
Pulmonary Stretch and Lung Mechanotransduction: Implications for Progression in the Fibrotic Lung


 ,
,  ,
, 
{kind=link}
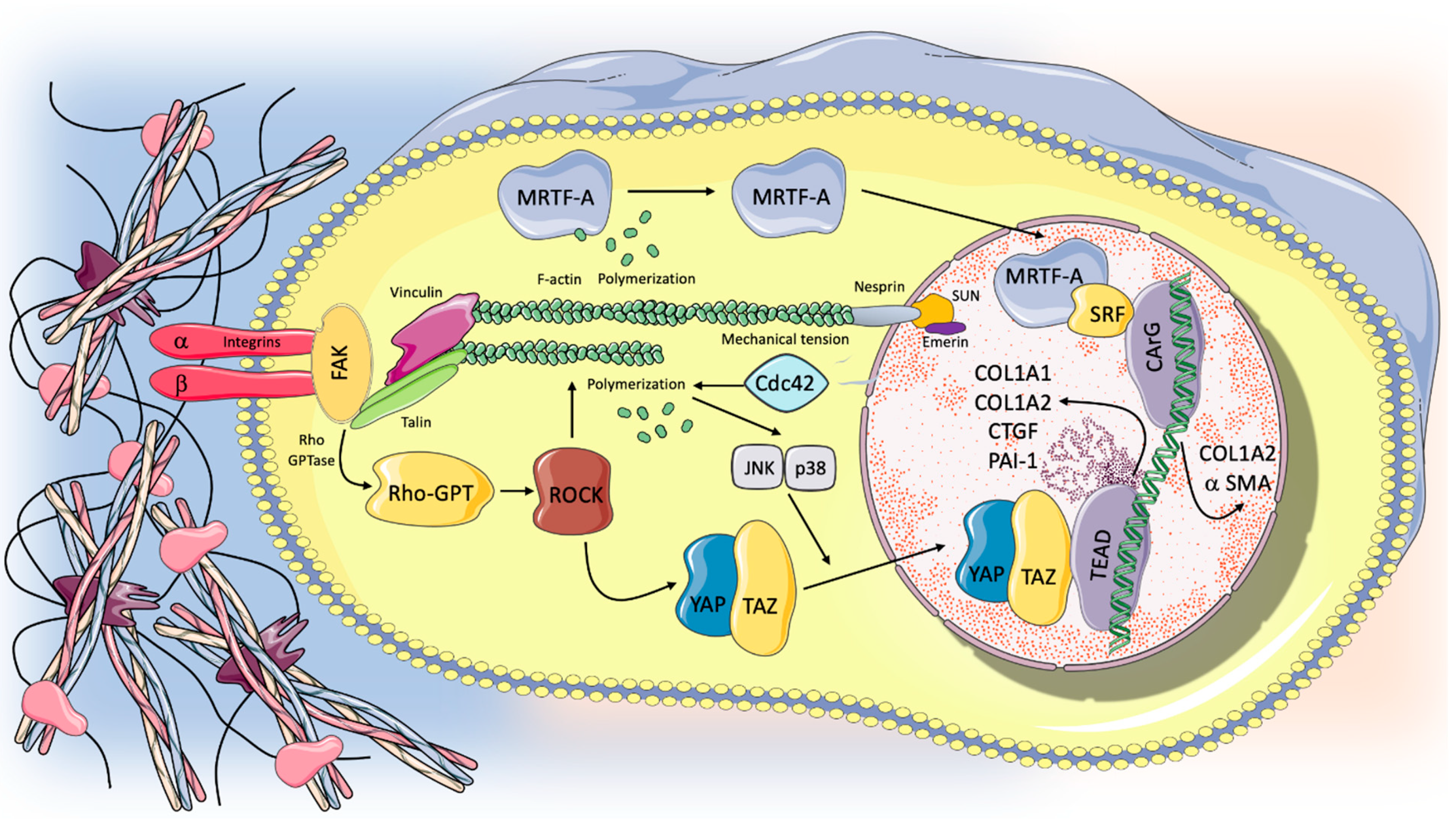{kind=link}
Abstract
1. Background
2. Matrix Abnormalities and Mechanical Behavior in Pulmonary Fibrosis
3. Stress and Strain Behavior of the Normal and Pathologic Lung
4. The Micro-Strain Concept in the Fibrotic Lung
5. The Mechanotransduction Process: Biological Response to Stretch and Progression in the Fibrotic Lung
6. Role of Alveolar Type 2 Cells in the Progression of Lung Fibrosis
7. Conclusions and Clinical Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chen, C.S. Mechanotransduction—A field pulling together? J. Cell Sci. 2008, 121, 3285–3292. [Google Scholar] [CrossRef]
- Moessinger, A.C.; Harding, R.; Adamson, T.M.; Singh, M.; Kiu, G.T. Role of lung fluid volume in growth and maturation of the fetal sheep lung. J. Clin. Investig. 1990, 86, 1270–1277. [Google Scholar] [CrossRef]
- Schmitt, S.; Hendricks, P.; Weir, J.; Somasundaram, R.; Sittampalam, G.S.; Nirmalanandhan, V.S. Stretching Mechanotransduction from the Lung to the Lab: Approaches and Physiological Relevance in Drug Discovery. ASSAY Drug Dev. Technol. 2012, 10, 137–147. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Fear, M.W.; Choi, Y.S.; Wood, F.M.; Allahham, A.; Mutsaers, S.E.; Prêle, C.M. The extracellular matrix and mechanotransduction in pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2020, 126, 105802. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Sgalla, G.; Cocconcelli, E.; Tonelli, R.; Richeldi, L. Novel drug targets for idiopathic pulmonary fibrosis. Expert Rev. Respir. Med. 2016, 10, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Marchioni, A.; Tonelli, R.; Ball, L.; Fantini, R.; Castaniere, I.; Cerri, S.; Luppi, F.; Malerba, M.; Pelosi, P.; Clini, E. Acute exacerbation of idiopathic pulmonary fibrosis: Lessons learned from acute respiratory distress syndrome? Crit. Care 2018, 22, 80. [Google Scholar] [CrossRef] [PubMed]
- Burgstaller, G.; Oehrle, B.; Gerckens, M.; White, E.S.; Schiller, H.B.; Eickelberg, O. The instructive extracellular matrix of the lung: Basic composition and alterations in chronic lung disease. Eur. Respir. J. 2017, 50, 1601805. [Google Scholar] [CrossRef]
- White, E. Lung Extracellular Matrix and Fibroblast Function. Ann. Am. Thorac. Soc. 2015, 12, S30–S33. [Google Scholar] [CrossRef]
- Gattinoni, L.; Marini, J.J.; Pesenti, A.; Quintel, M.; Mancebo, J.; Brochard, L. The "baby lung" became an adult. Intensiv. Care Med. 2016, 42, 663–673. [Google Scholar] [CrossRef]
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27, 180033. [Google Scholar] [CrossRef] [PubMed]
- Todd, N.W.; Atamas, S.P.; Luzina, I.G.; Galvin, J.R. Permanent alveolar collapse is the predominant mechanism in idiopathic pulmonary fibrosis. Expert Rev. Respir. Med. 2015, 9, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Decaris, M.L.; Gatmaitan, M.; FlorCruz, S.; Luo, F.; Blisnick, T.; Holmes, W.E.; Hellerstein, M.K.; Turner, S.M.; Emson, C.L.; Subota, I.; et al. Proteomic Analysis of Altered Extracellular Matrix Turnover in Bleomycin-induced Pulmonary Fibrosis. Mol. Cell. Proteom. 2014, 13, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Rozin, G.F.; Gomes, M.M.; Parra, E.R.; A Kairalla, R.; De Carvalho, C.R.R.; Capelozzi, V.L. Collagen and elastic system in the remodelling process of major types of idiopathic interstitial pneumonias (IIP). Histopathology 2005, 46, 413–421. [Google Scholar] [CrossRef]
- Blaauboer, M.E.; Boeijen, F.R.; Emson, C.L.; Turner, S.M.; Doulabi, B.Z.; Hanemaaijer, R.; Smit, T.H.; Stoop, R.; Everts, V. Extracellular matrix proteins: A positive feedback loop in lung fibrosis? Matrix Biol. 2014, 34, 170–178. [Google Scholar] [CrossRef]
- Serini, G.; Bochaton-Piallat, M.-L.; Ropraz, P.; Geinoz, A.; Borsi, L.; Zardi, L.; Gabbiani, G. The Fibronectin Domain ED-A Is Crucial for Myofibroblastic Phenotype Induction by Transforming Growth Factor-β1. J. Cell Biol. 1998, 142, 873–881. [Google Scholar] [CrossRef]
- Marinković, A.; Liu, F.; Tschumperlin, D.J. Matrices of Physiologic Stiffness Potently Inactivate Idiopathic Pulmonary Fibrosis Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2013, 48, 422–430. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Rodarte, J.R. Stress-strain analysis and the lung. Fed. Proc. 1982, 41, 130–135. [Google Scholar] [PubMed]
- Vatankhah-Varnosfaderani, M.; Daniel, W.F.M.; Everhart, M.H.; Pandya, A.A.; Liang, H.; Matyjaszewski, K.; Dobrynin, A.V.; Sheiko, S.S. Mimicking biological stress–strain behaviour with synthetic elastomers. Nat. Cell Biol. 2017, 549, 497–501. [Google Scholar] [CrossRef]
- Booth, A.J.; Hadley, R.; Cornett, A.M.; Dreffs, A.A.; Matthes, S.A.; Tsui, J.L.; Weiss, K.; Horowitz, J.C.; Fiore, V.F.; Barker, T.H.; et al. Acellular Normal and Fibrotic Human Lung Matrices as a Culture System forIn VitroInvestigation. Am. J. Respir. Crit. Care Med. 2012, 186, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Protti, A.; Cressoni, M.; Santini, A.; Langer, T.; Mietto, C.; Febres, D.; Chierichetti, M.; Coppola, S.; Conte, G.; Gatti, S.; et al. Lung Stress and Strain during Mechanical Ventilation. Am. J. Respir. Crit. Care Med. 2011, 183, 1354–1362. [Google Scholar] [CrossRef]
- Chiumello, D.; Carlesso, E.; Cadringher, P.; Caironi, P.; Valenza, F.; Polli, F.; Tallarini, F.; Cozzi, P.; Cressoni, M.; Colombo, A.; et al. Lung Stress and Strain during Mechanical Ventilation for Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2008, 178, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Chiumello, D.A.; Chidini, G.; Calderini, E.; Colombo, A.; Crimella, F.; Brioni, M. Respiratory mechanics and lung stress/strain in children with acute respiratory distress syndrome. Ann. Intensiv. Care 2016, 6, 11. [Google Scholar] [CrossRef]
- González-López, A.; García-Prieto, E.; Batalla-Solís, E.; Amado-Rodríguez, L.; Avello, N.; Blanch, L.; Albaiceta, G.M. Lung strain and biological response in mechanically ventilated patients. Intensiv. Care Med. 2012, 38, 240–247. [Google Scholar] [CrossRef]
- Koshiyama, K.; Nishimoto, K.; Ii, S.; Sera, T.; Wada, S. Heterogeneous structure and surface tension effects on mechanical response in pulmonary acinus: A finite element analysis. Clin. Biomech. 2019, 66, 32–39. [Google Scholar] [CrossRef]
- Denny, E.; Schroter, R. A model of non-uniform lung parenchyma distortion. J. Biomech. 2006, 39, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Marchioni, A.; Tonelli, R.; Rossi, G.; Spagnolo, P.; Luppi, F.; Cerri, S.; Cocconcelli, E.; Pellegrino, M.R.; Fantini, R.; Tabbì, L.; et al. Ventilatory support and mechanical properties of the fibrotic lung acting as a “squishy ball”. Ann. Intensiv. Care 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Albert, R.K.; Smith, B.; Perlman, C.E.; Schwartz, D.A. Is Progression of Pulmonary Fibrosis due to Ventilation-induced Lung Injury? Am. J. Respir. Crit. Care Med. 2019, 200, 140–151. [Google Scholar] [CrossRef]
- Yen, S.; Preissner, M.; Bennett, E.; Dubsky, S.; Carnibella, R.; O’Toole, R.; Roddam, L.; Jones, H.; Dargaville, P.A.; Fouras, A.; et al. The Link between Regional Tidal Stretch and Lung Injury during Mechanical Ventilation. Am. J. Respir. Cell Mol. Biol. 2019, 60, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Torsani, V.; Gomes, S.; De Santis, R.R.; Beraldo, M.A.; Costa, E.L.V.; Tucci, M.R.; Zin, W.A.; Kavanagh, B.P.; Amato, M.B.P. Spontaneous Effort Causes Occult Pendelluft during Mechanical Ventilation. Am. J. Respir. Crit. Care Med. 2013, 188, 1420–1427. [Google Scholar] [CrossRef]
- D’Angelo, E.; Sant’Ambrogio, G.; Agostoni, E. Effect of diaphragm activity or paralysis on distribution of pleural pressure. J. Appl. Physiol. 1974, 37, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Edwards, Y.S. Stretch stimulation: Its effects on alveolar type II cell function in the lung. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2001, 129, 245–260. [Google Scholar] [CrossRef]
- Duscher, D.; Maan, Z.N.; Wong, V.W.; Rennert, R.C.; Januszyk, M.; Rodrigues, M.; Hu, M.; Whitmore, A.J.; Whittam, A.J.; Longaker, M.T.; et al. Mechanotransduction and fibrosis. J. Biomech. 2014, 47, 1997–2005. [Google Scholar] [CrossRef]
- Martino, F.; Perestrelo, A.R.; Vinarský, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Irianto, J.; Discher, D.E. Mechanosensing by the nucleus: From pathways to scaling relationships. J. Cell Biol. 2017, 216, 305–315. [Google Scholar] [CrossRef]
- Rajgor, D.; Shanahan, C.M. Nesprins: From the nuclear envelope and beyond. Expert Rev. Mol. Med. 2013, 15, e5. [Google Scholar] [CrossRef]
- Maurer, M.; Lammerding, J. The Driving Force: Nuclear Mechanotransduction in Cellular Function, Fate, and Disease. Annu. Rev. Biomed. Eng. 2019, 21, 443–468. [Google Scholar] [CrossRef] [PubMed]
- Finch-Edmondson, M.; Sudol, M. Framework to function: Mechanosensitive regulators of gene transcription. Cell. Mol. Biol. Lett. 2016, 21, 28. [Google Scholar] [CrossRef]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, X.; Hecker, L.; Kurundkar, D.; Kurundkar, A.; Liu, H.; Jin, T.-H.; Desai, L.; Bernard, K.; Thannickal, V.J. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J. Clin. Investig. 2013, 123, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Huang, H.; Liu, J.; Wang, Y.; Lu, Z.; Xu, Z. Fasudil, a Rho-Kinase Inhibitor, Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice. Int. J. Mol. Sci. 2012, 13, 8293–8307. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Dobashi, K.; Iizuka, K.; Horie, T.; Suzuki, K.; Tukagoshi, H.; Nakazawa, T.; Nakazato, Y.; Mori, M. Contribution of Small GTPase Rho and Its Target Protein ROCK in a Murine Model of Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2001, 163, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Knipe, R.S.; Probst, C.K.; Lagares, D.; Franklin, A.; Spinney, J.J.; Brazee, P.L.; Grasberger, P.; Zhang, L.; Black, K.E.; Sakai, N.; et al. The Rho Kinase Isoforms ROCK1 and ROCK2 Each Contribute to the Development of Experimental Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Dong, Z.; Han, W.; Kondrikov, D.; Su, Y. The role of RhoA and cytoskeleton in myofibroblast transformation in hyperoxic lung fibrosis. Free Radic. Biol. Med. 2013, 61, 26–39. [Google Scholar] [CrossRef]
- Sisson, T.H.; Ajayi, I.O.; Subbotina, N.; Dodi, A.E.; Rodansky, E.S.; Chibucos, L.N.; Kim, K.K.; Keshamouni, V.G.; White, E.; Zhou, Y.; et al. Inhibition of Myocardin-Related Transcription Factor/Serum Response Factor Signaling Decreases Lung Fibrosis and Promotes Mesenchymal Cell Apoptosis. Am. J. Pathol. 2015, 185, 969–986. [Google Scholar] [CrossRef]
- Yu, F.-X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Varelas, X. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 2014, 141, 1614–1626. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Gokey, J.J.; Sridharan, A.; Xu, Y.; Green, J.; Carraro, G.; Stripp, B.R.; Perl, A.-K.T.; Whitsett, J.A. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef]
- Haak, A.J.; Ducharme, M.T.; Espinosa, A.M.D.; Tschumperlin, D.J. Targeting GPCR Signaling for Idiopathic Pulmonary Fibrosis Therapies. Trends Pharmacol. Sci. 2020, 41, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Zmajkovicova, K.; Menyhart, K.; Bauer, Y.; Studer, R.; Renault, B.; Schnoebelen, M.; Bolinger, M.; Nayler, O.; Gatfield, J. The Antifibrotic Activity of Prostacyclin Receptor Agonism Is Mediated through Inhibition of YAP/TAZ. Am. J. Respir. Cell Mol. Biol. 2019, 60, 578–591. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, Y.; Takahashi, H.; Chiba, H.; Akino, T. Surfactant proteins A and D: Disease markers. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1998, 1408, 334–345. [Google Scholar] [CrossRef]
- Selman, M.; Lin, H.-M.; Jenkins, A.L.; Estrada, A.; Lin, Z.; Wang, G.; DiAngelo, S.L.; Guo, X.; Umstead, T.M.; Lang, C.M.; et al. Surfactant protein A and B genetic variants predispose to idiopathic pulmonary fibrosis. Qual. Life Res. 2003, 113, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Pearson, C.; Chinchilli, V.; Pietschmann, S.; Luo, J.; Pison, U.; Floros, J. Polymorphisms of humanSP-A, SP-B, andSP-Dgenes: Association ofSP-BThr131Ile with ARDS. Clin. Genet. 2000, 58, 181–191. [Google Scholar] [CrossRef]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic Defects in Surfactant Protein A2 Are Associated with Pulmonary Fibrosis and Lung Cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Mariencheck, W.I.; Alcorn, J.F.; Palmer, S.M.; Wright, J.R. Pseudomonas aeruginosaElastase Degrades Surfactant Proteins A and D. Am. J. Respir. Cell Mol. Biol. 2003, 28, 528–537. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, J.; Henderson, F.C.; Ryan, A.J.; Yahr, T.L.; Mallampalli, R.K. Chronic Pseudomonas aeruginosa infection reduces surfactant levels by inhibiting its biosynthesis. Cell. Microbiol. 2007, 9, 1062–1072. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef]
- Kropski, J.A.; Blackwell, T.S. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J. Clin. Investig. 2018, 128, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, E.; Stinson, A.; Shan, L.; Yang, J.; Gietl, D.; Albino, A.P. Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer 2008, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, K.B.; Samet, J.M.; A Stidley, C.; Colby, T.V.; A Waldron, J. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef]
- Kulkarni, T.; De Andrade, J.; Zhou, Y.; Luckhardt, T.; Thannickal, V.J. Alveolar epithelial disintegrity in pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L185–L191. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell. Signal. 2020, 66, 109482. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, H.; Jiang, K.; Wang, Y.; Zhang, W.; Chu, Q.; Li, J.; Huang, H.; Cai, T.; Ji, H.; et al. MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep. 2016, 16, 1810–1819. [Google Scholar] [CrossRef]
- Chen, F.; Ma, L.; Parrini, M.; Mao, X.; Lopez, M.; Wu, C.; Marks, P.; Davidson, L.; Kwiatkowski, D.; Kirchhausen, T.; et al. Cdc42 is required for PIP2-induced actin polymerization and early development but not for cell viability. Curr. Biol. 2000, 10, 758–765. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Debidda, M.; Witte, D.; Zheng, Y. Cdc42 GTPase-activating protein deficiency promotes genomic instability and premature aging-like phenotypes. Proc. Natl. Acad. Sci. USA 2007, 104, 1248–1253. [Google Scholar] [CrossRef]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121.e17. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.R.; Shimbori, C.; Bellaye, P.-S.; Inman, M.; Obex, S.; Fatima, S.; Jenkins, G.; Gauldie, J.; Ask, K.; Kolb, M. Stretch-induced Activation of Transforming Growth Factor-β1in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 194, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ryerson, C.J.; Cottin, V.; Brown, K.K.; Collard, H.R. Acute exacerbation of idiopathic pulmonary fibrosis: Shifting the paradigm. Eur. Respir. J. 2015, 46, 512–520. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchioni, A.; Tonelli, R.; Cerri, S.; Castaniere, I.; Andrisani, D.; Gozzi, F.; Bruzzi, G.; Manicardi, L.; Moretti, A.; Demurtas, J.; et al. Pulmonary Stretch and Lung Mechanotransduction: Implications for Progression in the Fibrotic Lung. Int. J. Mol. Sci. 2021, 22, 6443. https://doi.org/10.3390/ijms22126443
Marchioni A, Tonelli R, Cerri S, Castaniere I, Andrisani D, Gozzi F, Bruzzi G, Manicardi L, Moretti A, Demurtas J, et al. Pulmonary Stretch and Lung Mechanotransduction: Implications for Progression in the Fibrotic Lung. International Journal of Molecular Sciences. 2021; 22(12):6443. https://doi.org/10.3390/ijms22126443
Chicago/Turabian StyleMarchioni, Alessandro, Roberto Tonelli, Stefania Cerri, Ivana Castaniere, Dario Andrisani, Filippo Gozzi, Giulia Bruzzi, Linda Manicardi, Antonio Moretti, Jacopo Demurtas, and et al. 2021. "Pulmonary Stretch and Lung Mechanotransduction: Implications for Progression in the Fibrotic Lung" International Journal of Molecular Sciences 22, no. 12: 6443. https://doi.org/10.3390/ijms22126443
APA StyleMarchioni, A., Tonelli, R., Cerri, S., Castaniere, I., Andrisani, D., Gozzi, F., Bruzzi, G., Manicardi, L., Moretti, A., Demurtas, J., Baroncini, S., Andreani, A., Cappiello, G. F., Busani, S., Fantini, R., Tabbì, L., Samarelli, A. V., & Clini, E. (2021). Pulmonary Stretch and Lung Mechanotransduction: Implications for Progression in the Fibrotic Lung. International Journal of Molecular Sciences, 22(12), 6443. https://doi.org/10.3390/ijms22126443