
New Structural Perspectives in G Protein-Coupled Receptor-Mediated Src Family Kinase Activation



Abstract
1. Introduction
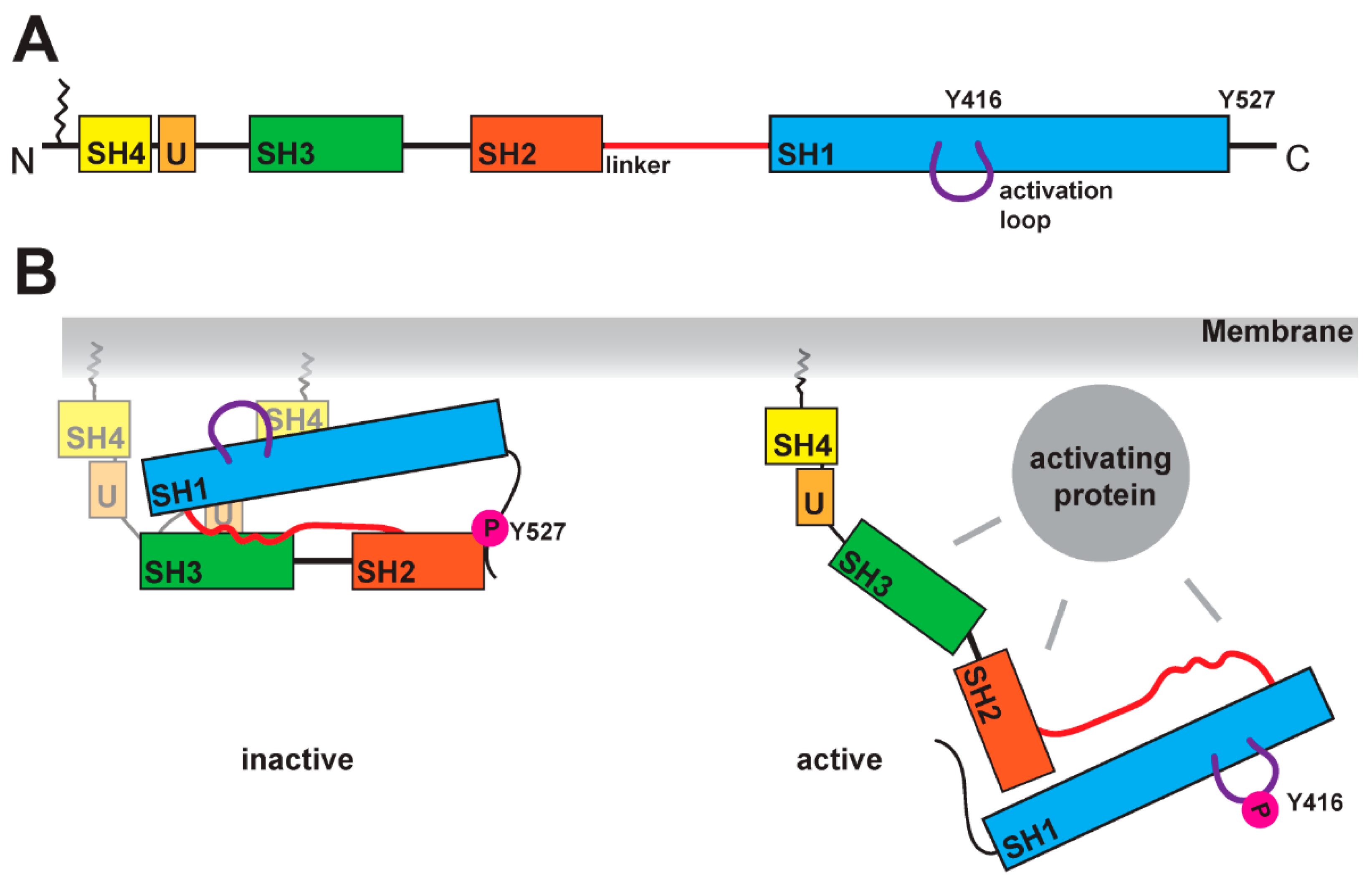
2. Structural Hallmarks of SFKs
3. Modes of SFK Activation
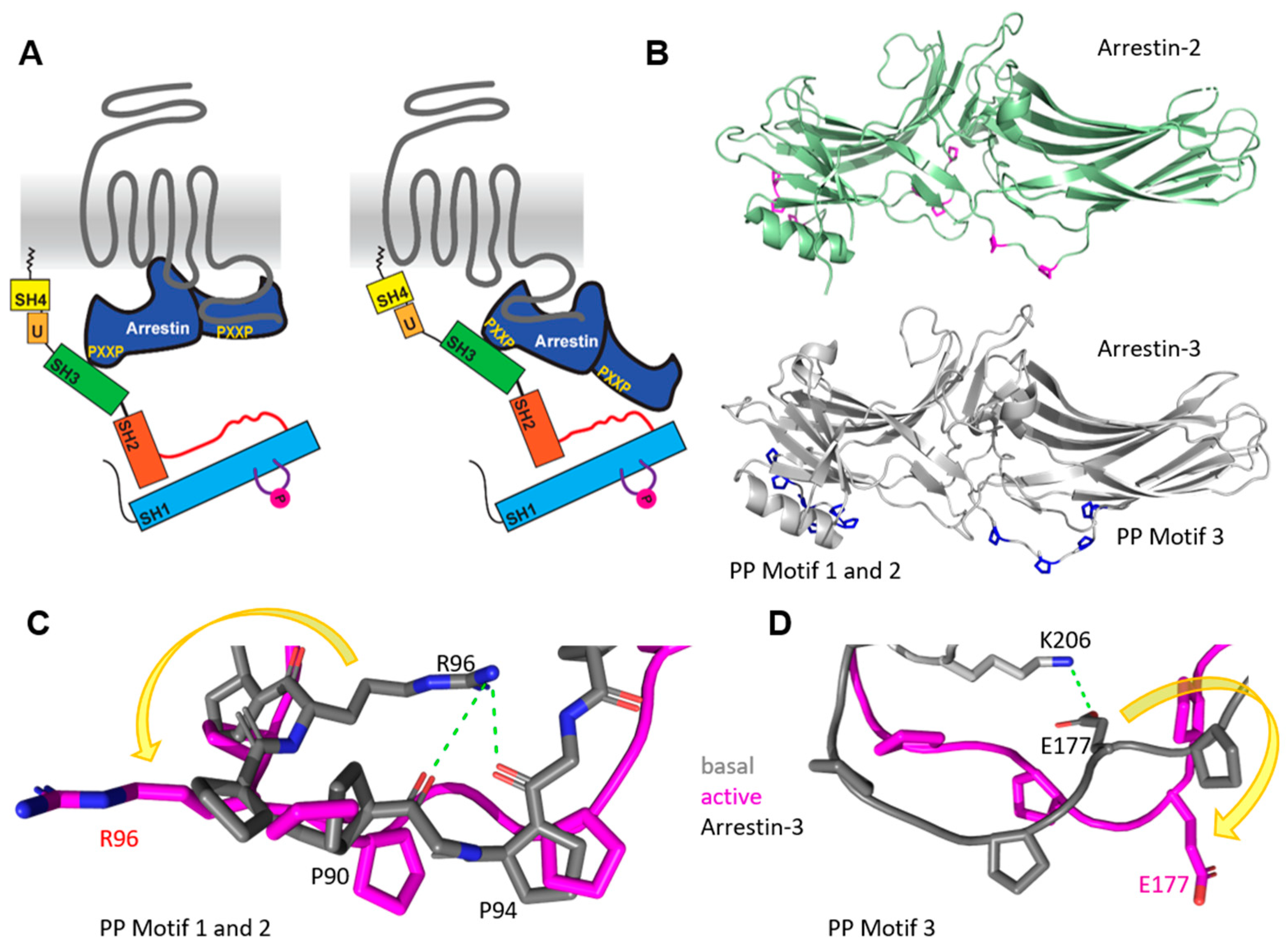
4. Src Activation through a GPCR–Arrestin Complex
5. Src Activation by G Proteins
6. Src Family Kinases as Direct Effectors of GPCRs
7. GPCR-Mediated Src Signaling with Undefined Mediators
Funding
Conflicts of Interest
References
- Tice, D.A.; Biscardi, J.S.; Nickles, A.L.; Parsons, S.J. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 1415–1420. [Google Scholar] [CrossRef]
- Cartwright, C.A.; Kamps, M.P.; Meisler, A.I.; Pipas, J.M.; Eckhart, W. pp60c-src activation in human colon carcinoma. J. Clin. Investig. 1989, 83, 2025–2033. [Google Scholar] [CrossRef]
- Talamonti, M.S.; Roh, M.S.; Curley, S.A.; Gallick, G.E. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J. Clin. Investig. 1993, 91, 53–60. [Google Scholar] [CrossRef]
- Guarino, M. Src signaling in cancer invasion. J. Cell. Physiol. 2010, 223, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, D.K.; Lee, A.; Lansing, T.J.; Crosby, R.M.; Jung, K.D.; Willard, D.; Luther, M.; Rodriguez, M.; Berman, J.; Gilmer, T.M. Involvement of pp60c-src with two major signaling pathways in human breast cancer. Proc. Natl. Acad. Sci. USA 1994, 91, 83–87. [Google Scholar] [CrossRef]
- Garmendia, I.; Pajares, M.J.; Hermida-Prado, F.; Ajona, D.; Bértolo, C.; Sainz, C.; Lavín, A.; Remírez, A.B.; Valencia, K.; Moreno, H.; et al. YES1 Drives Lung Cancer Growth and Progression and Predicts Sensitivity to Dasatinib. Am. J. Respir. Crit. Care Med. 2019, 200, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.A.; Parikh, N.U.; Gallick, G.E. Decreased tumorigenicity of a human colon adenocarcinoma cell line by an antisense expression vector specific for c-Src. Cell Growth Differ. 1997, 8, 269–274. [Google Scholar] [PubMed]
- Stettner, M.R.; Wang, W.; Nabors, L.B.; Bharara, S.; Flynn, D.C.; Grammer, J.R.; Gillespie, G.Y.; Gladson, C.L. Lyn kinase activity is the predominant cellular SRC kinase activity in glioblastoma tumor cells. Cancer Res. 2005, 65, 5535–5543. [Google Scholar] [CrossRef] [PubMed]
- Alcalá, S.; Mayoral-Varo, V.; Ruiz-Cañas, L.; López-Gil, J.C.; Heeschen, C.; Martín-Pérez, J.; Sainz, B. Targeting SRC Kinase Signaling in Pancreatic Cancer Stem Cells. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Patel, R.K.; Weir, M.C.; Shen, K.; Snyder, D.; Cooper, V.S.; Smithgall, T.E. Expression of myeloid Src-family kinases is associated with poor prognosis in AML and influences Flt3-ITD kinase inhibitor acquired resistance. PLoS ONE 2019, 14, e0225887. [Google Scholar] [CrossRef]
- Han, N.M.; Curley, S.A.; Gallick, G.E. Differential activation of pp60(c-src) and pp62(c-yes) in human colorectal carcinoma liver metastases. Clin. Cancer Res. 1996, 2, 1397–1404. [Google Scholar]
- Qiu, H.; Miller, W.T. Regulation of the nonreceptor tyrosine kinase Brk by autophosphorylation and by autoinhibition. J. Biol. Chem. 2002, 277, 34634–34641. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef]
- Stein, P.L.; Vogel, H.; Soriano, P. Combined deficiencies of Src, Fyn, and Yes tyrosine kinases in mutant mice. Genes Dev. 1994, 8, 1999–2007. [Google Scholar] [CrossRef]
- Kovács, M.; Németh, T.; Jakus, Z.; Sitaru, C.; Simon, E.; Futosi, K.; Botz, B.; Helyes, Z.; Lowell, C.A.; Mócsai, A. The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J. Exp. Med. 2014, 211, 1993–2011. [Google Scholar] [CrossRef] [PubMed]
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Biscardi, J.S.; Tice, D.A.; Parsons, S.J. c-Src, receptor tyrosine kinases, and human cancer. Adv. Cancer Res. 1999, 76, 61–119. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.M.; Costa, D.B.; Heist, R.S.; Garcia, E.; Lindeman, N.I.; Sholl, L.M.; Oxnard, G.R.; Johnson, B.E.; Hammerman, P.S. Treatment-related toxicities in a phase II trial of dasatinib in patients with squamous cell carcinoma of the lung. J. Thorac. Oncol. 2013, 8, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Koegl, M.; Zlatkine, P.; Ley, S.C.; Courtneidge, S.A.; Magee, A.I. Palmitoylation of multiple Src-family kinases at a homologous N-terminal motif. Biochem. J. 1994, 303, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Resh, M.D. Fatty acylation of proteins: New insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim. Biophys. Acta 1999, 1451, 1–16. [Google Scholar] [CrossRef]
- Pérez, Y.; Gairí, M.; Pons, M.; Bernadó, P. Structural characterization of the natively unfolded N-terminal domain of human c-Src kinase: Insights into the role of phosphorylation of the unique domain. J. Mol. Biol. 2009, 391, 136–148. [Google Scholar] [CrossRef]
- Arbesú, M.; Maffei, M.; Cordeiro, T.N.; Teixeira, J.M.C.; Pérez, Y.; Bernadó, P.; Roche, S.; Pons, M. The Unique Domain Forms a Fuzzy Intramolecular Complex in Src Family Kinases. Structure 2017, 25, 630–640.e4. [Google Scholar] [CrossRef]
- Teixeira, J.M.C.; Fuentes, H.; Bielskutė, S.; Gairi, M.; Żerko, S.; Koźmiński, W.; Pons, M. The Two Isoforms of Lyn Display Different Intramolecular Fuzzy Complexes with the SH3 Domain. Molecules 2018, 23. [Google Scholar] [CrossRef]
- Kurochkina, N.; Guha, U. SH3 domains: Modules of protein-protein interactions. Biophys. Rev. 2013, 5, 29–39. [Google Scholar] [CrossRef]
- Cooper, J.A.; Gould, K.L.; Cartwright, C.A.; Hunter, T. Tyr527 is phosphorylated in pp60c-src: Implications for regulation. Science 1986, 231, 1431–1434. [Google Scholar] [CrossRef]
- Smart, J.E.; Oppermann, H.; Czernilofsky, A.P.; Purchio, A.F.; Erikson, R.L.; Bishop, J.M. Characterization of sites for tyrosine phosphorylation in the transforming protein of Rous sarcoma virus (pp60v-src) and its normal cellular homologue (pp60c-src). Proc. Natl. Acad. Sci. USA 1981, 78, 6013–6017. [Google Scholar] [CrossRef]
- Nada, S.; Okada, M.; MacAuley, A.; Cooper, J.A.; Nakagawa, H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature 1991, 351, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, I.; Yamaguchi, N.; Suda, J.; Iwama, A.; Hirao, A.; Hashiyama, M.; Aizawa, S.; Suda, T. Analysis of CSK homologous kinase (CHK/HYL) in hematopoiesis by utilizing gene knockout mice. Biochem. Biophys. Res. Commun. 1996, 224, 172–179. [Google Scholar] [CrossRef]
- Sicheri, F.; Moarefi, I.; Kuriyan, J. Crystal structure of the Src family tyrosine kinase Hck. Nature 1997, 385, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.C.; Weijland, A.; Gonfloni, S.; Thompson, A.; Courtneidge, S.A.; Superti-Furga, G.; Wierenga, R.K. The 2.35 A crystal structure of the inactivated form of chicken Src: A dynamic molecule with multiple regulatory interactions. J. Mol. Biol. 1997, 274, 757–775. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Harrison, S.C.; Eck, M.J. Three-dimensional structure of the tyrosine kinase c-Src. Nature 1997, 385, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Ahler, E.; Register, A.C.; Chakraborty, S.; Fang, L.; Dieter, E.M.; Sitko, K.A.; Vidadala, R.S.R.; Trevillian, B.M.; Golkowski, M.; Gelman, H.; et al. A Combined Approach Reveals a Regulatory Mechanism Coupling Src’s Kinase Activity, Localization, and Phosphotransferase-Independent Functions. Mol. Cell 2019, 74, 393–408.e20. [Google Scholar] [CrossRef]
- Somani, A.K.; Bignon, J.S.; Mills, G.B.; Siminovitch, K.A.; Branch, D.R. Src kinase activity is regulated by the SHP-1 protein-tyrosine phosphatase. J. Biol. Chem. 1997, 272, 21113–21119. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, G.S.; Alexander, D.B.; Pellicena, P.; Zhang, Z.-Y.; Tsuda, H.; Miller, W.T. Src phosphorylates Cas on tyrosine 253 to promote migration of transformed cells. J. Biol. Chem. 2003, 278, 46533–46540. [Google Scholar] [CrossRef] [PubMed]
- Bjorge, J.D.; Pang, A.; Fujita, D.J. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J. Biol. Chem. 2000, 275, 41439–41446. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.; Maeda, S. Neuron-specific Cdk5 kinase is responsible for mitosis-independent phosphorylation of c-Src at Ser75 in human Y79 retinoblastoma cells. J. Biochem. 1999, 126, 957–961. [Google Scholar] [CrossRef]
- Tiran, Z.; Peretz, A.; Attali, B.; Elson, A. Phosphorylation-dependent regulation of Kv2.1 Channel activity at tyrosine 124 by Src and by protein-tyrosine phosphatase epsilon. J. Biol. Chem. 2003, 278, 17509–17514. [Google Scholar] [CrossRef]
- Renkema, G.H.; Pulkkinen, K.; Saksela, K. Cdc42/Rac1-mediated activation primes PAK2 for superactivation by tyrosine phosphorylation. Mol. Cell. Biol. 2002, 22, 6719–6725. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Z.; Izaguirre, G.; Lin, S.-Y.; Lee, H.Y.; Schaefer, E.; Haimovich, B. The phosphorylation of vinculin on tyrosine residues 100 and 1065, mediated by SRC kinases, affects cell spreading. Mol. Biol. Cell 2004, 15, 4234–4247. [Google Scholar] [CrossRef]
- Jones, D.A.; Benjamin, C.W. Phosphorylation of growth factor receptor binding protein-2 by pp60c-src tyrosine kinase. Arch. Biochem. Biophys. 1997, 337, 143–148. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Hawes, B.E.; van Biesen, T.; Luttrell, D.K.; Lansing, T.J.; Lefkowitz, R.J. Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gbetagamma subunit-mediated activation of mitogen-activated protein kinases. J. Biol. Chem. 1996, 271, 19443–19450. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, R.; Zhao, J.; Limbird, L.E. Arrestin serves as a molecular switch, linking endogenous alpha2-adrenergic receptor to SRC-dependent, but not SRC-independent, ERK activation. J. Biol. Chem. 2006, 281, 25948–25955. [Google Scholar] [CrossRef] [PubMed]
- Pakharukova, N.; Masoudi, A.; Pani, B.; Staus, D.P.; Lefkowitz, R.J. Allosteric activation of proto-oncogene kinase Src by GPCR-beta-arrestin complexes. J. Biol. Chem. 2020, 295, 16773–16784. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Luttrell, L.M.; Medvedev, A.V.; Pierce, K.L.; Daniel, K.W.; Dixon, T.M.; Lefkowitz, R.J.; Collins, S. Direct binding of activated c-Src to the beta 3-adrenergic receptor is required for MAP kinase activation. J. Biol. Chem. 2000, 275, 38131–38134. [Google Scholar] [CrossRef] [PubMed]
- Kaya, A.I.; Perry, N.A.; Gurevich, V.V.; Iverson, T.M. Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 14139–14149. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.-M.; Wang, J.Q. Dopaminergic and cholinergic regulation of Fyn tyrosine kinase phosphorylation in the rat striatum in vivo. Neuropharmacology 2015, 99, 491–499. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, H.; Voyno-Yasenetskaya, T.; Ye, R.D. Requirement of Gbetagamma and c-Src in D2 dopamine receptor-mediated nuclear factor-kappaB activation. Mol. Pharmacol. 2003, 64, 447–455. [Google Scholar] [CrossRef]
- Hattori, K.; Uchino, S.; Isosaka, T.; Maekawa, M.; Iyo, M.; Sato, T.; Kohsaka, S.; Yagi, T.; Yuasa, S. Fyn is required for haloperidol-induced catalepsy in mice. J. Biol. Chem. 2006, 281, 7129–7135. [Google Scholar] [CrossRef]
- Oldenhof, J.; Vickery, R.; Anafi, M.; Oak, J.; Ray, A.; Schoots, O.; Pawson, T.; von Zastrow, M.; van Tol, H.H. SH3 binding domains in the dopamine D4 receptor. Biochemistry 1998, 37, 15726–15736. [Google Scholar] [CrossRef][Green Version]
- Perkovska, S.; Méjean, C.; Ayoub, M.A.; Li, J.; Hemery, F.; Corbani, M.; Laguette, N.; Ventura, M.-A.; Orcel, H.; Durroux, T.; et al. V1b vasopressin receptor trafficking and signaling: Role of arrestins, G proteins and Src kinase. Traffic 2018, 19, 58–82. [Google Scholar] [CrossRef]
- Kraus, S.; Levy, G.; Hanoch, T.; Naor, Z.; Seger, R. Gonadotropin-releasing hormone induces apoptosis of prostate cancer cells: Role of c-Jun NH2-terminal kinase, protein kinase B, and extracellular signal-regulated kinase pathways. Cancer Res. 2004, 64, 5736–5744. [Google Scholar] [CrossRef] [PubMed]
- Kraus, S.; Benard, O.; Naor, Z.; Seger, R. C-Src is Activated by the EGF Receptor in a Pathway that Mediates JNK and ERK Activation by Gonadotropin-Releasing Hormone in COS7 Cells. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Xu, J.; Lei, G.; Lombroso, P.J.; Jackson, M.F.; MacDonald, J.F. STEP activation by Gαq coupled GPCRs opposes Src regulation of NMDA receptors containing the GluN2A subunit. Sci. Rep. 2016, 6, 36684. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.-M.; Faris, H.J.; Wang, J.Q. Muscarinic Acetylcholine Receptors Inhibit Fyn Activity in the Rat Striatum In Vivo. J. Mol. Neurosci. 2018, 64, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Pronin, A.N.; Wang, Q.; Slepak, V.Z. Teaching an Old Drug New Tricks: Agonism, Antagonism, and Biased Signaling of Pilocarpine through M3 Muscarinic Acetylcholine Receptor. Mol. Pharmacol. 2017, 92, 601–612. [Google Scholar] [CrossRef]
- Cheng, C.-Y.; Tseng, H.-C.; Yang, C.-M. Bradykinin-mediated cell proliferation depends on transactivation of EGF receptor in corneal fibroblasts. J. Cell. Physiol. 2012, 227, 1367–1381. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-S.; Lin, T.-H.; Tang, C.-H. Bradykinin enhances cell migration in human prostate cancer cells through B2 receptor/PKCδ/c-Src dependent signaling pathway. Prostate 2013, 73, 89–100. [Google Scholar] [CrossRef]
- Simo-Cheyou, E.R.; Vardatsikos, G.; Srivastava, A.K. Src tyrosine kinase mediates endothelin-1-induced early growth response protein-1 expression via MAP kinase-dependent pathways in vascular smooth muscle cells. Int. J. Mol. Med. 2016, 38, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Mima, A.; Matsubara, T.; Arai, H.; Abe, H.; Nagai, K.; Kanamori, H.; Sumi, E.; Takahashi, T.; Iehara, N.; Fukatsu, A.; et al. Angiotensin II-dependent Src and Smad1 signaling pathway is crucial for the development of diabetic nephropathy. Lab. Investig. 2006, 86, 927–939. [Google Scholar] [CrossRef]
- Touyz, R.M.; Wu, X.-H.; He, G.; Salomon, S.; Schiffrin, E.L. Increased angiotensin II-mediated Src signaling via epidermal growth factor receptor transactivation is associated with decreased C-terminal Src kinase activity in vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension 2002, 39, 479–485. [Google Scholar] [CrossRef]
- Lee, C.-S.; Cho, H.-J.; Lee, J.-W.; Son, H.; Chai, J.; Kim, H.-S. Adhesion GPCR Latrophilin-2 Specifies Cardiac Lineage Commitment through CDK5, Src, and P38MAPK. Stem Cell Rep. 2021, 16, 868–882. [Google Scholar] [CrossRef]
- Chatterjee, T.; Zhang, S.; Posey, T.A.; Jacob, J.; Wu, L.; Yu, W.; Francisco, L.E.; Liu, Q.J.; Carmon, K.S. Anti-GPR56 monoclonal antibody potentiates GPR56-mediated Src-Fak signaling to modulate cell adhesion. J. Biol. Chem. 2021, 296, 100261. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Ferguson, S.S.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, V.V.; Benovic, J.L. Mechanism of phosphorylation-recognition by visual arrestin and the transition of arrestin into a high affinity binding state. Mol. Pharmacol. 1997, 51, 161–169. [Google Scholar] [CrossRef]
- Goodman, O.B.; Krupnick, J.G.; Santini, F.; Gurevich, V.V.; Penn, R.B.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 1996, 383, 447–450. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Chen, Q.; Gurevich, E.V. Arrestins: Introducing Signaling Bias into Multifunctional Proteins. Prog. Mol. Biol. Transl. Sci. 2018, 160, 47–61. [Google Scholar] [CrossRef]
- Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Merten, N.; Malfacini, D.; Inoue, A.; Preis, P.; Simon, K.; Rüttiger, N.; Ziegler, N.; Benkel, T.; Schmitt, N.K.; et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 2015, 523, 561–567. [Google Scholar] [CrossRef]
- Shukla, A.K.; Westfield, G.H.; Xiao, K.; Reis, R.I.; Huang, L.-Y.; Tripathi-Shukla, P.; Qian, J.; Li, S.; Blanc, A.; Oleskie, A.N.; et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 2014, 512, 218–222. [Google Scholar] [CrossRef]
- Chen, Q.; Perry, N.A.; Vishnivetskiy, S.A.; Berndt, S.; Gilbert, N.C.; Zhuo, Y.; Singh, P.K.; Tholen, J.; Ohi, M.D.; Gurevich, E.V.; et al. Structural basis of arrestin-3 activation and signaling. Nat. Commun. 2017, 8, 1427. [Google Scholar] [CrossRef]
- Shukla, A.K.; Manglik, A.; Kruse, A.C.; Xiao, K.; Reis, R.I.; Tseng, W.-C.; Staus, D.P.; Hilger, D.; Uysal, S.; Huang, L.-Y.; et al. Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 2013, 497, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular basis of β-arrestin coupling to formoterol-bound β1-adrenoceptor. Nature 2020, 583, 862–866. [Google Scholar] [CrossRef]
- Cahill, T.J.; Thomsen, A.R.B.; Tarrasch, J.T.; Plouffe, B.; Nguyen, A.H.; Yang, F.; Huang, L.-Y.; Kahsai, A.W.; Bassoni, D.L.; Gavino, B.J.; et al. Distinct conformations of GPCR-β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. USA 2017, 114, 2562–2567. [Google Scholar] [CrossRef]
- Chen, Q.; Iverson, T.M.; Gurevich, V.V. Structural Basis of Arrestin-Dependent Signal Transduction. Trends Biochem. Sci. 2018, 43, 412–423. [Google Scholar] [CrossRef]
- Yang, F.; Xiao, P.; Qu, C.-X.; Liu, Q.; Wang, L.-Y.; Liu, Z.-X.; He, Q.-T.; Liu, C.; Xu, J.-Y.; Li, R.-R.; et al. Allosteric mechanisms underlie GPCR signaling to SH3-domain proteins through arrestin. Nat. Chem. Biol. 2018, 14, 876–886. [Google Scholar] [CrossRef]
- Milano, S.K.; Pace, H.C.; Kim, Y.-M.; Brenner, C.; Benovic, J.L. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry 2002, 41, 3321–3328. [Google Scholar] [CrossRef]
- Zhan, X.; Gimenez, L.E.; Gurevich, V.V.; Spiller, B.W. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J. Mol. Biol. 2011, 406, 467–478. [Google Scholar] [CrossRef]
- Kuo, F.-T.; Lu, T.-L.; Fu, H.-W. Opposing effects of beta-arrestin1 and beta-arrestin2 on activation and degradation of Src induced by protease-activated receptor 1. Cell. Signal. 2006, 18, 1914–1923. [Google Scholar] [CrossRef]
- Cesareni, G.; Panni, S.; Nardelli, G.; Castagnoli, L. Can we infer peptide recognition specificity mediated by SH3 domains? FEBS Lett. 2002, 513, 38–44. [Google Scholar] [CrossRef]
- Feng, S.; Kasahara, C.; Rickles, R.J.; Schreiber, S.L. Specific interactions outside the proline-rich core of two classes of Src homology 3 ligands. Proc. Natl. Acad. Sci. USA 1995, 92, 12408–12415. [Google Scholar] [CrossRef]
- Grabs, D.; Slepnev, V.I.; Songyang, Z.; David, C.; Lynch, M.; Cantley, L.C.; de Camilli, P. The SH3 domain of amphiphysin binds the proline-rich domain of dynamin at a single site that defines a new SH3 binding consensus sequence. J. Biol. Chem. 1997, 272, 13419–13425. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor-β-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.I.; Strathmann, M.P.; Gautam, N. Diversity of G proteins in signal transduction. Science 1991, 252, 802–808. [Google Scholar] [CrossRef]
- Ma, Y.C.; Huang, J.; Ali, S.; Lowry, W.; Huang, X.Y. Src tyrosine kinase is a novel direct effector of G proteins. Cell 2000, 102, 635–646. [Google Scholar] [CrossRef]
- Berlot, C.H.; Bourne, H.R. Identification of effector-activating residues of Gs alpha. Cell 1992, 68, 911–922. [Google Scholar] [CrossRef]
- Tesmer, J.J.; Sunahara, R.K.; Gilman, A.G.; Sprang, S.R. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha.GTPgammaS. Science 1997, 278, 1907–1916. [Google Scholar] [CrossRef]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.-Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef]
- Parra-Mercado, G.K.; Fuentes-Gonzalez, A.M.; Hernandez-Aranda, J.; Diaz-Coranguez, M.; Dautzenberg, F.M.; Catt, K.J.; Hauger, R.L.; Olivares-Reyes, J.A. CRF1 Receptor Signaling via the ERK1/2-MAP and Akt Kinase Cascades: Roles of Src, EGF Receptor, and PI3-Kinase Mechanisms. Front. Endocrinol. 2019, 10, 869. [Google Scholar] [CrossRef]
- Zhang, F.; Steinberg, S.F. S49G and R389G polymorphisms of the β₁-adrenergic receptor influence signaling via the cAMP-PKA and ERK pathways. Physiol. Genom. 2013, 45, 1186–1192. [Google Scholar] [CrossRef]
- Brovkovych, V.; Zhang, Y.; Brovkovych, S.; Minshall, R.D.; Skidgel, R.A. A novel pathway for receptor-mediated post-translational activation of inducible nitric oxide synthase. J. Cell. Mol. Med. 2011, 15, 258–269. [Google Scholar] [CrossRef]
- Moyers, J.S.; Linder, M.E.; Shannon, J.D.; Parsons, S.J. Identification of the in vitro phosphorylation sites on Gs alpha mediated by pp60c-src. Biochem. J. 1995, 305, 411–417. [Google Scholar] [CrossRef]
- Hausdorff, W.P.; Pitcher, J.A.; Luttrell, D.K.; Linder, M.E.; Kurose, H.; Parsons, S.J.; Caron, M.G.; Lefkowitz, R.J. Tyrosine phosphorylation of G protein alpha subunits by pp60c-src. Proc. Natl. Acad. Sci. USA 1992, 89, 5720–5724. [Google Scholar] [CrossRef]
- Chakravorty, D.; Assmann, S.M. G protein subunit phosphorylation as a regulatory mechanism in heterotrimeric G protein signaling in mammals, yeast, and plants. Biochem. J. 2018, 475, 3331–3357. [Google Scholar] [CrossRef]
- Bell, M.W.; Desai, N.; Guo, X.X.; Ghalayini, A.J. Tyrosine phosphorylation of the alpha subunit of transducin and its association with Src in photoreceptor rod outer segments. J. Neurochem. 2000, 75, 2006–2019. [Google Scholar] [CrossRef]
- Romero, G.; von Zastrow, M.; Friedman, P.A. Role of PDZ proteins in regulating trafficking, signaling, and function of GPCRs: Means, motif, and opportunity. Adv. Pharmacol. 2011, 62, 279–314. [Google Scholar] [CrossRef]
- Liu, J.; Liao, Z.; Camden, J.; Griffin, K.D.; Garrad, R.C.; Santiago-Pérez, L.I.; González, F.A.; Seye, C.I.; Weisman, G.A.; Erb, L. Src homology 3 binding sites in the P2Y2 nucleotide receptor interact with Src and regulate activities of Src, proline-rich tyrosine kinase 2, and growth factor receptors. J. Biol. Chem. 2004, 279, 8212–8218. [Google Scholar] [CrossRef]
- Fan, G.; Shumay, E.; Malbon, C.C.; Wang, H. c-Src tyrosine kinase binds the beta 2-adrenergic receptor via phospho-Tyr-350, phosphorylates G-protein-linked receptor kinase 2, and mediates agonist-induced receptor desensitization. J. Biol. Chem. 2001, 276, 13240–13247. [Google Scholar] [CrossRef] [PubMed]
- Oldenhof, J.; Ray, A.; Vickery, R.; van Tol, H.H. SH3 ligands in the dopamine D3 receptor. Cell. Signal. 2001, 13, 411–416. [Google Scholar] [CrossRef]
- Nair, V.D.; Sealfon, S.C. Agonist-specific transactivation of phosphoinositide 3-kinase signaling pathway mediated by the dopamine D2 receptor. J. Biol. Chem. 2003, 278, 47053–47061. [Google Scholar] [CrossRef]
- Zhen, X.; Zhang, J.; Johnson, G.P.; Friedman, E. D(4) dopamine receptor differentially regulates Akt/nuclear factor-kappa b and extracellular signal-regulated kinase pathways in D(4)MN9D cells. Mol. Pharmacol. 2001, 60, 857–864. [Google Scholar]
- Oak, J.N.; Lavine, N.; van Tol, H.H. Dopamine D(4) and D(2L) Receptor Stimulation of the Mitogen-Activated Protein Kinase Pathway Is Dependent on trans-Activation of the Platelet-Derived Growth Factor Receptor. Mol. Pharmacol. 2001, 60, 92–103. [Google Scholar] [CrossRef]
- Liu, P.; Yin, Y.-L.; Wang, T.; Hou, L.; Wang, X.-X.; Wang, M.; Zhao, G.-G.; Shi, Y.; Xu, H.E.; Jiang, Y. Ligand-induced activation of ERK1/2 signaling by constitutively active Gs-coupled 5-HT receptors. Acta Pharmacol. Sin. 2019, 40, 1157–1167. [Google Scholar] [CrossRef]
- Kundu, K.; Costa, F.; Backofen, R. A graph kernel approach for alignment-free domain-peptide interaction prediction with an application to human SH3 domains. Bioinformatics 2013, 29, i335–i343. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| GPCR | G-Protein | Arrestin | Direct | Other | References |
|---|---|---|---|---|---|
| α2AR | Arrestin-2/3 | [42] | |||
| β2AR | Arrestin-2 | [43,44] | |||
| β3AR | 3rd ICL | [44] | |||
| D1R | Arresin-3 | unknown | [45,46] | ||
| D2R | Arrestin-2 | unknown | [47,48] | ||
| D4R | 3rd ICL | [49] | |||
| V1bR | unknown | [50] | |||
| V2R | Indication of direct | [50] | |||
| GnRH-a | Indication of Gβγ protein | [51,52] | |||
| M1R | Indication of Gαq protein | [53] | |||
| M2R | Arrestin-2 | [43,54] | |||
| M3R | Arrestin | [55] | |||
| M4R | unknown | [54] | |||
| B1R | Gαi | unknown | [56,57] | ||
| ETAR | unknown | [58] | |||
| ATR2 | unknown | [59,60] | |||
| Latrophilin-2 | unknown | [61] | |||
| GPR56 (ADGRG1) | unknown | [62] |
| 3ICL | C-Terminus | Predicted SFK SH3 Domain Interactions | |
|---|---|---|---|
| b1AR | REAQKQVKKIDSCERRFLGGPARPPSPSPSPVPAPAPPPGPPRPAAAAATAPLANGRAGKRRPSRLVALRE | CRSPDFRKAFQRLLCCARRAARRRHATHGDRPRASGCLARPGPPPSPGAASDDDDDDVVGATPPARLLEPWAGCNGGAAADSDSSLDEPCRPGFASESKV | FGR, LYN |
| b2AR | RVFQEAKRQLQKIDKSEGRFHVQNLSQVEQDGRTGHGLRRSSKFCLKEHKALKT | PDFRIAFQELLCLRRSSLKAYGNGYSSNGNTGEQSGYHVEQEKENKLLCEDLPGTEDFVGHQGTVPSDNIDSQGRNCSTNDSLL | - |
| b3AR | RVFVVATRQLRLLRGELGRFPPEESPPAPSRSLAPAPVGTCAPPEGVPACGRRPARLLPLREHRALC | RSPDFRSAFRRLLCRCGRRLPPEPCAAARPALFPSGVPAARSSPAQPRLCQRLDGASWGVS | SRC, FGR, LYN, HCK, LCK, FYN |
| P2Y2 | MARRLLKPAYGTSGGLPRAKRKSVRT | GQRLVRFARDAKPPTGPSPATPARRRLGLRRSDRTDMQRIEDVLGSSEDSRRTESTPAGSENTKDIRL | FGR |
| 5HT6 | CRILLAARKQAVQVASLTTGMASQASETLQVPRTPRPGVESADSRRLATKHSRKALK | PLFMRDFKRALGRFLPCPRCPRERQASLASPSLRTSHSGPRPGLSLQQVLPLPLPPDSDSDSDAGSGGSSGLRLTAQLLLPGEATQDPPLPTRAAAAVNFFNIDPAEPELRPHPLGIPTN | LYN |
| D1R | RIAQKQIRRIAALERAAVHAKNCQTTTGNGKPVECSQPESSFKMSFKRETKVLK | RKAFSTLLGCYRLCPATNNAIETVSINNNGAAMFSSHHEPRGSISKECNLVYLIPHAVGSSEDLKKEEAAGIARPLEKLSPALSVILDYDTDVSLEKIQPITQNGQHPT | - |
| D2R | IVLRRRRKRVNTKRSSRAFRAHLRAPLKGNCTHPEDMKLCTVIMKSNGSFPVNRRRVEAARRAQELEMEMLSSTSPPERTRYSPIPPSHHQLTLPDPSHHGLHSTPDSPAKPEKNGHAKDHPKIAKIFEIQTMPNGKTRTSLKTMSRRKLSQQKEKKATQ | EFRKAFLKILHC | - |
| D3R | RIYVVLKQRRRKRILTRQNSQCNSVRPGFPQQTLSPDPAHLELKRYYSICQDTALGGPGFQERGGELKREEKTRNSLSPTIAPKLSLEVRKLSNGRLSTSLKLGPLQPRGVPLREKKATQ | NIEFRKAFLKILSC | LYN |
| D4R | ATFRGLQRWEVARRAKLHGRAPRRPSGPGPPSPTPPAPRLPQDPCGPDCAPPAPGLPRGPCGPDCAPAAPSLPQDPCGPDCAPPAPGLPPDPCGSNCAPPDAVRAAALPPQTPPQTRRRRRAKITGRERKAMR | NAEFRNVFRKALRACC | SRC, FGR, HCK, LYN, |
| D5R | RIYRIAQVQIRRISSLERAAEHAQSCRSSAACAPDTSLRASIKKETKVLK | FNADFQKVFAQLLGCSHFCSRTPVETVNISNELISYNQDIVFHKEIAAAYIHMMPNAVTPGNREVDNDEEEGPFDRMFQIYQTSPDGDPVAESVWELDCEGEISLDKITPFTPNGFH | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berndt, S.; Liebscher, I. New Structural Perspectives in G Protein-Coupled Receptor-Mediated Src Family Kinase Activation. Int. J. Mol. Sci. 2021, 22, 6489. https://doi.org/10.3390/ijms22126489
Berndt S, Liebscher I. New Structural Perspectives in G Protein-Coupled Receptor-Mediated Src Family Kinase Activation. International Journal of Molecular Sciences. 2021; 22(12):6489. https://doi.org/10.3390/ijms22126489
Chicago/Turabian StyleBerndt, Sandra, and Ines Liebscher. 2021. "New Structural Perspectives in G Protein-Coupled Receptor-Mediated Src Family Kinase Activation" International Journal of Molecular Sciences 22, no. 12: 6489. https://doi.org/10.3390/ijms22126489
APA StyleBerndt, S., & Liebscher, I. (2021). New Structural Perspectives in G Protein-Coupled Receptor-Mediated Src Family Kinase Activation. International Journal of Molecular Sciences, 22(12), 6489. https://doi.org/10.3390/ijms22126489