
Cardiomyocyte Proliferation as a Source of New Myocyte Development in the Adult Heart


Abstract
1. Introduction
2. Main Text
2.1. Targeting Cell Cycle Regulators to Induce Cardiomyocyte Proliferation
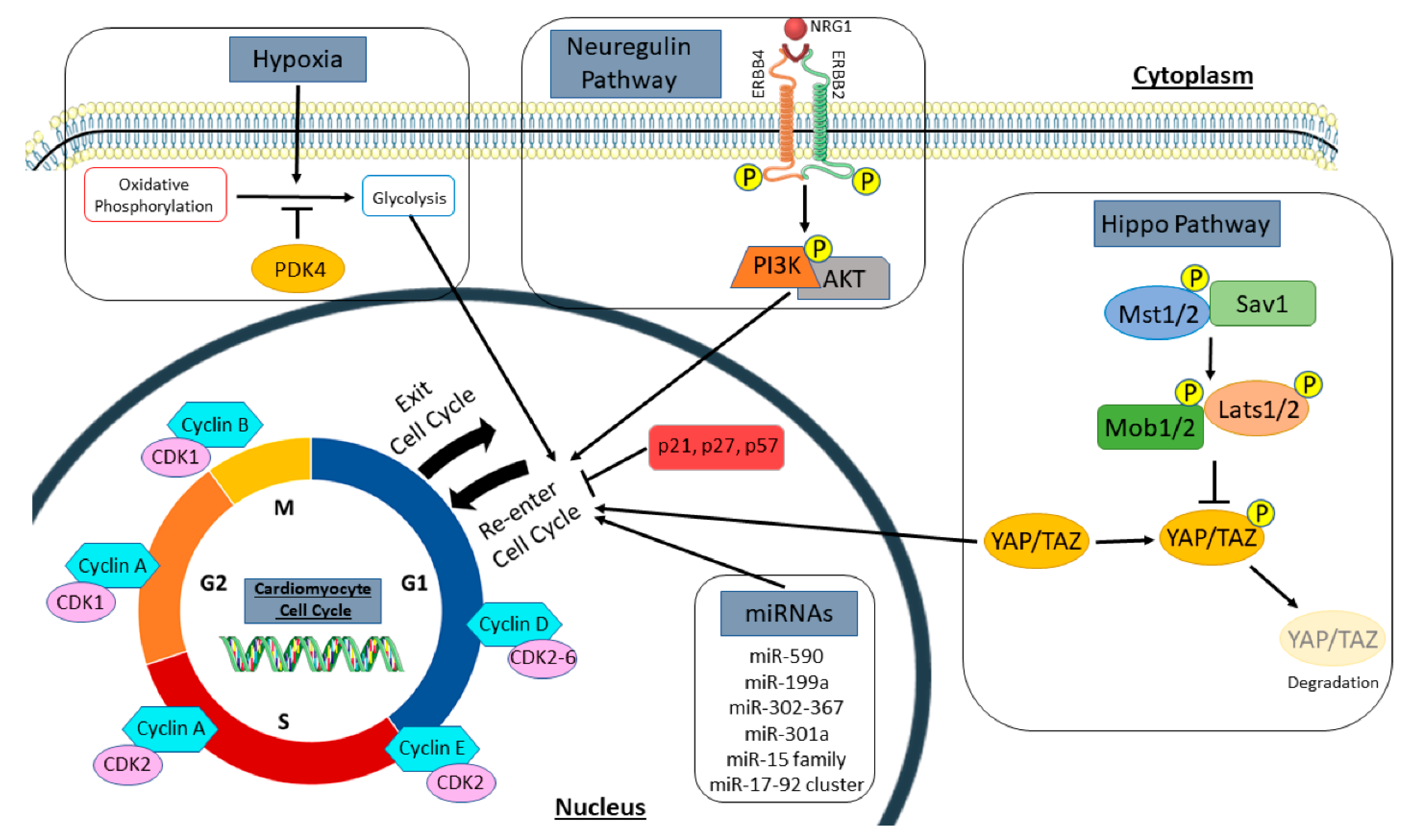
2.2. Manipulation of Signaling Pathways to Promote New Myocyte Formation
2.3. Selective Induction/Inhibition of Gene Expression can Induce Cell Cycle Re-Activation in Cardiac Myocytes
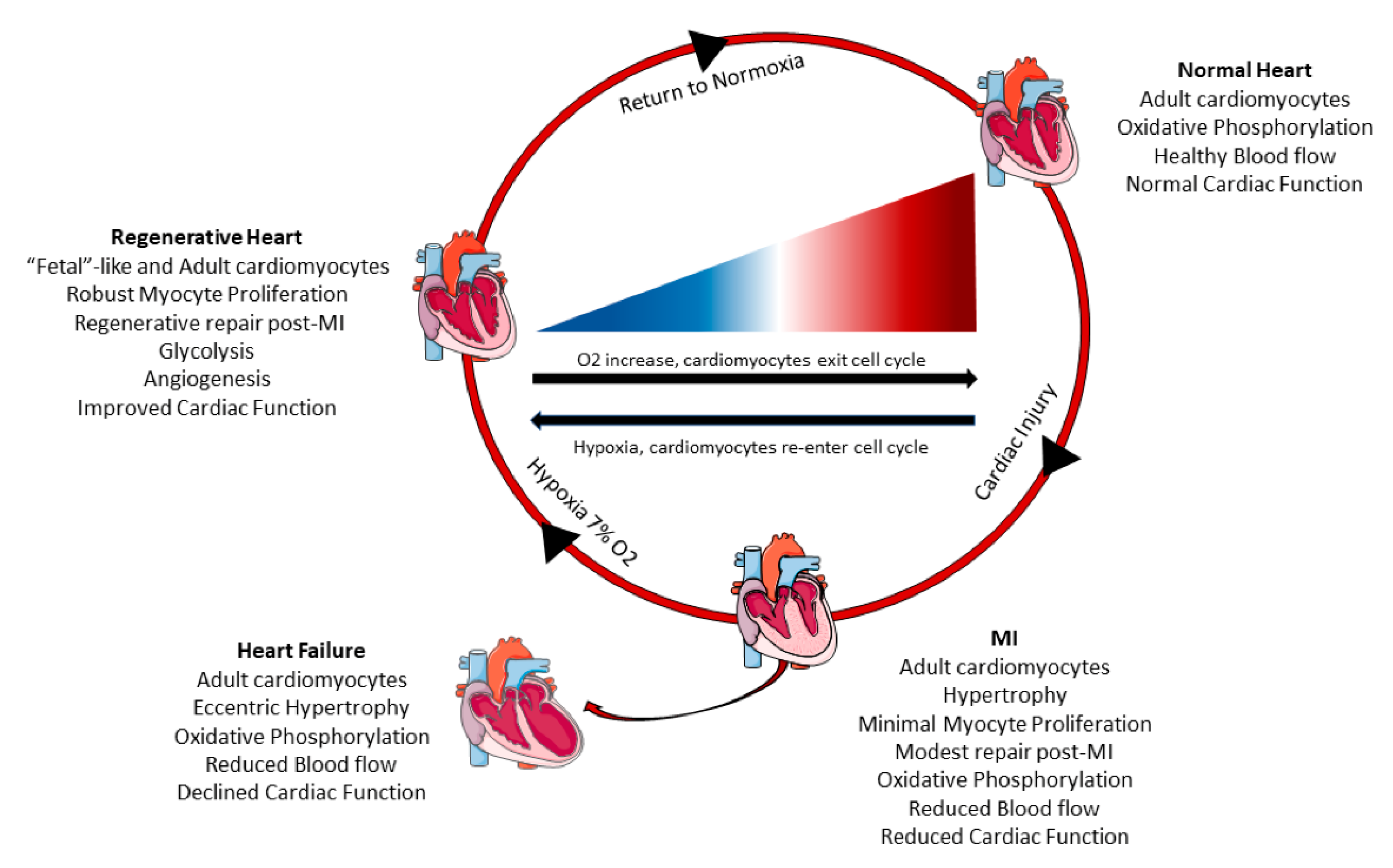
2.4. Changes in Cardiac Myocyte Metabolism Can Induce Cell Cycle Re-Entry
2.5. Limitations in Cardiomyocyte Proliferation Techniques
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Sutton, M.G.; Sharpe, N. Left ventricular remodeling after Myocardial infarction: Pathophysiology and therapy. Circulation 2000, 101, 2981–2988. [Google Scholar] [CrossRef]
- Gajarsa, J.J.; Kloner, R.A. Left ventricular remodeling in the post-infarction heart: A review of cellular, molecular mechanisms, and therapeutic modalities. Hear. Fail. Rev. 2011, 16, 13–21. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair after Myocardial infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Prabhu, S.D. Post-infarction ventricular remodeling: An array of molecular events. J. Mol. Cell. Cardiol. 2005, 38, 547–550. [Google Scholar] [CrossRef]
- Antonelli, L.; Katz, M.; Bacal, F.; Makdisse, M.R.P.; Correa, A.G.; Pereira, C.; Franken, M.; Fava, A.N.; Júnior, C.V.S.; Pesaro, A.E.P. Heart Failure with Preserved Left Ventricular Ejection Fraction in Patients with Acute Myocardial Infarction. Arq. Bras. Cardiol. 2015, 105, 145–150. [Google Scholar] [CrossRef]
- Hausenloy, D.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Uygur, A.; Lee, R.T. Mechanisms of Cardiac Regeneration. Dev. Cell 2016, 36, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.R.; Hippenmeyer, S.; Saadat, L.V.; Luo, L.; Weissman, I.L.; Ardehali, R. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 8850–8855. [Google Scholar] [CrossRef] [PubMed]
- Malliaras, K.; Zhang, Y.; Seinfeld, J.; Galang, G.; Tseliou, E.; Cheng, K.; Sun, B.; Aminzadeh, M.; Marbán, E. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol. Med. 2013, 5, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Senyo, S.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.-D.; Guerquin-Kern, J.-L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nat. Cell Biol. 2013, 493, 433–436. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, T.-S.; Lee, S.-T.; Wawrowsky, K.A.; Cheng, K.; Galang, G.; Malliaras, K.; Abraham, M.R.; Wang, C.; Marbán, E. Dedifferentiation and Proliferation of Mammalian Cardiomyocytes. PLoS ONE 2010, 5, e12559. [Google Scholar] [CrossRef]
- Zhang, Y.; López, N.G.; Li, N.; Zhang, Z.; Alver, N.; Liu, Y.; Martinson, A.M.; Mehri, A.; MacLellan, W.R. Single-cell imaging and transcriptomic analyses of endogenous cardiomyocyte dedifferentiation and cycling. Cell Discov. 2019, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.E.; Li, L.; Xia, X.; Fu, W.; Liao, Q.; Lan, C.; Yang, D.; Chen, H.; Yue, R.; Zen, C.; et al. Dedifferentiation, Proliferation, and Redifferentiation of Adult Mammalian Cardiomyocytes After Ischemic Injury. Circulation 2017, 136, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H. Transient Regenerative Potential of the Neonatal Mouse Heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef]
- Poolman, R.A.; Gilchrist, R.; Brooks, G. Cell cycle profiles and expressions of p21CIP1 AND P27KIP1 during myocyte development. Int. J. Cardiol. 1998, 67, 133–142. [Google Scholar] [CrossRef]
- Soonpaa, M.H.; Koh, G.Y.; Pajak, L.; Jing, S.; Wang, H.; Franklin, M.T.; Kim, K.K.; Field, L.J. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J. Clin. Investig. 1997, 99, 2644–2654. [Google Scholar] [CrossRef] [PubMed]
- Tane, S.; Kubota, M.; Okayama, H.; Ikenishi, A.; Yoshitome, S.; Iwamoto, N.; Satoh, Y.; Kusakabe, A.; Ogawa, S.; Kanai, A.; et al. Repression of cyclin D1 expression is necessary for the maintenance of cell cycle exit in adult mammalian cardiomyocytes. J. Biol. Chem. 2014, 289, 18033–18044. [Google Scholar] [CrossRef] [PubMed]
- Pasumarthi, K.B.; Nakajima, H.; Hisako, O.N.; Mark, H.S.; Loren, J.F. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ. Res. 2005, 96, 110–118. [Google Scholar] [CrossRef]
- Toischer, K.; Zhu, W.; Hünlich, M.; Mohamed, B.A.; Khadjeh, S.; Reuter, S.P.; Schäfer, K.; Ramanujam, D.; Engelhardt, S.; Field, L.J.; et al. Cardiomyocyte proliferation prevents failure in pressure overload but not volume overload. J. Clin. Investig. 2017, 127, 4285–4296. [Google Scholar] [CrossRef]
- Chaudhry, H.W.; Dashoush, N.H.; Tang, H.; Zhang, L.; Wang, X.; Wu, E.X.; Wolgemuth, D.J. Cyclin A2 Mediates Cardiomyocyte Mitosis in the Postmitotic Myocardium. J. Biol. Chem. 2004, 279, 35858–35866. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Asai, T.; Tang, H.; Dashoush, N.H.; Kara, R.J.; Costa, K.D.; Naka, Y.; Wu, E.X.; Wolgemuth, D.J.; Chaudhry, H.W. Cyclin A2 Induces Cardiac Regeneration After Myocardial Infarction and Prevents Heart Failure. Circ. Res. 2007, 100, 1741–1748. [Google Scholar] [CrossRef]
- Woo, Y.J.; Panlilio, C.M.; Cheng, R.; Liao, G.P.; Atluri, P.; Hsu, V.M.; Cohen, J.E.; Chaudhry, H.W. Therapeutic Delivery of Cyclin A2 Induces Myocardial Regeneration and Enhances Cardiac Function in Ischemic Heart Failure. Circulation 2006, 114, I-206. [Google Scholar] [CrossRef]
- Shapiro, S.D.; Ranjan, A.; Kawase, Y.; Cheng, R.; Kara, R.J.; Bhattacharya, R.; Guzmán-Martínez, G.; Sanz, J.; Garcia, M.J.; Chaudhry, H.W. Cyclin A2 Induces Cardiac Regeneration After Myocardial Infarction Through Cytokinesis of Adult Cardiomyocytes. Sci. Transl. Med. 2014, 6, 224ra27. [Google Scholar] [CrossRef]
- Bicknell, K.A.; Coxon, C.H.; Brooks, G. Forced expression of the cyclin B1-CDC2 complex induces proliferation in adult rat cardiomyocytes. Biochem. J. 2004, 382, 411–416. [Google Scholar] [CrossRef]
- Liao, H.-S.; Kang, P.M.; Nagashima, H.; Yamasaki, N.; Usheva, A.; Ding, B.; Lorell, B.H.; Izumo, S. Cardiac-Specific Overexpression of Cyclin-Dependent Kinase 2 Increases Smaller Mononuclear Cardiomyocytes. Circ. Res. 2001, 88, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, V.; Giacca, M.; Capogrossi, M.C.; Crescenzi, M.; Martelli, F. Knockdown of Cyclin-dependent Kinase Inhibitors Induces Cardiomyocyte Re-entry in the Cell Cycle. J. Biol. Chem. 2011, 286, 8644–8654. [Google Scholar] [CrossRef]
- Hatzistergos, K.E.; Williams, A.R.; Dykxhoorn, D.M.; Bellio, M.A.; Yu, W.; Hare, J.M. Tumor Suppressors RB1 and CDKN2a Cooperatively Regulate Cell-Cycle Progression and Differentiation during Cardiomyocyte Development and Repair. Circ. Res. 2019, 124, 1184–1197. [Google Scholar] [CrossRef] [PubMed]
- Volland, C.; Schott, P.; Didié, M.; Männer, J.; Unsöld, B.; Toischer, K.; Schmidt, C.; Urlaub, H.; Nickels, K.; Knöll, R.; et al. Control of p21Cip by BRCA1-associated protein is critical for cardiomyocyte cell cycle progression and survival. Cardiovasc. Res. 2019, 116, 592–604. [Google Scholar] [CrossRef]
- Agah, R.; Kirshenbaum, L.A.; Abdellatif, M.; Truong, L.D.; Chakraborty, S.; Michael, L.H.; Schneider, M.D. Adenoviral delivery of E2F-1 directs cell cycle reentry and p53-independent apoptosis in postmitotic adult myocar-dium in vivo. J. Clin. Investig. 1997, 100, 2722–2728. [Google Scholar] [CrossRef] [PubMed]
- Ebelt, H.; Zhang, Y.; Kampke, A.; Xu, J.; Schlitt, A.; Buerke, M.; Müller-Werdan, U.; Werdan, K.; Braun, T. E2F2 expression induces proliferation of terminally differentiated cardiomyocytes in vivo. Cardiovasc. Res. 2008, 80, 219–226. [Google Scholar] [CrossRef]
- Mahmoud, A.I.; Kocabas, F.; Muralidhar, S.A.; Kimura, W.; Koura, A.S.; Thet, S.; Porrello, E.; Sadek, H.A. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nat. Cell Biol. 2013, 497, 249–253. [Google Scholar] [CrossRef]
- Xiang, F.-L.; Guo, M.; Yutzey, K.E. Overexpression of Tbx20 in Adult Cardiomyocytes Promotes Proliferation and Improves Cardiac Function after Myocardial Infarction. Circulation 2016, 133, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Haginiwa, S.; Sadahiro, T.; Kojima, H.; Isomi, M.; Tamura, F.; Kurotsu, S.; Tani, H.; Muraoka, N.; Miyake, N.; Miyake, K.; et al. Tbx6 induces cardiomyocyte proliferation in postnatal and adult mouse hearts. Biochem. Biophys. Res. Commun. 2019, 513, 1041–1047. [Google Scholar] [CrossRef]
- Nguyen, N.U.N.; Canseco, D.C.; Xiao, F.; Nakada, Y.; Li, S.; Lam, N.T.; Muralidhar, S.A.; Savla, J.J.; Hill, J.A.; Le, V.; et al. A calcineurin–Hoxb13 axis regulates growth mode of mammalian cardiomyocytes. Nat. Cell Biol. 2020, 582, 271–276. [Google Scholar] [CrossRef]
- Mohamed, T.M.; Ang, Y.-S.; Radzinsky, E.; Zhou, P.; Huang, Y.; Elfenbein, A.; Foley, A.; Magnitsky, S.; Srivastava, D. Regulation of Cell Cycle to Stimulate Adult Cardiomyocyte Proliferation and Cardiac Regeneration. Cell 2018, 173, 104–116.e12. [Google Scholar] [CrossRef]
- Lin, Z.; von Gise, A.; Zhou, P.; Ma, Q.; Chen, J.; Jiang, J.; Seidman, J.G.; Wang, D.-Z.; Pu, W.T. Abstract 10: Cardiac-specific Yap Activation Improve Cardiac Function And Survival In An Experimental Murine Mi Model. Circ. Res. 2014, 115, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.; Morikawa, Y.; Leach, J.; Tao, G.; Willerson, J.T.; Johnson, R.L.; Martin, J.F. Hippo signaling impedes adult heart regeneration. Development 2013, 140, 4683–4690. [Google Scholar] [CrossRef] [PubMed]
- Leach, J.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nat. Cell Biol. 2017, 550, 260–264. [Google Scholar] [CrossRef]
- Bersell, K.; Shima, A.; Bernhard, H.; Bernhard, K. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 2009, 138, 257–270. [Google Scholar] [CrossRef] [PubMed]
- D’Uva, G.; Aharonov, A.; Lauriola, M.; Kain, D.; Yahalom-Ronen, Y.; Carvalho, S.; Weisinger, K.; Bassat, E.; Rajchman, D.; Yifa, O.; et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 2015, 17, 627–638. [Google Scholar] [CrossRef]
- Lesizza, P.; Prosdocimo, G.; Martinelli, V.; Sinagra, G.; Zacchigna, S.; Giacca, M. Single-Dose Intracardiac Injection of Pro-Regenerative MicroRNAs Improves Cardiac Function After Myocardial Infarction. Circ. Res. 2017, 120, 1298–1304. [Google Scholar] [CrossRef]
- Gabisonia, K.; Prosdocimo, G.; Aquaro, G.D.; Carlucci, L.; Zentilin, L.; Secco, I.; Ali, H.; Braga, L.; Gorgodze, N.; Bernini, F.; et al. MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature 2019, 569, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Z.-P.; Seok, H.Y.; Ding, J.; Kataoka, M.; Zhang, Z.; Hu, X.; Wang, G.; Lin, Z.; Wang, S.; et al. mir-17–92 Cluster Is Required for and Sufficient to Induce Cardiomyocyte Proliferation in Postnatal and Adult Hearts. Circ. Res. 2013, 112, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Kataoka, M.; Liu, N.; Liang, T.; Huang, Z.-P.; Gu, F.; Ding, J.; Liu, J.; Zhang, F.; Ma, Q.; et al. Therapeutic role of miR-19a/19b in cardiac regeneration and protection from myocardial infarction. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. USA 2013, 110, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, Y.; Wang, T.; Zhou, N.; Kong, J.; Chen, L.; Snitow, M.; Morley, M.; Li, D.; Petrenko, N.; et al. A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci. Transl. Med. 2015, 7, 279ra38. [Google Scholar] [CrossRef]
- Zhen, L.; Zhao, Q.; Lü, J.; Deng, S.; Xu, Z.; Zhang, L.; Zhang, Y.; Fan, H.; Chen, X.; Liu, Z.; et al. miR-301a-PTEN-AKT Signaling Induces Cardiomyocyte Proliferation and Promotes Cardiac Repair Post-MI. Mol. Ther. Nucleic Acids 2020, 22, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.; et al. Hypoxia induces heart regeneration in adult mice. Nat. Cell Biol. 2017, 541, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.C.; Lam, N.T.; Savla, J.J.; Nakada, Y.; Pereira, A.H.M.; Elnwasany, A.; Menendez-Montes, I.; Ensley, E.L.; Petric, U.B.; Sharma, G.; et al. Mitochondrial substrate utilization regulates cardiomyocyte cell-cycle progression. Nat. Metab. 2020, 2, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo Pathway Inhibits Wnt Signaling to Restrain Cardiomyocyte Proliferation and Heart Size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Kim, Y.; Lillian, B.S.; Qi, X.; McAnally, J.; Robert, J.S.; James, A.R.; Bassel, D.R.; Eric, N.O. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal. 2011, 4, ra70. [Google Scholar] [CrossRef]
- Von Gise, A.; Lin, Z.; Schlegelmilch, K.; Leah, B.H.; Gina, M.P.; Jessica, N.B.; Ma, Q.; Ishiwata, T.; Zhou, B.; Fernando, D.C.; et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. USA 2012, 109, 2394–2399. [Google Scholar] [CrossRef]
- Liu, R.; Jagannathan, R.; Li, F.; Lee, J.; Balasubramanyam, N.; Kim, B.S.; Yang, P.; Yechoor, V.K.; Moulik, M. Tead1 is required for perinatal cardiomyocyte proliferation. PLoS ONE 2019, 14, e0212017. [Google Scholar] [CrossRef]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar] [CrossRef]
- Del Re, D.P.; Yang, Y.; Nakano, N.; Cho, J.; Zhai, P.; Yamamoto, T.; Zhang, N.; Yabuta, N.; Nojima, H.; Pan, D.; et al. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J. Biol. Chem. 2013, 288, 3977–3988. [Google Scholar] [CrossRef]
- Monroe, T.O.; Matthew, C.H.; Morikawa, Y.; John, P.L.; Heallen, T.; Cao, S.; Peter, H.L.K.; Laat, W.; Xander, H.T.W.; George, G.R.; et al. YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In Vivo. Dev. Cell 2019, 48, 765–779. [Google Scholar] [CrossRef]
- Gründl, M.; Walz, S.; Hauf, L.; Schwab, M.; Werner, K.M.; Spahr, S.; Schulte, C.; Maric, H.M.; Carsten, P.A.; Gaubatz, S. Interaction of YAP with the Myb-MuvB (MMB) complex defines a transcriptional program to promote the proliferation of cardiomyocytes. PLoS Genet. 2020, 16, e1008818. [Google Scholar] [CrossRef]
- Honkoop, H.; de Bakker, D.E.; Aharonov, A.; Kruse, F.; Shakked, A.; Nguyen, P.D.; de Heus, C.; Garric, L.; Muraro, M.J.; Shoffner, A.; et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. eLife 2019, 8, e50163. [Google Scholar] [CrossRef]
- Beigi, F.; Schmeckpeper, J.; Pow-Anpongkul, P.; Payne, J.A.; Zhang, L.; Zhang, Z.; Huang, J.; Mirotsou, M.; Dzau, V.J. C3orf58, a Novel Paracrine Protein, Stimulates Cardiomyocyte Cell-Cycle Progression Through the PI3K–AKT–CDK7 Pathway. Circ. Res. 2013, 113, 372–380. [Google Scholar] [CrossRef]
- Fan, Y.; Ho, B.X.; Pang, J.K.S.; Pek, N.M.Q.; Hor, J.H.; Ng, S.-Y.; Soh, B.-S. Wnt/β-catenin-mediated signaling re-activates proliferation of matured cardiomyocytes. Stem. Cell Res. Ther. 2018, 9, 338. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Chen, J.; Lian, H.; Pei, J.; Li, Y.; Chen, X.; Song, S.; Xia, J.; Zhou, B.; Feng, J.; et al. PDGFR-β Signaling Regulates Cardiomyocyte Proliferation and Myocardial Regeneration. Cell Rep. 2019, 28, 966–978.e4. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Borikova, A.L.; Ben-Yair, R.; Guner-Ataman, B.; MacRae, C.A.; Lee, R.T.; Burns, C.G. Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc. Natl. Acad. Sci. USA 2014, 111, 1403–1408. [Google Scholar] [CrossRef] [PubMed]
- Lan, C.; Cao, N.; Chen, C.; Qu, S.; Fan, C.; Luo, H.; Zeng, A.; Yu, C.; Xue, Y.; Ren, H.; et al. Progesterone, via yes-associated protein, promotes cardiomyocyte proliferation and cardiac repair. Cell Prolif. 2020, 53, 12910. [Google Scholar] [CrossRef]
- Lin, Z.; Zhou, P.; Von Gise, A.; Gu, F.; Ma, Q.; Chen, J.; Guo, H.; Van Gorp, P.R.; Wang, D.-Z.; Pu, W.T. Pi3kcbLinks Hippo-YAP and PI3K-AKT Signaling Pathways to Promote Cardiomyocyte Proliferation and Survival. Circ. Res. 2015, 116, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Blankesteijn, W.M. Interventions in WNT Signaling to Induce Cardiomyocyte Proliferation: Crosstalk with Other Pathways. Mol. Pharmacol. 2019, 97, 90–101. [Google Scholar] [CrossRef]
- Aharonov, A.; Shakked, A.; Umansky, K.B.; Savidor, A.; Genzelinakh, A.; Kain, D.; Lendengolts, D.; Revach, O.-Y.; Morikawa, Y.; Dong, J.; et al. ERBB2 drives YAP activation and EMT-like processes during cardiac regeneration. Nat. Cell Biol. 2020, 22, 1–11. [Google Scholar] [CrossRef]
- Ikeda, S.; Mizushima, W.; Sciarretta, S.; Abdellatif, M.; Zhai, P.; Mukai, R.; Fefelova, N.; Oka, S.-I.; Nakamura, M.; Del Re, D.P.; et al. Hippo Deficiency Leads to Cardiac Dysfunction Accompanied by Cardiomyocyte Dedifferentiation During Pressure Overload. Circ. Res. 2019, 124, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Mano, M.; Ferro, M.D.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nat. Cell Biol. 2012, 492, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.; Johnson, B.A.; Aurora, A.B.; Simpson, E.; Nam, Y.-J.; Matkovich, S.; Dorn, G.W.; Van Rooij, E.; Olson, E.N. miR-15 Family Regulates Postnatal Mitotic Arrest of Cardiomyocytes. Circ. Res. 2011, 109, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Torrini, C.; Cubero, R.J.; Dirkx, E.; Braga, L.; Ali, H.; Prosdocimo, G.; Gutierrez, M.I.; Collesi, C.; Licastro, D.; Zentilin, L.; et al. Common Regulatory Pathways Mediate Activity of MicroRNAs Inducing Cardiomyocyte Proliferation. Cell Rep. 2019, 27, 2759–2771.e5. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.-L.; Zhu, B.-L.; Sun, Y.-Y.; Qiu, G.-R.; Fu, W.-N.; Jiang, H.-K. MicroRNA-144 Regulates Cardiomyocyte Proliferation and Apoptosis by Targeting TBX1 through the JAK2/STAT1 Pathway. Cytogenet. Genome Res. 2019, 159, 190–200. [Google Scholar] [CrossRef]
- Ding, Y.; Bi, L.; Wang, J. MiR-1180 promotes cardiomyocyte cell cycle re-entry after injury through the NKIRAS2–NFκB pathway. Biochem. Cell Biol. 2020, 98, 449–457. [Google Scholar] [CrossRef]
- Borden, A.; Kurian, J.; Nickoloff, E.; Yang, Y.; Troupes, C.D.; Ibetti, J.; Lucchese, A.M.; Gao, E.; Mohsin, S.; Koch, W.J.; et al. Transient Introduction of miR-294 in the Heart Promotes Cardiomyocyte Cell Cycle Reentry after Injury. Circ. Res. 2019, 125, 14–25. [Google Scholar] [CrossRef]
- Qin, X.; Gao, S.; Yang, Y.; Wu, L.; Wang, L. microRNA-25 promotes cardiomyocytes proliferation and migration via targeting Bim. J. Cell. Physiol. 2019, 234, 22103–22115. [Google Scholar] [CrossRef]
- Xiao, J.; Liu, H.; Cretoiu, D.; Toader, D.O.; Suciu, N.; Shi, J.; Shen, S.; Bei, Y.; Sluijter, J.P.; Das, S.; et al. miR-31a-5p promotes postnatal cardiomyocyte proliferation by targeting RhoBTB1. Exp. Mol. Med. 2017, 49, e386. [Google Scholar] [CrossRef]
- Huang, W.; Feng, Y.; Liang, J.; Yu, H.; Wang, C.; Wang, B.; Wang, M.; Jiang, L.; Meng, W.; Cai, W.; et al. Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. Nat. Commun. 2018, 9, 700. [Google Scholar] [CrossRef]
- Puente, B.N.; Wataru, K.; Shalini, A.M.; Moon, J.; James, F.A.; Kate, L.P.; Grinsfelder, D.; Beverly, A.R.; Chen, R.; Joseph, A.G.; et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef]
- Guimarães-Camboa, N.; Stowe, J.; Aneas, I.; Sakabe, N.; Cattaneo, P.; Henderson, L.; Kilberg, M.S.; Johnson, R.; Chen, J.; McCulloch, A.D.; et al. HIF1α Represses Cell Stress Pathways to Allow Proliferation of Hypoxic Fetal Cardiomyocytes. Dev. Cell 2015, 33, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Kimura, W.; Xiao, F.; Canseco, D.C.; Muralidhar, S.; Thet, S.; Zhang, H.M.; Abderrahman, Y.; Chen, R.; Garcia, J.A.; Shelton, J.M.; et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nat. Cell Biol. 2015, 523, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zheng, H.; Wu, J.; Ma, H.; Yu, J.; Yiliyaer, M. The key role of microtubules in hypoxia preconditioning-induced nuclear translocation of HIF-1α in rat cardiomyocytes. PeerJ 2017, 5, e3662. [Google Scholar] [CrossRef]
- Karliner, J.S.; Honbo, N.; Epstein, C.J.; Xian, M.; Lau, Y.F.C.; Gray, M.O. Neonatal mouse cardiac myocytes exhibit cardioprotection induced by hypoxic and pharmacologic preconditioning and by transgenic overexpression of human Cu/Zn superoxide dismutase. J. Mol. Cell. Cardiol. 2000, 32, 1779–1786. [Google Scholar] [CrossRef]
- Przyklenk, K.; Bauer, B.; Ovize, M.; A Kloner, R.; Whittaker, P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993, 87, 893–899. [Google Scholar] [CrossRef]
- Gaspar, A.; Lourenço, A.P.; Pereira, M.Á.; Azevedo, P.; Roncon-Albuquerque, R.; Marques, J.; Leite-Moreira, A.F. Randomized controlled trial of remote ischaemic conditioning in ST-elevation myocardial infarction as adjuvant to primary angioplasty (RIC-STEMI). Basic Res. Cardiol. 2018, 113, 14. [Google Scholar] [CrossRef]
- Günaydin, B.; Çakici, I.; Soncul, H.; Kalaycioglu, S.; Çevik, C.; Sancak, B.; Kanzik, I.; Karadenizli, Y. Does remote organ ischaemia trigger cardiac preconditioning during coronary artery surgery? Pharm. Res. 2000, 41, 493–496. [Google Scholar] [CrossRef]
- Ye, L.; Qiu, L.; Feng, B.; Jiang, C.; Huang, Y.; Zhang, H.; Hong, H.; Liu, J. Role of Blood Oxygen Saturation During Post-Natal Human Cardiomyocyte Cell Cycle Activities. JACC Basic Transl. Sci. 2020, 5, 447–460. [Google Scholar] [CrossRef]
- Mortimer, E.A.; Monson, R.R.; MacMahon, B. Reduction in Mortality from Coronary Heart Disease in Men Residing at High Altitude. N. Engl. J. Med. 1977, 296, 581–585. [Google Scholar] [CrossRef]
- Faeh, D.; Gutzwiller, F.; Bopp, M. Lower mortality from coronary heart disease and stroke at higher altitudes in Switzerland. Circulation 2009, 120, 495–501. [Google Scholar] [CrossRef]
- Faeh, D.; Moser, A.; Panczak, R.; Bopp, M.; Röösli, M.; Spoerri, A.; for the Swiss National Cohort Study Group. Independent at heart: Persistent association of altitude with ischaemic heart disease mortality after consideration of climate, topography and built environment. J. Epidemiol. Community Health 2016, 70, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Zebrowski, D.C.; Becker, R.; Engel, F.B. Towards regenerating the mammalian heart: Challenges in evaluating experimentally induced adult mammalian cardiomyocyte proliferation. Am. J. Physiol. Circ. Physiol. 2016, 310, H1045–H1054. [Google Scholar] [CrossRef]
- Tasic, B.; Miyamichi, K.; Hippenmeyer, S.; Vardhan, S.D.; Zeng, H.; Joo, W.; Zong, H.; Chen-Tsai, Y.; Luo, L. Extensions of MADM (mosaic analysis with double markers) in mice. PLoS ONE 2012, 7, e33332. [Google Scholar] [CrossRef]
- Sereti, K.-I.; Nguyen, N.B.; Kamran, P.; Zhao, P.; Ranjbarvaziri, S.; Park, S.; Sabri, S.; Engel, J.L.; Sung, K.; Kulkarni, R.P.; et al. Analysis of cardiomyocyte clonal expansion during mouse heart development and injury. Nat. Commun. 2018, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R., Jr.; Bingyan, J.W.; Pearl, J.Q.; Avitabile, D.; Ho, T.; Shaitrit, M.; Chavarria, M.; Firouzi, F.; Ebeid, D.; Megan, M.M.; et al. Cardiomyocyte cell cycle dynamics and proliferation revealed through cardiac-specific transgenesis of fluorescent ubiquitinated cell cycle indicator (FUCCI). J. Mol. Cell. Cardiol. 2019, 127, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Liao, Q.; Li, L.; Shi, Y.; Zeng, A.; Zeng, C.; Wang, W.E. An Aurora Kinase B–Based Mouse System to Efficiently Identify and Analyze Proliferating Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 570252. [Google Scholar] [CrossRef] [PubMed]
- Milliron, H.-Y.Y.; Weiland, M.J.; Kort, E.J.; Jovinge, S. Isolation of Cardiomyocytes Undergoing Mitosis With Complete Cytokinesis. Circ. Res. 2019, 125, 1070–1086. [Google Scholar] [CrossRef] [PubMed]
- Sampaio-Pinto, V.; Elsa, D.S.; Tiago, L.L.; Martins, P.D.; Pinto-do-Ó, P.; Diana, S.N. Stereological estimation of cardiomyocyte number and proliferation. Methods 2020, 190, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhang, W.; Mu, T.; Song, T.; Li, D. Aurora B kinase is required for cytokinesis through effecting spindle structure. Cell Biol. Int. 2013, 37, 436–442. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Pathway | Molecular Factor | Species | Injury | Completed Division | Results | Reference |
|---|---|---|---|---|---|---|
| Cell cycle promoters | Cyclin D1 | Mouse | No Injury | No | >40% Adult cardiomyocytes re-entered the cell cycle | [20] |
| Cyclin D2 | Mouse | MI | Yes | Cardiomyocyte DNA synthesis up to 5 months post-MI | [21] | |
| Cyclin A2 | Mouse | MI | Yes, in vitro | Cardiac Hyperplasia develops in postnatal hearts | [24] | |
| Cyclin A2 | Rat, Pig | HF, MI | Yes | ~18% increase in porcine ejection fraction post MI | [25,26] | |
| Cyclin B1-CDC2 | Rat | No Injury | No * | Increased number of small mononucleated myocytes | [27] | |
| CDK2 | Mouse | Pressure Overload | No * | Increased number of small less-differentiated mononuclear cardiomyocytes | [28] | |
| Cell cycle inhibitors | p21, p27, p57 (tKD) | Mouse, Rat | No Injury | Yes | Adult cardiomyocytes completed cytokinesis, expressed neonatal genes, and resembled neonatal morphology | [29] |
| p21 | Mouse | No Injury | No | Induced DNA synthesis without completion of mitosis | [31] | |
| Cell cycle transcription factors | Meis1, Hoxb13 (dKO) | Mouse | MI | Yes | Increased cytokinetic and mononucleated cardiomyocytes and improved EF post MI | [37] |
| Combination of cell cycle regulators | 4F, 2F-2i | Mouse, Rat, Human | MI | Yes | 15–20% Of adult mouse myocytes re-entered the cell cycle and completed cytokinesis | [38] |
| Hippo | human YAP | Mouse | MI | No | Increased cardiomyocyte proliferation post MI | [39] |
| hippo (KO) | Mouse | MI | Yes | Adult cardiomyocytes re-entered cell cycle | [40] | |
| Salv (KO, KD) | Mouse | HF | Yes * | DNA synthesis in adult myocytes up to 9 weeks post MI | [41] | |
| Neuregulin | NRG1, ERBB4 | Mouse, Rat | MI | Yes | Only mononuclear cardiomyocytes completed division | [42] |
| ERBB2 | Mouse | MI | Yes | Induced myocyte proliferation and redifferentiation | [43] | |
| miRNAs | human miR-590, human miR-199a | Mouse | MI | Yes | Induced cardiomyocyte proliferation and division, and cardiac repair post MI | [44] |
| human miR-199a | Pig | IR | Yes | Increased myocyte proliferation at 12 days post IR | [45] | |
| miR-17-92 cluster | Mouse | MI | Yes | Promoted cardiomyocyte DNA synthesis in adult mice | [46] | |
| miR-19a/19b | Mouse | HF, MI | Yes | Induced cardiac regeneration up to 12 months post-MI | [47] | |
| miR-15 family (KD) | Mouse | IR | No | Increased proliferation of adult cardiomyocytes post IR | [48] | |
| miR-302-367 | Mouse | MI | Yes | Robust cardiomyocyte proliferation in the adult heart | [49] | |
| miR-301a | Mouse | MI | Yes, in vitro | Induced cell cycle re-entry of cardiomyocytes after MI | [50] | |
| Metabolic regulators | Hypoxia | Mouse | MI | Yes | Increased cardiomyocyte division and cardiac repair | [51] |
| PDK4 (KO, KD) | Mouse | MI | Yes | Enhanced cardiomyocyte proliferation post MI | [52] |
| Approach | Labeling Strategy | Advantages | Cell Cycle Phase | Limitations |
|---|---|---|---|---|
| Markers | EdU | Labeling cardiomyocytes that undergo DNA synthesis; EdU processing is easy, rapid, and sensitive | S | EdU also labels cardiomyocytes undergoing DNA damage and repair; no evidence of whether myocytes continue to mitosis and complete division |
| Ki67 | Identifies cardiomyocytes that re-entered the cell cycle | G1, S, G2 | Does not identify if cardiomyocytes undergo cytokinesis | |
| pHH3 | Labeling of cardiomyocytes that have entered Mitosis | G2, M | Unable to distinguish whether cardiac myocytes underwent cytokinesis, endoreplication, or poly-nucleation; short expression time during mitosis so low detection | |
| Aurora B | Present between two daughter cells during cytokinesis | G2, M, cytokinesis | Expressed during shortest phases of the cell cycle (M and cytokinesis) so low detection | |
| Molecular Beacons | Sorts live, isolated cardiomyocytes that are in anaphase and telophase/cytokinesis | M, cytokinesis | Unable to sort isolated cardiomyocytes from large animal models by flow cytometry | |
| Mouse Models | MADM | Labeling of cardiomyocytes yellow (GFP + RFP) when they enter the cell cycle and upon completion of cell division, daughter cells are labeled as only green (GFP) and only red (RFP) | GFP + RFP: G1-M, only GFP and only RFP: cytokinesis | Daily or frequent tamoxifen injections can be cytotoxic for the animal |
| Rainbow | Clonal expansion of labeled cardiomyocytes identifies myocytes that proliferated from pre-existing myocytes | Indirectly measures cytokinesis | Indirect way to measure cardiomyocyte division | |
| FUCCI | Identifies which stages of the cell cycle cardiomyocytes are present, by oscillations of an orange (mKO) and green (AzG) color within cardiomyocyte nuclei | mKO: G1, AzG: S, G2, M; mKO + AzG: G1/S cell cycle arrest | AzG labeling unable to distinguish whether myocytes are in S phase, G2 phase, or M phase; inability to visualize cytokinesis | |
| Aurkb | Cardiomyocytes are labeled red when they undergo proliferation and division; Cytokinesis visualized by live time lapse imaging | S, G2, M, cytokinesis | Inability to distinguish whether myocytes in vivo are in S phase, G2 phase, M phase, or cytokinesis; completed cytokinesis limited to live cell imaging | |
| 3D Imaging | Stereology | Estimates the number of cardiomyocytes in the heart using representative thick sections | Indirectly measures new myocyte numbers | Accuracy of cardiomyocyte number is limited |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, J.; Mohsin, S.; Houser, S.R. Cardiomyocyte Proliferation as a Source of New Myocyte Development in the Adult Heart. Int. J. Mol. Sci. 2021, 22, 7764. https://doi.org/10.3390/ijms22157764
Johnson J, Mohsin S, Houser SR. Cardiomyocyte Proliferation as a Source of New Myocyte Development in the Adult Heart. International Journal of Molecular Sciences. 2021; 22(15):7764. https://doi.org/10.3390/ijms22157764
Chicago/Turabian StyleJohnson, Jaslyn, Sadia Mohsin, and Steven R. Houser. 2021. "Cardiomyocyte Proliferation as a Source of New Myocyte Development in the Adult Heart" International Journal of Molecular Sciences 22, no. 15: 7764. https://doi.org/10.3390/ijms22157764
APA StyleJohnson, J., Mohsin, S., & Houser, S. R. (2021). Cardiomyocyte Proliferation as a Source of New Myocyte Development in the Adult Heart. International Journal of Molecular Sciences, 22(15), 7764. https://doi.org/10.3390/ijms22157764