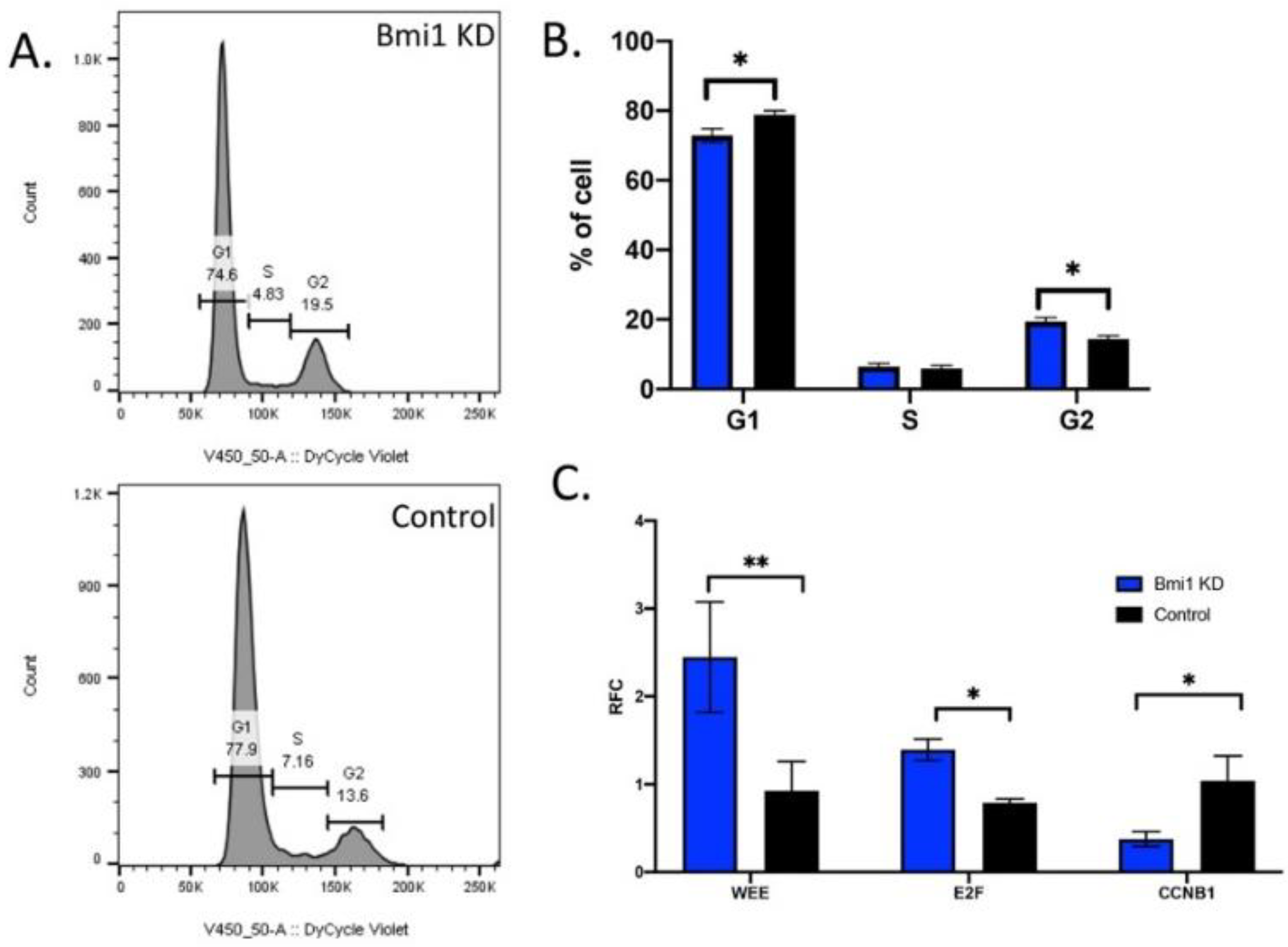
2.4. Bmi1 Knockdown Causes Cell Cycle Arrest in CBSCs
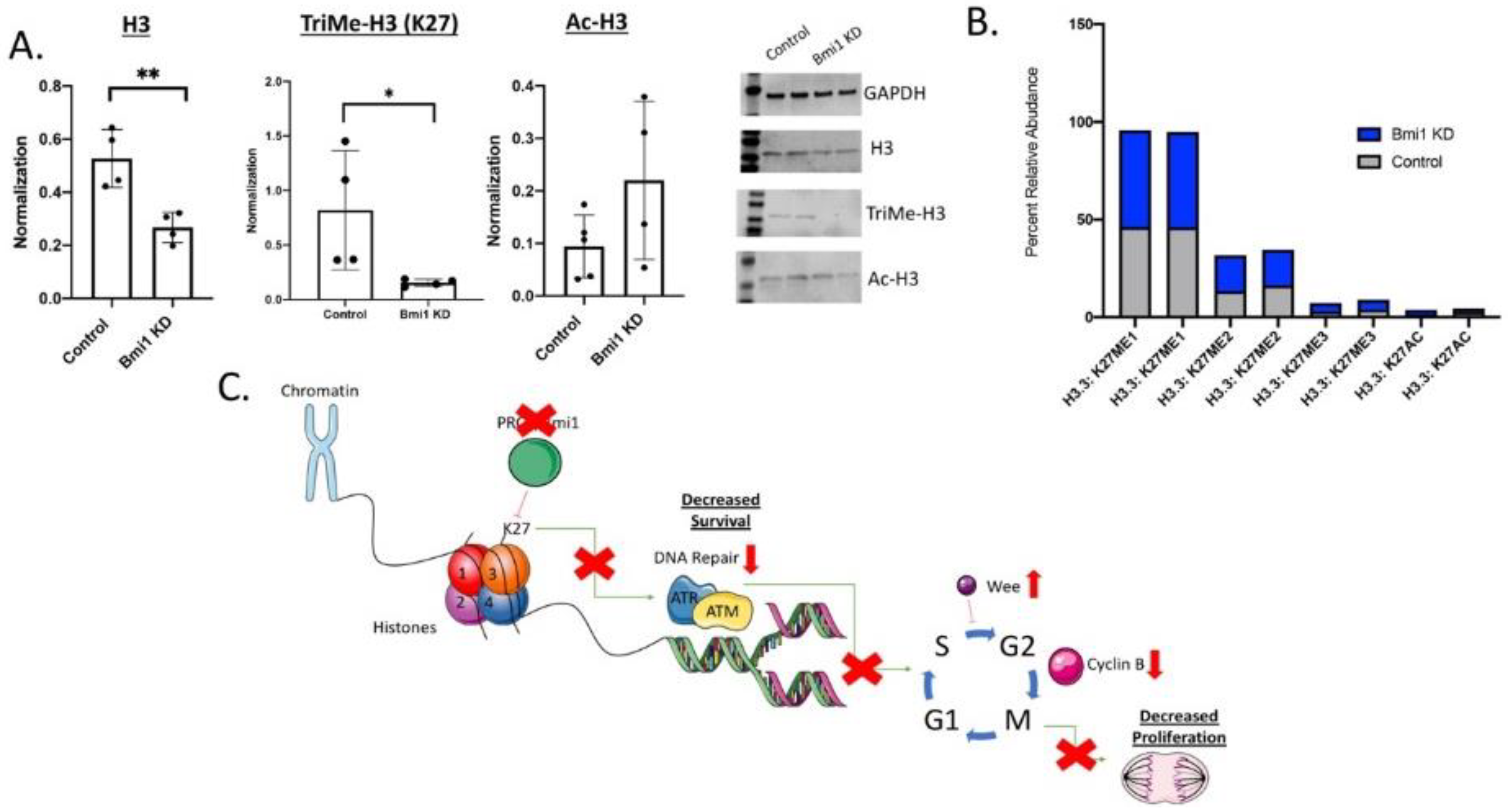
2.5. Bmi1 Modifies Epigenetic Regulation in CBSCs
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Bmi1 knockdown
4.2. Heat Map Comparison
4.3. Lentivirus Transduction
4.4. RT-PCR and Western Blotting
4.5. Proliferation Assays
4.6. Cell Death and Apoptosis Assays
4.7. Cell Cycle Assays
4.8. Histone Modification Assays
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Nomenclature
| ATM | Ataxia-Telangiectasia Mutated |
| ATR | ATM and Rad3-Related |
| CBSC | Cortical Bone Stem Cell |
| CCND | D type cyclins |
| CDC | Cardiac-Derived Stem Cell |
| CPC | Cardiac Progenitor Cell |
| E2F | E2F Transcription Factor 1 |
| GAPDH | Glyceraldehyde 3-Phosphate Dehydrogenase |
| MSC | Mesenchymal Stem Cell |
| PRC1 | Polycomb Repressive Complex 1 |
| PRC2 | Polycomb Repressive Complex 2 |
| WEE | WEE1 G2 Checkpoint Kinase |
References
- WHO | Prevention of Recurrences of Myocardial Infarction and Stroke Study. Available online: https://www.who.int/cardiovascular_diseases/priorities/secondary_prevention/country/en/index1.html (accessed on 21 July 2020).
- Coles, J.A.; Sigg, D.C.; Iaizzo, P.A. Reversible and Irreversible Damage of the Myocardium. In Handbook of Cardiac Anatomy, Physiology, and Devices; Iaizzo, P.A., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 161–170. [Google Scholar] [CrossRef]
- Heide, R.S.V.; Steenbergen, C. Cardioprotection and Myocardial Reperfusion: Pitfalls to Clinical Application. Circ. Res. 2013, 113, 464–477. [Google Scholar] [CrossRef]
- Stem Cell Therapy for Cardiac Repair: Benefits and Barriers | Expert Reviews in Molecular Medicine | Cambridge Core. Available online: https://www.cambridge.org/core/journals/expert-reviews-in-molecular-medicine/article/stem-cell-therapy-for-cardiac-repair-benefits-and-barriers/A84F2EB15990EFA8FBF433E428DA3CD3 (accessed on 11 December 2020).
- Jadczyk, T.; Faulkner, A.; Madeddu, P. Stem cell therapy for cardiovascular disease: The demise of alchemy and rise of pharmacology: Cardiovascular cell therapy. Br. J. Pharmacol. 2013, 169, 247–268. [Google Scholar] [CrossRef] [Green Version]
- Watt, F.M.; Driskell, R.R. The therapeutic potential of stem cells. Philos. Trans. R Soc. Lond. B Biol. Sci. 2010, 365, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Pré, B.C.D.; Doevendans, P.A.; van Laake, L.W. Stem cells for cardiac repair: An introduction. J. Geriatr. Cardiol. 2013, 10, 186–197. [Google Scholar] [CrossRef]
- Abbott, J.D.; Giordano, F.J. Stem cells and cardiovascular disease. J. Nucl. Cardiol. 2003, 10, 403–412. [Google Scholar] [CrossRef]
- Li, M.D.; Atkins, H.; Bubela, T. The global landscape of stem cell clinical trials. Regen. Med. 2013, 9, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Trounson, A.; McDonald, C. Stem Cell Therapies in Clinical Trials: Progress and Challenges. Cell Stem Cell 2015, 17, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, J.M.; Makarewich, C.A.; Sharp, T.E.; Starosta, T.; Zhu, F.; Hoffman, N.E.; Chiba, Y.; Madesh, M.; Berretta, R.M.; Kubo, H.; et al. Bone-derived stem cells repair the heart after myocardial infarction through transdifferentiation and paracrine signaling mechanisms. Circ. Res. 2013, 113, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Sharp, T.E., III; Schena, G.J.; Hobby, A.R.; Starosta, T.; Berretta, R.M.; Wallner, M.; Borghetti, G.; Gross, P.; Yu, D.; Johnson, J.; et al. Cortical Bone Stem Cell Therapy Preserves Cardiac Structure and Function After Myocardial Infarction. Circ. Res. 2017, 121, 1263–1278. [Google Scholar] [CrossRef]
- Mohsin, S.; Houser, S.R. Cortical Bone Derived Stem Cells for Cardiac Wound Healing. Korean Circ. J. 2019, 49, 314–325. [Google Scholar] [CrossRef]
- Kraus, L.; Ma, L.; Yang, Y.; Nguyen, F.; Hoy, R.C.; Okuno, T.; Khan, M.; Mohsin, S. Cortical Bone Derived Stem Cells Modulate Cardiac Fibroblast Response via miR-18a in the Heart After Injury. Front. Cell Dev. Biol. 2020, 8, 494. [Google Scholar] [CrossRef] [PubMed]
- Baraniak, P.R.; McDevitt, T.C. Stem cell paracrine actions and tissue regeneration. Regen. Med. 2010, 5, 121–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagnocchi, L.; Mazzoleni, S.; Zippo, A. Integration of Signaling Pathways with the Epigenetic Machinery in the Maintenance of Stem Cells. Stem Cells Int. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Lunyak, V.V.; Rosenfeld, M.G. Epigenetic regulation of stem cell fate. Hum. Mol. Genet. 2008, 17, R28–R36. [Google Scholar] [CrossRef]
- Sauvageau, M.; Sauvageau, G. Polycomb Group Proteins: Multi-Faceted Regulators of Somatic Stem Cells and Cancer. Cell Stem Cell 2010, 7, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Pivetti, S.; Fernandez-Perez, D.; D’Ambrosio, A.; Barbieri, C.M.; Manganaro, D.; Rossi, A.; Barnabei, L.; Zanotti, M.; Scelfo, A.; Chiacchiera, F.; et al. Loss of PRC1 activity in different stem cell compartments activates a common transcriptional program with cell type–dependent outcomes. Sci. Adv. 2019, 5, eaav1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, J.; O’Loghlen, A. PRC1 complex diversity: Where is it taking us? Trends Cell Biol. 2014, 24, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, L.; Vaseghi, H.R.; Liu, Z.; Lu, R.; Alimohamadi, S.; Yin, C.; Fu, J.D.; Wang, G.G.; Liu, J.; et al. Bmi1 Is a Key Epigenetic Barrier to Direct Cardiac Reprogramming. Cell Stem Cell 2016, 18, 382–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, J.J.L.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef]
- Kranc, K.R.; Bamforth, S.D.; Bragança, J.; Norbury, C.; van Lohuizen, M.; Bhattacharya, S. Transcriptional Coactivator Cited2 Induces Bmi1 and Mel18 and Controls Fibroblast Proliferation via Ink4a/ARF. Mol. Cell. Biol. 2003, 23, 7658–7666. [Google Scholar] [CrossRef] [Green Version]
- Park, I.-K.; Morrison, S.J.; Clarke, M.F. Bmi1, stem cells, and senescence regulation. J. Clin. Investig. 2004, 113, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Ganapathi, M.; Boles, N.C.; Charniga, C.; Lotz, S.; Campbell, M.; Temple, S.; Morse, R.H. Effect of Bmi1 over-expression on gene expression in adult and embryonic murine neural stem cells. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Herrero, D.; Tomé, M.; Cañón, S.; Cruz, F.M.; Carmona, R.M.; Fuster, E.; Roche, E.; Bernad, A. Redox-dependent BMI1 activity drives in vivo adult cardiac progenitor cell differentiation. Cell Death Differ. 2018, 25, 809–822. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Liu, H.-Q.; Yang, Z.; Fan, D.; Tang, Q.-Z. BMI1 in the heart: Novel functions beyond tumorigenesis. EBioMedicine 2021, 63, 103193. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Cao, R.; Viatour, P.; Sage, J.; Zhang, Y.; Xiong, Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4a tumor suppressor gene. Genes Dev. 2007, 21, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, I.H.; Andrin, C.; McDonald, D.; Hendzel, M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 2010, 191, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, W.; Dobrzyński, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res Ther. 2019, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Oshima, M.; Yuan, J.; Saraya, A.; Miyagi, S.; Konuma, T.; Yamazaki, S.; Osawa, M.; Nakauchi, H.; Koseki, H. Bmi1 confers resistance to oxidative stress on hematopoietic stem cells. PLoS ONE 2012, 7, e36209. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Oshima, M.; Yuan, J.; Saraya, A.; Miyagi, S.; Konuma, T.; Yamazaki, S.; Osawa, M.; Nakauchi, H.; Koseki, H.; et al. BMI-1, a promising therapeutic target for human cancer. Oncol. Lett. 2015, 10, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Dhawan, S.; Tschen, S.-I.; Bhushan, A. Bmi1 regulates the INK4a/ARF locus to control pancreatic beta-cell proliferation. Genes Dev. 2009, 23, 906–911. [Google Scholar] [CrossRef] [Green Version]
- Biehs, B.; Hu, J.K.H.; Strauli, N.B.; Sangiorgi, E.; Jung, H.; Heber, R.P.; Ho, S.; Goodwin, A.F.; Dasen, J.S.; Capecchi, M.R.; et al. BMI1 represses Ink4a/Arf and Hox genes to regulate stem cells in the rodent incisor. Nat. Cell Biol. 2013, 15, 846–852. [Google Scholar] [CrossRef]
- Chen, H.; Gu, X.; Su, I.H.; Bottino, R.; Contreras, J.L.; Tarakhovsky, A.; Kim, S.K. Polycomb protein Ezh2 regulates pancreatic β-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009, 23, 975–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valiente-Alandi, I.; Albo-Castellanos, C.; Herrero, D.; Arza, E.; Garcia-Gomez, M.; Segovia, J.C.; Capecchi, M.; Bernad, A. Cardiac Bmi1(+) cells contribute to myocardial renewal in the murine adult heart. Stem Cell Res. Ther. 2015, 6, 1–16. Available online: https://pubmed.ncbi.nlm.nih.gov/26503423/ (accessed on 21 July 2020). [CrossRef] [PubMed] [Green Version]
- Pan, M.-R.; Hsu, M.-C.; Chen, L.-T.; Hung, W.-C. Orchestration of H3K27 methylation: Mechanisms and therapeutic implication. Cell Mol. Life Sci. 2018, 75, 209–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdouh, M.; Hanna, R.; el Hajjar, J.; Flamier, A.; Bernier, G. The Polycomb Repressive Complex 1 Protein BMI1 Is Required for Constitutive Heterochromatin Formation and Silencing in Mammalian Somatic Cells. J. Biol. Chem. 2016, 291, 182–197. [Google Scholar] [CrossRef] [Green Version]
- Mohsin, S.; Troupes, C.D.; Starosta, T.; Sharp, T.E.; Agra, E.J.; Smith, S.; Duran, J.M.; Zalavadia, N.; Zhou, Y.; Kubo, H.; et al. Unique Features of Cortical Bone Stem Cells Associated with Repair of the Injured Heart. Circ. Res. 2015, 117, 1024–1033. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Liu, Y.; Yang, C. Evaluating In Vitro DNA Damage Using Comet Assay. J. Vis. Exp. 2017, 128, e56450. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward | Reverse |
|---|---|---|
| Bmi1 | CCAGGGCTTTTCAAAAATGA | CGGGTGAGCTGCATAAAAAT |
| ATM | GCTAGTTCTGTGAGCGATGC | GCAGCTAAAGGACTCATGGC |
| ATR | GCGAATCATGACCCCTTTCC | CACATCATCGAAGCCTGCAA |
| CCNB1 | AGCGAAGAGCTACAGGCAAG | TTCCACCTCTGGTTCACACA |
| E2F | CGAGGCCCTTGACTATCACT | AGGTCCCCAAAGTCACAGTC |
| WEE | AACCCCTTTACTCCGGATCC | TACGCTCTCTTTCTCCCACG |
| GAPDH | AGCGAGACCCCACTAACATC | TACGGCCAAATCCGTTCACA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraus, L.; Bryan, C.; Wagner, M.; Kino, T.; Gunchenko, M.; Jalal, W.; Khan, M.; Mohsin, S. Bmi1 Augments Proliferation and Survival of Cortical Bone-Derived Stem Cells after Injury through Novel Epigenetic Signaling via Histone 3 Regulation. Int. J. Mol. Sci. 2021, 22, 7813. https://doi.org/10.3390/ijms22157813
Kraus L, Bryan C, Wagner M, Kino T, Gunchenko M, Jalal W, Khan M, Mohsin S. Bmi1 Augments Proliferation and Survival of Cortical Bone-Derived Stem Cells after Injury through Novel Epigenetic Signaling via Histone 3 Regulation. International Journal of Molecular Sciences. 2021; 22(15):7813. https://doi.org/10.3390/ijms22157813
Chicago/Turabian StyleKraus, Lindsay, Chris Bryan, Marcus Wagner, Tabito Kino, Melissa Gunchenko, Wassy Jalal, Mohsin Khan, and Sadia Mohsin. 2021. "Bmi1 Augments Proliferation and Survival of Cortical Bone-Derived Stem Cells after Injury through Novel Epigenetic Signaling via Histone 3 Regulation" International Journal of Molecular Sciences 22, no. 15: 7813. https://doi.org/10.3390/ijms22157813
APA StyleKraus, L., Bryan, C., Wagner, M., Kino, T., Gunchenko, M., Jalal, W., Khan, M., & Mohsin, S. (2021). Bmi1 Augments Proliferation and Survival of Cortical Bone-Derived Stem Cells after Injury through Novel Epigenetic Signaling via Histone 3 Regulation. International Journal of Molecular Sciences, 22(15), 7813. https://doi.org/10.3390/ijms22157813