
Molecular Biomarkers in Idiopathic Pulmonary Fibrosis: State of the Art and Future Directions



Abstract
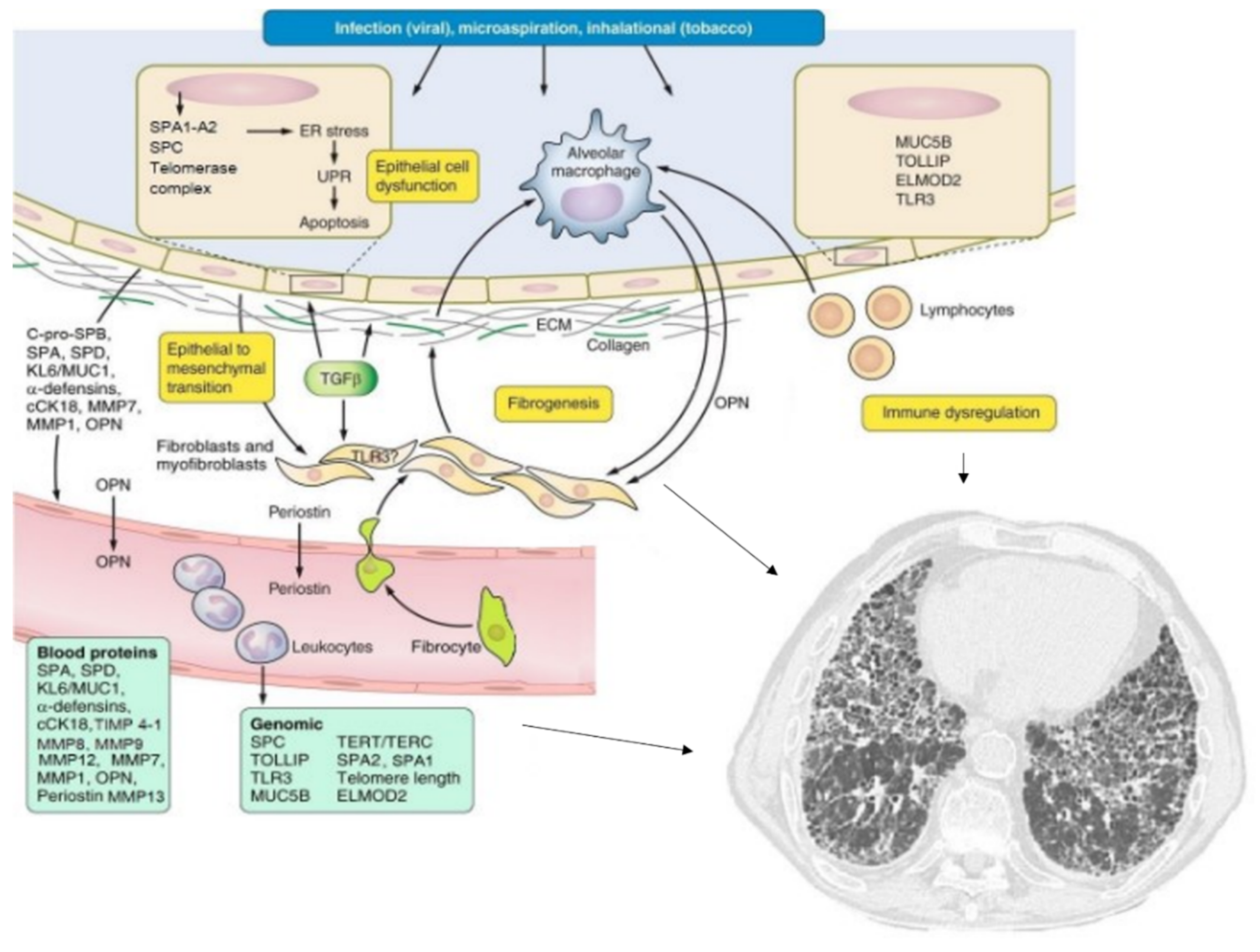
1. Introduction
2. Definition of a Biomarker
3. Molecular Biomarkers in IPF
4. Predisposition Biomarkers
5. Diagnostic Biomarkers
6. Prognostic Biomarkers
7. Therapeutic Biomarkers
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IPF | idiopathic pulmonary fibrosis |
| UIP | usual interstitial pneumonia |
| ATS | American Thoracic Society |
| ERS | European Respiratory Society |
| JRS | Japanese Respiratory Society |
| ALAT | American Latin Thoracic Association |
| ILD | interstitial lung disease |
| BAL | broncho-alveolar lavage |
| AECs | alveolar epithelial cells |
| SP-C | surfactant protein C |
| SP-A2 | surfactant protein A2 |
| SP-A1 | surfactant protein A1 |
| BALF | bronchoalveolar lavage fluid |
| SNP | single nucleotide polymorphism |
| MUC5B | mucin 5 B |
| ILA | interstitial lung abnormalities |
| TERT | telomerase reverse transcriptase |
| TIN2 | telomere binding protein 2 |
| TERF1 | telomerase repeat binding factor 1 |
| TERC | telomerase RNA component |
| COPD | chronic obstructive pulmonary disease |
| TLR | Toll-like receptors |
| TGFB1 | transforming growth factor beta 1 |
| PGE2 | prostaglandin E2 |
| KL-6 | Krebs von den Lungen-6 |
| MUC | 1 mucin 1 |
| cCK-18 | circulating caspase-cleaved cytokeratin-18 |
| CK-18 | cytokeratin-18 |
| MMPs | metalloproteases |
| BUILD | Bosentan Use in Interstitial Lung Disease |
| OPN | osteopontin |
| IL-6 | interleukin 6 |
| TIMPs | tissue inhibitors of MMPs |
| FVC | forced vital capacity |
References
- Luppi, F.; Spagnolo, P.; Cerri, S.; Richeldi, L. The big clinical trials in idiopathic pulmonary fibrosis. Curr. Opin. Pulm. Med. 2012, 18, 428–432. [Google Scholar] [CrossRef]
- Hutchinson, J.P.; Fogarty, A.W.; Hubbard, R.B.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef]
- Duchemann, B.; Annesi-Maesano, I.; De Naurois, C.J.; Sanyal, S.; Brillet, P.-Y.; Brauner, M.; Kambouchner, M.; Huynh, S.; Naccache, J.M.; Borie, R.; et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- Richeldi, L.; Rubin, A.S.; Avdeev, S.; Udwadia, Z.F.; Xu, Z.J. Idiopathic pulmonary fibrosis in BRIC countries: The cases of Brazil, Russia, India, and China. BMC Med. 2015, 13, 1–9. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- American Thoracic Society. Idiopathic pulmonary fibrosis: Diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am. J. Respir. Crit. Care Med. 2000, 161, 646–664. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Buendía-Roldán, I.; Mejía, M.; Navarro, C.; Selman, M. Idiopathic pulmonary fibrosis: Clinical behavior and aging associated comorbidities. Respir. Med. 2017, 129, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Fell, C.D. Idiopathic Pulmonary Fibrosis: Phenotypes and Comorbidities. Clin. Chest Med. 2012, 33, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R.; King, T.E. Clinical Course and Prediction of Survival in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Cuello-Garcia, C.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef] [PubMed]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Ley, B.; Brown, K.K.; Collard, H.R. Molecular biomarkers in idiopathic pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L681–L691. [Google Scholar] [CrossRef] [PubMed]
- Luppi, F.; Cerri, S.; Beghè, B.; Fabbri, L.; Richeldi, L. Corticosteroid and immunomodulatory agents in idiopathic pulmonary fibrosis. Respir. Med. 2004, 98, 1035–1044. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet Lond. Engl. 2017, 13, 1941–1952. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Wert, S.E.; Weaver, T.E. Alveolar Surfactant Homeostasis and the Pathogenesis of Pulmonary Disease. Annu. Rev. Med. 2010, 61, 105–119. [Google Scholar] [CrossRef]
- Van Moorsel, C.H.M.; van Oosterhout, M.F.M.; Barlo, N.P.; de Jong, P.A.; van der Vis, J.J.; Ruven, H.J.T.; van Es, H.W.; van den Bosch, J.M.M.; Grutters, J. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am. J. Respir. Crit. Care Med. 2010, 182, 1419–1425. [Google Scholar] [CrossRef]
- Lawson, W.E.; Grant, S.W.; Ambrosini, V.; Womble, K.E.; Dawson, E.P.; Lane, K.B.; Markin, C.; Renzoni, E.; Lympany, P.; Thomas, A.Q.; et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax 2004, 59, 977–980. [Google Scholar] [CrossRef]
- Nogee, L.M.; Dunbar, A.E.; Wert, S.E.; Askin, F.; Hamvas, A.; Whitsett, J.A. A Mutation in the Surfactant Protein C Gene Associated with Familial Interstitial Lung Disease. N. Engl. J. Med. 2001, 344, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Thouvenin, G.; Taam, R.A.; Flamein, F.; Guillot, L.; Le Bourgeois, M.; Reix, P.; Fayon, M.; Counil, F.; Depontbriand, U.; Feldmann, D.; et al. Characteristics of disorders associated with genetic mutations of surfactant protein C. Arch. Dis. Child. 2010, 95, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Van Moorsel, C.H.M.; Ten Klooster, L.; van Oosterhout, M.F.M.; de Jong, P.A.; Adams, H.; Wouter van Es, H.; Ruven, H.J.T.; van der Vis, J.J.; Grutters, J. SFTPA2 Mutations in Familial and Sporadic Idiopathic Interstitial Pneumonia. Am. J. Respir. Crit. Care Med. 2015, 192, 1249–1252. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic Defects in Surfactant Protein A2 Are Associated with Pulmonary Fibrosis and Lung Cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Maitra, M.; Wang, Y.; Gerard, R.D.; Mendelson, C.R.; Garcia, C.K. Surfactant Protein A2 Mutations Associated with Pulmonary Fibrosis Lead to Protein Instability and Endoplasmic Reticulum Stress. J. Biol. Chem. 2010, 285, 22103–22113. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Qin, J.; Guo, T.; Chen, P.; Ouyang, R.; Peng, H.; Luo, H. Identification and functional characterization of a novel surfactant protein A2 mutation (p.N207Y) in a Chinese family with idiopathic pulmonary fibrosis. Mol. Genet. Genom. Med. 2020, 8, e1393. [Google Scholar] [CrossRef] [PubMed]
- Nathan, N.; Giraud, V.; Picard, C.; Nunes, H.; Dastot-Le Moal, F.; Copin, B.; Galeron, L.; de Ligniville, A.; Kuziner, N.; Reynaud-Gaubert, M. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum. Mol. Genet. 2016, 25, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef]
- Borie, R.; Crestani, B.; Dieude, P.; Nunes, H.; Allanore, Y.; Kannengiesser, C.; Airo, P.; Matucci-Cerinic, M.; Wallaert, B.; Israel-Biet, D.; et al. The MUC5B Variant Is Associated with Idiopathic Pulmonary Fibrosis but Not with Systemic Sclerosis Interstitial Lung Disease in the European Caucasian Population. PLoS ONE 2013, 8, e70621. [Google Scholar] [CrossRef]
- Zhang, Y.; Noth, I.; Garcia, J.G.N.; Kaminski, N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1576–1577. [Google Scholar] [CrossRef]
- Stock, C.J.; Sato, H.; Fonseca, C.; Banya, W.A.S.; Molyneaux, P.L.; Adamali, H.; Russell, A.-M.; Denton, C.P.; Abraham, D.J.; Hansell, D.M.; et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax 2013, 68, 436–441. [Google Scholar] [CrossRef]
- Noth, I.; Zhang, Y.; Ma, S.-F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef]
- Wei, R.; Li, C.; Zhang, M.; Jones-Hall, Y.L.; Myers, J.L.; Noth, I.; Liu, W. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl. Res. 2014, 163, 494–502. [Google Scholar] [CrossRef]
- Horimasu, Y.; Ohshimo, S.; Bonella, F.; Tanaka, S.; Ishikawa, N.; Hattori, N.; Kohno, N.; Guzman, J.; Costabel, U. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirol. Carlton. Vic. 2015, 20, 439–444. [Google Scholar] [CrossRef]
- Peljto, A.L.; Selman, M.; Kim, D.S.; Murphy, E.; Tucker, L.; Pardo, A.; Lee, J.S.; Ji, W.; Schwarz, M.I.; Yang, I.V.; et al. The MUC5B Promoter Polymorphism Is Associated With Idiopathic Pulmonary Fibrosis in a Mexican Cohort but Is Rare Among Asian Ancestries. Chest 2015, 147, 460–464. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, X.; Zhang, S.; Tang, S.; Min, H.; Yi, L.; Xu, B.; Song, Y. Association Between the MUC5B Promoter Polymorphism rs35705950 and Idiopathic Pulmonary Fibrosis: A Meta-analysis and Trial Sequential Analysis in Caucasian and Asian Populations. Medicine 2015, 94, e1901. [Google Scholar] [CrossRef]
- Hunninghake, G.M.; Hatabu, H.; Okajima, Y.; Gao, W.; Dupuis, J.; Latourelle, J.C.; Nishino, M.; Araki, T.; Zazueta, O.E.; Kurugol, S.; et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N. Engl. J. Med. 2013, 368, 2192–2200. [Google Scholar] [CrossRef]
- Calado, R.T.; Young, N.S. Telomere diseases. N. Engl. J. Med. 2009, 361, 2353–2365. [Google Scholar] [CrossRef]
- Borie, R.; Kannengiesser, C.; Dupin, C.; Debray, M.-P.; Cazes, A.; Crestani, B. Impact of genetic factors on fibrosing interstitial lung diseases. Incidence and clinical presentation in adults. Presse Méd. 2020, 49. [Google Scholar] [CrossRef]
- Alder, J.K.; Chen, J.J.-L.; Lancaster, L.; Danoff, S.; Su, S.-C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [PubMed]
- Rode, L.; Bojesen, S.E.; Weischer, M.; Vestbo, J.; Nordestgaard, B.G. Short telomere length, lung function and chronic obstructive pulmonary disease in 46 396 individuals. Thorax 2012, 68, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Karampitsakos, T.; Woolard, T.; Bouros, D.; Tzouvelekis, A. Toll-like receptors in the pathogenesis of pulmonary fibrosis. Eur. J. Pharmacol. 2017, 808, 35–43. [Google Scholar] [CrossRef]
- Hanson, K.M.; Hernady, E.B.; Reed, C.K.; Johnston, C.J.; Groves, A.M.; Finkelstein, J.N. Apoptosis Resistance in Fibroblasts Precedes Progressive Scarring in Pulmonary Fibrosis and Is Partially Mediated by Toll-Like Receptor 4 Activation. Toxicol. Sci. 2019, 170, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Ebener, S.; Barnowski, S.; Wotzkow, C.; Marti, T.M.; Lopez-Rodriguez, E.; Crestani, B.; Blank, F.; Schmid, R.A.; Geiser, T.; Funke, M. Toll-like receptor 4 activation attenuates profibrotic response in control lung fibroblasts but not in fibroblasts from patients with IPF. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L42–L55. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhang, Y.; Xie, T.; Liu, N.; Chen, H.; Geng, Y.; Kurkciyan, A.; Mena, J.M.; Stripp, B.R.; Jiang, D.; et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat. Med. 2016, 22, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, U.; Pulkkinen, V.; Dixon, M.; Peyrard-Janvid, M.; Rehn, M.; Lahermo, P.; Ollikainen, V.; Salmenkivi, K.; Kinnula, V.; Kere, J.; et al. ELMOD2 Is a Candidate Gene for Familial Idiopathic Pulmonary Fibrosis. Am. J. Hum. Genet. 2006, 79, 149–154. [Google Scholar] [CrossRef]
- Barlo, N.P.; Van Moorsel, C.H.M.; Ruven, H.J.T.; Zanen, P.; Bosch, J.M.M.V.D.; Grutters, J.C. Surfactant protein-D predicts survival in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2009, 26, 155–161. [Google Scholar]
- Ishii, H.; Mukae, H.; Kadota, J.; Kaida, H.; Nagata, T.; Abe, K.; Matsukura, S.; Kohno, S. High serum concentrations of surfactant protein A in usual interstitial pneumonia compared with non-specific interstitial pneumonia. Thorax 2003, 58, 52–57. [Google Scholar] [CrossRef]
- Greene, K.; King, T.; Kuroki, Y.; Bucher-Bartelson, B.; Hunninghake, G.; Newman, L.; Nagae, H.; Mason, R. Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur. Respir. J. 2002, 19, 439–446. [Google Scholar] [CrossRef]
- Ohnishi, H.; Yokoyama, A.; Kondo, K.; Hamada, H.; Abe, M.; Nishimura, K.; Hiwada, K.; Kohno, N. Comparative Study of KL-6, Surfactant Protein-A, Surfactant Protein-D, and Monocyte Chemoattractant Protein-1 as Serum Markers for Interstitial Lung Diseases. Am. J. Respir. Crit. Care Med. 2002, 165, 378–381. [Google Scholar] [CrossRef]
- Wang, K.; Ju, Q.; Cao, J.; Tang, W.; Zhang, J. Impact of serum SP-A and SP-D levels on comparison and prognosis of idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Medicine 2017, 96, e7083. [Google Scholar] [CrossRef]
- Kahn, N.; Rossler, A.; Hornemann, K.; Muley, T.; Grünig, E.; Schmidt, W.; Herth, F.J.F.; Kreuter, M. C-proSP-B: A Possible Biomarker for Pulmonary Diseases? Respir. Int. Rev. Thorac. Dis. 2018, 96, 117–126. [Google Scholar] [CrossRef]
- Collard, H.R.; Calfee, C.S.; Wolters, P.J.; Song, J.W.; Hong, S.-B.; Brady, S.; Ishizaka, A.; Jones, K.D.; King, T.E.; Matthay, M.A.; et al. Plasma biomarker profiles in acute exacerbation of idiopathic pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2010, 299, L3–L7. [Google Scholar] [CrossRef]
- Ishikawa, N.; Hattori, N.; Yokoyama, A.; Kohno, N. Utility of KL-6/MUC1 in the clinical management of interstitial lung diseases. Respir. Investig. 2012, 50, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, Y.; Fujita, J.; Hachisuka, Y.; Uomoto, M.; Okada, Y.; Yoshinouchi, T.; Lee, G.-H.; Furihata, M.; Kohno, N. Immunohistochemical and immunoelectron microscopic studies of the localization of KL-6 and epithelial membrane antigen (EMA) in presumably normal pulmonary tissue and in interstitial pneumonia. Med. Mol. Morphol. 2007, 40, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Guo, Z.; Zhou, L.; Sun, B.; Wang, H. Evaluation of the Diagnostic Efficacies of Serological Markers Kl-6, SP-A, SP-D, CCL2, and CXCL13 in Idiopathic Interstitial Pneumonia A67. Struct. Funct. Relat. 2020, 98, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.; Salvini, M.; Fui, A.; Cillis, G.; Cameli, P.; Mazzei, M.A.; Fossi, A.; Refini, R.M.; Rottoli, P. Calgranulin B and KL-6 in Bronchoalveolar Lavage of Patients with IPF and NSIP. Inflammation 2019, 42, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Luo, Q.; Han, Q.; Huang, J.; Ou, Y.; Chen, M.; Wen, Y.; Mosha, S.S.; Deng, K.; Chen, R. Sequential changes of serum KL-6 predict the progression of interstitial lung disease. J. Thorac. Dis. 2018, 10, 4705–4714. [Google Scholar] [CrossRef]
- Zhu, C.; Zhao, Y.B.; Kong, L.F.; Li, Z.H.; Kang, J. The expression and clinical role of KL-6 in serum and BALF of patients with different diffuse interstitial lung diseases. Chin. J. Tuberc. Respir. Dis. 2016, 39. [Google Scholar] [CrossRef]
- Cha, S.; Ryerson, C.J.; Lee, J.S.; Kukreja, J.; Barry, S.S.; Jones, K.D.; Elicker, B.M.; Kim, D.S.; Papa, F.R.; Collard, H.R. Cleaved cytokeratin-18 is a mechanistically informative biomarker in idiopathic pulmonary fibrosis. Respir. Res. 2012, 13. [Google Scholar] [CrossRef]
- Margaritopoulos, G.A.; Antoniou, K.M.; Karagiannis, K.; Samara, K.D.; Lasithiotaki, I.; Vassalou, E.; Lymbouridou, R.; Koutala, H.; Siafakas, N.M. Investigation of toll-like receptors in the pathogenesis of fibrotic and granulomatous disorders: A bronchoalveolar lavage study. Fibrogenes. Tiss. Repair. 2010, 3, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Mahalanobish, S.; Saha, S.; Dutta, S.; Sil, P.C. Matrix metalloproteinase: An upcoming therapeutic approach for idiopathic pulmonary fibrosis. Pharmacol. Res. 2020, 152. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Selman, M. Role of matrix metaloproteases in idiopathic pulmonary fibrosis. Fibrogenes. Tiss. Repair. 2012, 5, S9. [Google Scholar] [CrossRef]
- Konishi, K.; Gibson, K.F.; Lindell, K.O.; Richards, T.J.; Zhang, Y.; Dhir, R.; Bisceglia, M.; Gilbert, S.; Yousem, S.A.; Song, J.W.; et al. Gene Expression Profiles of Acute Exacerbations of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Morais, A.; Beltrão, M.; Sokhatska, O.; Costa, D.; Melo, N.; Mota, P.; Marques, A.; Delgado, L. Serum metalloproteinases 1 and 7 in the diagnosis of idiopathic pulmonary fibrosis and other interstitial pneumonias. Respir. Med. 2015, 109, 1063–1068. [Google Scholar] [CrossRef]
- Borensztajn, K.; Crestani, B.; Kolb, M. Idiopathic Pulmonary Fibrosis: From Epithelial Injury to Biomarkers—Insights from the Bench Side. Respiration 2013, 86, 441–452. [Google Scholar] [CrossRef]
- Bauer, Y.; White, E.S.; De Bernard, S.; Cornelisse, P.; Leconte, I.; Morganti, A.; Roux, S.; Nayler, O. MMP-7 is a predictive biomarker of disease progression in patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2017, 3, 74–2016. [Google Scholar] [CrossRef] [PubMed]
- White, E.S.; Xia, M.; Murray, S.; Dyal, R.; Flaherty, C.M.; Flaherty, K.R.; Moore, B.; Cheng, L.; Doyle, T.J.; Villalba, J.; et al. Plasma Surfactant Protein-D, Matrix Metalloproteinase-7, and Osteopontin Index Distinguishes Idiopathic Pulmonary Fibrosis from Other Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2016, 194, 1242–1251. [Google Scholar] [CrossRef]
- Denhardt, D.T.; Noda, M.; O’Regan, A.W.; Pavlin, D.; Berman, J.S. Osteopontin as a means to cope with environmental insults: Regulation of inflammation, tissue remodeling, and cell survival. J. Clin. Investig. 2001, 107, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, Y.; Matsuda, K.; Uehara, T. Osteopontin enhances the migration of lung fibroblasts via upregulation of interleukin-6 through the extracellular signal-regulated kinase (ERK) pathway. Biol. Chem. 2020, 401, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M.; et al. Up-Regulation and Profibrotic Role of Osteopontin in Human Idiopathic Pulmonary Fibrosis. PLoS Med. 2005, 2, e251. [Google Scholar] [CrossRef]
- Foster, M.W.; Morrison, L.D.; Todd, J.L.; Snyder, L.; Thompson, J.W.; Soderblom, E.J.; Plonk, K.; Weinhold, K.J.; Townsend, R.; Minnich, A.; et al. Quantitative Proteomics of Bronchoalveolar Lavage Fluid in Idiopathic Pulmonary Fibrosis. J. Proteome Res. 2015, 14, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Kadota, J.; Mizunoe, S.; Mito, K.; Mukae, H.; Yoshioka, S.; Kawakami, K.; Koguchi, Y.; Fukushima, K.; Kon, S.; Kohno, S.; et al. High plasma concentrations of osteopontin in patients with interstitial pneumonia. Respir. Med. 2005, 99, 111–117. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dai, J.; Cai, H.; Li, H.; Zhuang, Y.; Min, H.; Wen, Y.; Yang, J.; Gao, Q.; Shi, Y.; Yi, L. Association between telomere length and survival in patients with idiopathic pulmonary fibrosis. Respirology 2015, 20, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Stuart, B.D.; Lee, J.S.; Kozlitina, J.; Noth, I.; Devine, M.S.; Glazer, C.S.; Torres, F.; Kaza, V.; E Girod, C.; Jones, K.D.; et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: An observational cohort study with independent validation. Lancet Respir. Med. 2014, 2, 557–565. [Google Scholar] [CrossRef]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.-F.; Garcia, J.G.N.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.; et al. Association Between the MUC5B Promoter Polymorphism and Survival in Patients With Idiopathic Pulmonary Fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef]
- Wang, H.; Zhuang, Y.; Peng, H.; Cao, M.; Li, Y.; Xu, Q.; Xin, X.; Zhou, K.; Liang, G.; Cai, H.; et al. The relationship between MUC5B promoter, TERT polymorphisms and telomere lengths with radiographic extent and survival in a Chinese IPF cohort. Sci. Rep. 2019, 9, 15307. [Google Scholar] [CrossRef]
- Jiang, H.; Hu, Y.; Shang, L.; Li, Y.; Yang, L.; Chen, Y. Association between MUC5B polymorphism and susceptibility and severity of idiopathic pulmonary fibrosis. Int. J. Clin. Exp. Pathol. 2015, 8, 14953–14958. [Google Scholar]
- Bonella, F.; Campo, I.; Zorzetto, M.; Boerner, E.; Ohshimo, S.; Theegarten, D.; Taube, C.; Costabel, U. Potential clinical utility of MUC5B und TOLLIP single nucleotide polymorphisms (SNPs) in the management of patients with IPF. Orphanet. J. Rare Dis. 2021, 16, 1–9. [Google Scholar] [CrossRef]
- Kinder, B.W.; Brown, K.K.; McCormack, F.; Ix, J.H.; Kervitsky, A.; Schwarz, M.I.; King, T.E. Serum Surfactant Protein-A Is a Strong Predictor of Early Mortality in Idiopathic Pulmonary Fibrosis. Chest 2009, 135, 1557–1563. [Google Scholar] [CrossRef]
- Takahashi, H.; Fujishima, T.; Koba, H.; Murakami, S.; Kurokawa, K.; Shibuya, Y.; Shiratori, M.; Kuroki, Y.; Abe, S. Serum Surfactant Proteins A and D as Prognostic Factors in Idiopathic Pulmonary Fibrosis and Their Relationship to Disease Extent. Am. J. Respir. Crit. Care Med. 2000, 162, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Oballa, E.; Simpson, J.K.; Porte, J.; Habgood, A.; A Fahy, W.; Flynn, A.; Molyneaux, P.L.; Braybrooke, R.; Divyateja, H.; et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: An analysis from the multicentre PROFILE cohort study. Lancet Respir. Med. 2017, 5, 946–955. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kondo, K.; Nakajima, M.; Matsushima, T.; Takahashi, T.; Nishimura, M.; Bando, M.; Sugiyama, Y.; Totani, Y.; Ishizaki, T.; et al. Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology 2006, 11, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Kushima, H.; Kinoshita, Y.; Fujita, M.; Watanabe, K. The serum KL-6 levels in untreated idiopathic pulmonary fibrosis can naturally decline in association with disease progression. Clin. Respir. J. 2018, 12, 2411–2418. [Google Scholar] [CrossRef]
- Satoh, H.; Kurishima, K.; Ishikawa, H.; Ohtsuka, M. Increased levels of KL-6 and subsequent mortality in patients with interstitial lung diseases. J. Intern. Med. 2006, 260, 429–434. [Google Scholar] [CrossRef]
- Guo, L.; Yang, Y.; Liu, F.; Jiang, C.; Yang, Y.; Pu, H.; Li, W.; Zhong, Z. Clinical Research on Prognostic Evaluation of Subjects With IPF by Peripheral Blood Biomarkers, Quantitative Imaging Characteristics and Pulmonary Function Parameters. Arch. Bronconeumol. 2020, 56, 365–372. [Google Scholar] [CrossRef]
- Song, J.W.; Do, K.H.; Jang, S.J.; Colby, T.V.; Han, S.; Kim, D.S. Blood biomarkers MMP-7 and SP-A: Predictors of outcome in idiopathic pulmonary fibrosis. Chest 2013, 143, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Aloisio, E.; Braga, F.; Puricelli, C.; Panteghini, M. Prognostic role of Krebs von den Lungen-6 (KL-6) measurement in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Clin. Chem. Lab. Med. 2021. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Nagata, N.; Kumazoe, H.; Oda, K.; Ishimoto, H.; Yoshimi, M.; Takata, S.; Hamada, M.; Koreeda, Y.; Takakura, K.; et al. Prognostic value of serial serum KL-6 measurements in patients with idiopathic pulmonary fibrosis. Respir. Investig. 2017, 55, 16–23. [Google Scholar] [CrossRef]
- Ohshimo, S.; Ishikawa, N.; Horimasu, Y.; Hattori, N.; Hirohashi, N.; Tanigawa, K.; Kohno, N.; Bonella, F.; Guzman, J.; Costabel, U. Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir. Med. 2014, 108, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y.; Kawashima, T.; Kuwabara, R.; Hayakawa, S.; Irie, T.; Yoshida, T.; Rikitake, H.; Wakabayashi, T.; Okada, N.; Kawashima, K.; et al. Change in serum marker of oxidative stress in the progression of idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2015, 32, 1–6. [Google Scholar] [CrossRef]
- O’Dwyer, D.N.; Armstrong, M.E.; Trujillo, G.; Cooke, G.; Keane, M.P.; Fallon, P.G.; Simpson, A.J.; Millar, A.B.; McGrath, E.E.; Whyte, M.K.; et al. The Toll-like Receptor 3 L412F Polymorphism and Disease Progression in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 1442–1450. [Google Scholar] [CrossRef]
- O’Dwyer, D.N.; E Armstrong, M.; Kooblall, M.; Donnelly, S.C. Targeting defective Toll-like receptor-3 function and idiopathic pulmonary fibrosis. Exp. Opin. Ther. Targets 2014, 19, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Hamai, K.; Iwamoto, H.; Ishikawa, N.; Horimasu, Y.; Masuda, T.; Miyamoto, S.; Nakashima, T.; Ohshimo, S.; Fujitaka, K.; Hamada, H.; et al. Comparative Study of Circulating MMP-7, CCL18, KL-6, SP-A, and SP-D as Disease Markers of Idiopathic Pulmonary Fibrosis. Dis. Mark. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.L.; Vinisko, R.; Liu, Y.; Neely, M.L.; Overton, R.; Flaherty, K.R.; Noth, I.; Newby, L.K.; Lasky, J.A.; Olman, M.A.; et al. Circulating matrix metalloproteinases and tissue metalloproteinase inhibitors in patients with idiopathic pulmonary fibrosis in the multicenter IPF-PRO Registry cohort. BMC Pulm. Med. 2020, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Boon, K.; Bailey, N.W.; Yang, J.; Steel, M.P.; Groshong, S.; Kervitsky, L.; Brown, K.K.; Schwarz, M.I.; Schwartz, D.A. Molecular Phenotypes Distinguish Patients with Relatively Stable from Progressive Idiopathic Pulmonary Fibrosis (IPF). PLoS ONE 2009, 4, e5134. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Qiu, X.; Xie, M.; Tian, Y.; Min, C.; Huang, M.; HongYan, W.; Chen, T.; Zhang, X.; Chen, J.; et al. Prognostic Value of Serum Osteopontin in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. BioMed. Res. Int. 2020, 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Izuhara, K.; Ohta, S.; Ono, J.; Hoshino, T. Ability of Periostin as a New Biomarker of Idiopathic Pulmonary Fibrosis. Single Mol. Single Cell Seq. 2019, 1132, 79–87. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Otsuka, M.; Chiba, H.; Ikeda, K.; Mori, Y.; Umeda, Y.; Nishikiori, H.; Kuronuma, K.; Takahashi, H. Surfactant protein A as a biomarker of outcomes of anti-fibrotic drug therapy in patients with idiopathic pulmonary fibrosis. BMC Pulm. Med. 2020, 20, 27. [Google Scholar] [CrossRef]
- Ikeda, K.; Shiratori, M.; Nishikiori, H.; Yokoo, K.; Asai, Y.; Takahashi, Y.; Saito, A.; Kuronuma, K.; Otsuka, M.; Chiba, H.; et al. Serum surfactant protein D predicts the outcome of patients with idiopathic pulmonary fibrosis treated with pirfenidone. Respir. Med. 2017, 131, 184–191. [Google Scholar] [CrossRef]
- Ikeda, K.; Chiba, H.; Nishikiori, H.; Azuma, A.; Kondoh, Y.; Ogura, T.; Taguchi, Y.; Ebina, M.; Sakaguchi, H.; Miyazawa, S.; et al. Serum surfactant protein D as a predictive biomarker for the efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis: A post-hoc analysis of the phase 3 trial in Japan. Respir. Res. 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Bonella, F.; Ohshimo, S.; Boerner, E.; Guzman, J.; Wessendorf, T.E.; Costabel, U. Serum Kl-6 Levels Correlate with Response to Pirfenidone in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 191, A4398. [Google Scholar]
- Bergantini, L.; Bargagli, E.; Cameli, P.; Cekorja, B.; Lanzarone, N.; Pianigiani, L.; Vietri, L.; Bennett, D.; Sestini, P.; Rottoli, P. Serial KL-6 analysis in patients with idiopathic pulmonary fibrosis treated with nintedanib. Respir. Investig. 2019, 57, 290–291. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Okamoto, M.; Fujimoto, K.; Ebata, T.; Tominaga, M.; Nouno, T.; Zaizen, Y.; Kaieda, S.; Tsuda, T.; Kawayama, T.; et al. A retrospective study of the tolerability of nintedanib for severe idiopathic pulmonary fibrosis in the real world. Ann. Transl. Med. 2019, 7, 262. [Google Scholar] [CrossRef] [PubMed]
- Adegunsoye, A.; Alqalyoobi, S.; Linderholm, A.; Bowman, W.S.; Lee, C.T.; Pugashetti, J.V.; Sarma, N.; Ma, S.-F.; Haczku, A.; Sperling, A.; et al. Circulating Plasma Biomarkers of Survival in Antifibrotic-Treated Patients With Idiopathic Pulmonary Fibrosis. Chest 2020, 158, 1526–1534. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Biomarker | Predisposition | Diagnosis | Prognosis | Therapy Monitoring |
|---|---|---|---|---|
| SP-C SP-A SP-D C-pro-SP-B | Disease: ++ Disease: ++ | Disease: ++ Disease: + AE: ++ Disease: ++ | Disease: ++ Disease: ++ | Disease: + Disease: + |
| MUC5B | Disease: +++ | Disease: +/− | Disease: ++ | |
| Telomerase complex | Disease: + | Disease: ++ | Disease: ++ | |
| TLRs | Disease: + | Disease: + | Disease: ++ | |
| ELMOD-2 | Disease: + | |||
| KL-6/MUC1 | Disease: + | Disease: + AE: ++ | Disease: ++ | |
| cCK18 | Disease: ++ | Disease: − | ||
| MMPs: | Diagnosis: ++ | Disease: +++ | Disease: +++ | |
| OPN | Disease: + | Disease: − AE: ++ | ||
| TOLLIP | Disease: ++ | |||
| α-defensins | Disease: + AE: ++ | |||
| Periostin | Disease: ++ |
| Primary Outcome | Secondary Outcomes | Biomarkers Considered | Type of Biomarker | Status and Results | ||
|---|---|---|---|---|---|---|
| Biomarker Discovery for Novel Drug Development in IPF NCT01718990 Year: 2012 | Type: observational prospective longitudinal cohort trial N. part: 110 Patients with IPF vs. healthy volunteers | Dose on BAL, alveolar macrophages, and blood of mechanistically informative markers of alveolar epithelial cell ER stress, αvβ6-mediated TGFβ activation, and EMT | / | Mechanistically informative markers | Diagnostic Therapeutic | Status: completed Results:/ |
| Exhaled Breath Condensate Biomarkers and Cough in People with IPF NCT02630940 Year: 2015 | Type: cross-sectional cohort study N. part: 52 IPF cohort | Detection of 8-isoprostane levels in patients’ exhaled breath condensate samples | LCQ KBILD MRC dyspnoea scale Visual analogue scale for Cough Non-validated acceptability questionnaire | 8-isoprostane in exhaled breath condensate | Prognostic | Status: completed Results:/ |
| Prospective Evaluation of Biomarker Profiles in IPF NCT02151435 Year: 2014 | Type: Observational perspective N. part: 43 IPF cohort | Progression-free survival at 1 year | Longitudinal change in biomarker levels | Peripheral blood biomarkers based on extracellular matrix and matrix-modifying molecules | Prognostic | Status: completed Results:/ |
| COMET study NCT01071707 Year: 2010 | Type: Observational perspective N. part: 108 IPF cohort | Progression free survival as determined by time until any of: death, AE of IPF, relative change in FVC (liters) of at least 10% or DLCO (ml/min/mmHg) of 15% (min 16 weeks; max 80 weeks FU) | / | Multiple biomarkers at baseline (from blood, BAL, bioptic lung tissue) | Prognostic | Status: completed Results:
|
| PROFILE—Central England NCT01134822 Year: 2010 | Type: observational prospective N. part: 330 IPF/NSIP cohort | Discover biomarkers in IPF (discover and validate novel biomarkers, prospectively validate a panel of previously published biomarkers, investigate genetic associations and epigenetic modifications which affect disease severity and progression) | Survival from pulmonary fibrosis (up to 10 years) | Multiple biomarkers | Diagnostic Prognostic | Status: completed Results:
|
| PROFILE_Brompton Study NCT01110694 Year: 2010 | Type: observational prospective N. part: 230 IPF/NSIP cohort | Discover and validate novel biomarkers and gene expression profiles for use in subsequent clinical studies in patients with IPF. | Prospectively evaluate longitudinal disease behavior in patients with IPF and other fibrotic lung diseases with a view to developing composite clinical endpoints for subsequent use in clinical studies in patients with pulmonary fibrosis. Identify differences in the pathogenetic mechanisms involved in the development of different types of fibrosis | Multiple biomarkers | Diagnostic Prognostic | Status: completed Results: as above |
| It’s Not JUST IPF Study NCT03670576 Year: 2018 | Type: observational prospective N. part: 250 Fibrotic Lung disease cohort (4 categories: RA-UIP, Asbestosis, Chronic HP and Unclassifiable) vs. IPF | Disease progression defined as >10% relative decline in FVC Overall survival | Serum and Plasma Biomarkers (SP-D, MUC16, CA199, Nordic Neoepitopes), DLCO and QoL at 3,6,12 and 24 months Domiciliary spirometry | Plasma Biomarkers (SP-D, MUC16, CA199, Nordic Neoepitopes) | Prognostic | Status: suspended (due to COVID-19 pandemics) Results:/ |
| Exhaled Breath Analysis by Secondary Electrospray Ionization—Mass Spectrometry (SESI-MS) in patients with IPF NCT02437448 Year: 2015 | Type: prospective observational N. part: 40 20 IPF patients vs. 20 healthy controls | IPF specific mass spectrometric profile of volatile organic compounds of exhaled breath analysis (markers of IPF in exhaled breath) | / | Amino acids | Predisposition | Status: completed Results: exhaled breath of IPF patients showed higher levels of proline, 4-hydroxyproline, alanine, valine, leucine/isoleucine and allysine compared with healthy controls (p < 0.05) |
| IPFJES NCT03211507 Year: 2017 | Type: observational (case-control) prospective N. part: 960 IPF males vs. male controls | Association between asbestos exposure and IPF | Dose-response relationship between asbestos exposure and IPF Gene-environment interaction (for MUC5 B rs35705950 and asbestos exposure) odds ratio | MUC5B rs35705950 | Predisposition | Status: completed Results:/ |
| Microarray Analysis of Gene Expression in IPF (MAA) NCT00258544 Year: 2005 | Type: observational (cohort) N. prat: 80 | Identification of genetic markers of IPF | / | / | Predisposition | Status: actrive, not recruiting Results: Eighteen microRNAs including let-7d were significantly decreased in IPF (p < 0.05). The down-regulation of let-7d in IPF and the profibrotic effects of this down-regulation in vitro and in vivo suggest a key regulatory role for this microRNA in preventing lung fibrosis. DOI: 10.1164/rccm.200911-1698OC. Epub 2010 Apr 15. |
| Study to investigate longitudinal changes in breath biomarkers in IPF VOC (BI 1199-0311) ISRCTN18106574 Year: 2018 | Type: observational longitudinal cohort N. part: 88 | VOC, measured using mass spectrometry, that can distinguish between IPF patients based on their baseline GAP stage (I, II or III) | VOC, measured using mass spectrometry, that can distinguish between patients based on change in FVC after 12 months VOC which can distinguish between patients with an increase in MRC dyspnoea score of 1 or more after 12 months and those without a change VOC that can distinguish between patients with an increase in USCD, SOBQ scores of 5 or more after 12 months compared to those without a change VOC that can distinguish between patients that respond to antifibrotic treatments and those that do not VOC that can distinguish between patients having an AE of IPF and those who are not | Volatile Organic Compounds | Prognostic | Status: completed Results:/ |
| Primary Outcome | Secondary Outcomes | Biomarkers Considered | Status and Results | ||
|---|---|---|---|---|---|
| INMARK study NCT02788474 Year: 2016 | Type: RCT N. part: 347 Nintedanib vs. Placebo | The rate of change (slope) in blood CRPM from baseline to week 12. | Percentage of patients with disease progression The rate of change in blood C1M from baseline to week 12 The rate of change in blood C3M from baseline to week 12 | CRPM C3M C1M | Status: completed Results: rate of change in CRPM is not a marker of response to nintedanib in patients with IPF The rate of change in CRPM from baseline to week 12 was −2.57 × 10−3 ng/mL/month in the nintedanib group and −1.90 × 10−3 ng/mL/month in the placebo group (between-group difference −0.66 × 10−3 ng/mL/month [95% CI −6.21 × 10−3 to 4.88 × 10−3]; p = 0.8146). |
| A Randomized, Double-blind, Placebo-controlled, Crossover Study to Assess the Effect of 28 Day Treatment with Fostair® Pressurized Metered-dose Inhaler (pMDI) 200/12 on Biomarkers of Platelet Adhesion in Patients with IPF NCT02048644 Year: 2014 | Type: RCT N. part 20 beclomethasone/formoterol pMDI 100/6 mcg 2 puffs twice daily for 28 days vs. placebo | Platelet-monocyte complex formation platelet P-selectin expression platelet fibrinogen binding | FVC sputum eosinophils cells six minutes-walk d istance | Platelet derived markers | Status: completed Results: Change from baseline spirometric measurements of FEV1(L), FEV/FVC % pred FEF25–75 were significantly improved following 28 days B/F by (mean ± SD), 0.88 ± 0.16 L (p = 0.03), 0.03 ± 0.03 (p = 0.03), 12.4 ± 19.1% (p = 0.02) respectively when compared to placebo. There was no change in quality of life or exercise measures. The effects of beclomethasone/formoterol in this study may represent delivery of corticosteroid to the peripheral airways ameliorating local injury and altering platelet activation |
| Randomized, Double-Blind, Placebo-Controlled, Multiple Dose, Dose-Escalation Study of STX-100 in Patients With IPF NCT01371305 Year: 2011 | Type: RCT N. part: 41 SXT-100 vs. placebo in IPF | Number of Participants with adverse events | Pharmacodinamic and pharmacokinetic parameters of BG00011 (STX-100) Percentage change from baseline in biomarkers solated from BAL Number of participants with treatment emergent antibodies to BG00011 | The expression level of 7 genes; ALOX5, FN1, OLR1, PAI-1 (aka SERPINE 1), TGM2, TREM1, and ETS1 were assessed via BAL as well as a ratio of pSMAD2 to tSMAD2 levels. | Status: completed Results:/ |
| A Open-label, Multicenter Study, With a Single Intravenous Dose of QAX576 to Determine IL-13 Production in patients with IPF NCT00532233 Year: 2007 | Type: open label clinical trial N. part: 52 QAX576 in IPF cohort | To investigate the possibility that some IPF patients experience increased IL-13 production. Blood samples to be collected pre-dose and weekly after dosing. -To investigate the hypothesis that QAX576 will neutralize IL-13 in patients with IPF | To evaluate the changes in biomarkers in blood over time in patients with IPF. Serum samples will be obtained at pre-dose and 2 weeks post-dose. | IL-13 Other blood biomarkers | Status: completed Results: the study was terminated early after 31 patients were enrolled and randomized to receive QAX576 due to slow enrolment rate. |
| Randomized, Double-Blind, Parallel Group, Placebo-Controlled, Multicenter, Exploratory Phase IIa Study to Assess Safety, Tolerability, Pharmacokinetic and Pharmacodynamic Properties of GLPG1690 Administered for 12 Weeks in Subjects With IPF NCT02738801 Year: 2016 | Type: RCT N. part: 23 GLPG1690 capsules, administered at a dose of 600 mg, orally QD vs. placebo in IPF cohort. | Adverse events, pharmacodynamic and pharmacokinetics parameters, mean Peak Area Ratio of LPA C18:2 species in Blood and BALF | / | LPA C18:2 | Status: completed Results: concentrations of LPA C18:2 in plasma decreased after administration of GLPG1690 at the week 4 (p = 0.0001) and 12 (p = 0.0014) visits and return to baseline concentrations at the FU visit. In BALF, LPA C18:2 and LPA C20:4 concentrations are below the level of quantification for more than 25% of baseline samples obtained from patients in the GLPG1690 treatment group. |
| A Randomized, Double-Blind, Placebo-Controlled Phase II Clinical Trial of GKT137831 in Patients with IPF NCT03865927 Year: 2020 | Type: RCT N. part: 60 GKT137831 400 mg bid for 24 weeks vs. placebo | Surrogate biomarker of oxidative stress by mass spectroscopy through 24 weeks (changes in concentrations of circulating o,o’-dityrosine) | Collagen degradation product (serum C1M) by enzyme linked immunoabsorbant assay through 24 weeks LFT Ambulatory ability by measuring walk distance in six-minutes Evaluation of safety by adverse events | o,o’-dityrosine C1M | Status: ongoing |
| Non-Interventional Collecting Evidences For ILD in Taiwan: Optimized Novel Therapy NCT04614441 Year: 2020 | Type: observational prospective N. part: 500 IPF vs. PF-ILD vs. SSc-ILD on therapy with Nintedanib 150 mg bid | Annual percentage of decline from baseline in FVC, %, DLCO, % and resting and exercise oxygen saturation (SpO2, %) per cohort of IPF, SSc-ILD, or PF-ILD | Time to first AE of IPF; or time to ILD worsening for SSc-ILD/PF-ILD after study enrolment Annual change from baseline in SGRQ for IPF or K-BILD for other ILDs, CAT, Berlin questionnaire and 6MWT Change from baseline in quantification of biomarkers Mortality | Include but not limited to PDGF, VEGF, FGF, TGF-β1, HGF, MMPs: MMP-1, MMP-7, MMP-9, α-defensin 1, HMGB1, TIMP, HSP: HSP-27, bile acid conjugated, LPA, LPAR1, PGE2, IL: IL-1β, IL-4, IL-18, IL-13, IL-17, MCP-1, MIP-2, periostin, osteopontin, SP-A, SP-D, KL-6/MUC1, anti-HSP70, IgG BMP, CA-199, CRPM, CCL 2, CCL-18 | Status: ongoing |
| Targeted Removal of Pro-Inflammatory Cells: An Open Label Human Pilot Study in IPF NCT02874989 Year: 2016 | Type: RCT N. part: 26 Dasatinib + Quercetin vs. placebo in IPF cohort | Percentage of pro-inflammatory expressing cells (skin biopsy) Percentage of pro-inflammatory expressing cells (skin biopsy) BP, weight, HR, CBC, lipid panel, HBA1c, comprehensive metabolic panel, high sensitivity CRP, plasma IL-6, plasma PASP biomarkers, p16INK4a biomarker | / | high sensitivity CRP, plasma IL-6, plasma PASP biomarkers, p16INK4a biomarker | Status: ongoing |
| EXCHANGE-IPF NCT03584802 Year: 2018 | Type: RCT N. part: 40 Therapeutic plasma exchanges vs. conventional treatment in AE of IPF | Overall mortality at day 28 after initiation of therapy | […] Changes in lung injury biomarkers in plasma (KL-6, SP-D) between day 1 and day 90 Changes in circulating autoantibodies levels (anti-periplakin, anti-HSP70 and anti-vimentin antibodies) between day 1 and day 90 | Injury biomarkers Circulating fibrocytes Auto-antibodies | Status: ongoing |
| Primary Outcome | Secondary Outcomes | Biomarkers Considered | Type of Biomarker | ||
|---|---|---|---|---|---|
| Early Diagnosis of Pulmonary Fibrosis—Use of Biomarkers in IPF NCT02755441 Year: 2016 | Type: observational perspective N. part: 300 IPF cohort | Disease progression or mortality at 1 year | Hospitalizations Exacerbations LFTs Mortality QoL Combined endpoints of disease progression Progression in serum/plasma biomarker levels | Unspecified multiple biomarkers | Prognostic |
| Immunopathologic Profiles of the Lung Micro-Environment Using Cryobiopsies and Identification of Blood Biomarkers in Patients With IPF NCT04187079 Year: 2017 | Type: observational prospective N. part: 100 IPF cohort vs. other ILD cohort | Expression of PD-L1 in the epithelial cells in lungs | / | PD-L1, PD-L2, Beta- catenin, B-cell follicles and Tenascin- C in cryobiopsies from the lungs anti HSP 70, p-ANCA, c-ANCA, CD4+/CD28- and CD8+/CD28- cells in blood samples | Diagnostic |
| Development of Airway Absorption Sampling Methods for Biomarker Assessment in Probable IPF Patients NCT04494334 Year: 2020 | Type: observational cross-sectional study N. part: 60 IPF vs. sarcoidosis vs. healthy controls | Levels of the of biomarker/mediator SP-D, CCL18, CXCL13 and periostin in bronchial Lining fluid in IPF and sarcoidosis patients | Levels of Periostin, SP-D, CCL18 and CXCL13 in nasosorption samples within and across the 3 groups of participants Levels of Periostin, SPD, CCL18 and CXCL13 in blood within and across the 3 groups of participants | SP-D, CCL18, CXCL13 and periostin | Diagnostic |
| Pulmonary Fibrosis Biomarkers During Exacerbation N CT04442711 Year: 2020 | Type: observational prospective N. part: 50 IPF cohort | Mortality at 30 and 90 days | Biomarkers level, change in oxygen need, QoL, need for respiratory support, decline of LFTs at 30 days. Treatment during and after hospitalization | Multiple biomarkers on blood serum and plasma collected within 24 h of hospital admission | Diagnostic Prognostic |
| LOCK-IPF NCT04268485 Year: 2020 | Type: observational prospective N. part: 60 IPF cohort | Change in serum KL-6 level between baseline and 12 months | Change in serum KL-6 level between baseline and 3 and 6 months. Correlation of KL-6 and FVC, DLCO, symptoms, response to antifibrotic therapy and GAP stage at 3, 6 and 12 months to baseline Correlation between KL-6 levels and CPI Difference in KL-6 levels between patients with indeterminate, probable and definite UIP on HRCT | KL-6 on blood | Prognostic |
| Cardiovascular fibrosis in IPF NCT04177251 Year: 2019 | Type: observational case-control prospective study N. part: 168 IPF cohort vs. healthy controls | Presence of cardiac fibrosis in a population of patients with overt IPF at diagnosis in comparison with healthy controls Presence of vascular fibrosis in a population of patients with overt IPF at diagnosis in comparison with healthy controls | Levels of biomarkers analyzed (galectins-3, osteopontin and periostin) IPF progression after 1 year from diagnosis in IPF patients Blood proteomic and metabolomic biomarkers | galectins-3, osteopontin and periostin Proteomic and metabolomic biomarkers | Diagnostic Prognostic |
| The Role of the miR200 Family in the Restoration of Normal Lung Homeostasis and Detection of Early IPF NCT03457935 Year: 2018 | Type: observational prospective N. part: 450 IPF vs. non-IPF ILD vs. healthy controls | Determine miR200 levels (fold change) in blood samples to identify biomarkers for IPF | / | miR200 | Diagnostic |
| IPF and Serum Bank NCT04016168 Year: 2014 | Type: observational prospective N. part: 500 Diffuse idiopathic ILD cohort | Determination of circulating CD163 serum concentration | / | CD163 | n/a |
| Role of Genetics in IPF NCT01088217 Year: 2010 | Type: observational cross-sectional study (family based) N. part: 8000 IPF, familial pulmonary fibrosis cohort, Idiopathic Interstitial Pneumonia Familial Interstitial Pneumonia | Identify a group of genetic loci that play a role in the development of familial interstitial pneumonia and idiopathic interstitial pneumonia. | Develop biomarkers using proteomic and genomic approaches that will facilitate establishing the diagnosis and prognosis of both familial and sporadic forms of idiopathic interstitial pneumonia | Multiple biomarkers | Diagnostic Prognostic |
| ELFMEN Study NCT04016181 Year: 2007 | Type: observational prosepective N. part: 800 IPF and other ILDs | Time to death | Biomarkers that are associated with increased rate of decline in vital capacity, increased lung-related mortality and that predict rate of change in gas transfer | Multiple biomarkers | Prognostic |
| Genomic and Proteomic Analysis (GAP) of Disease Progression in IPF NCT00373841 Year: 2006 | Type: observational N. part: 500 IPF cohort | Identify genetic and biologic markers that may predict the loss of lung function due to idiopathic pulmonary fibrosis through comparison of genetic and biologic markers of samples to changes in symptoms | / | Multiple biomarkers | Prognostic |
| EXCHANGE-IPF NCT03584802 Year: 2018 | Type: RCT N. part: 40 Therapeutic plasma exchanges vs. conventional treatment in AE of IPF | Overall mortality at day 28 after initiation of therapy | […] Changes in lung injury biomarkers in plasma (KL-6, SP-D) between day 1 and day 90 Changes in circulating autoantibodies levels (anti-periplakin, anti-HSP70 and anti-vimentin antibodies) between day 1 and day 90 | Injury biomarkers Circulating fibrocytes Auto-antibodies | Therapeutic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stainer, A.; Faverio, P.; Busnelli, S.; Catalano, M.; Della Zoppa, M.; Marruchella, A.; Pesci, A.; Luppi, F. Molecular Biomarkers in Idiopathic Pulmonary Fibrosis: State of the Art and Future Directions. Int. J. Mol. Sci. 2021, 22, 6255. https://doi.org/10.3390/ijms22126255
Stainer A, Faverio P, Busnelli S, Catalano M, Della Zoppa M, Marruchella A, Pesci A, Luppi F. Molecular Biomarkers in Idiopathic Pulmonary Fibrosis: State of the Art and Future Directions. International Journal of Molecular Sciences. 2021; 22(12):6255. https://doi.org/10.3390/ijms22126255
Chicago/Turabian StyleStainer, Anna, Paola Faverio, Sara Busnelli, Martina Catalano, Matteo Della Zoppa, Almerico Marruchella, Alberto Pesci, and Fabrizio Luppi. 2021. "Molecular Biomarkers in Idiopathic Pulmonary Fibrosis: State of the Art and Future Directions" International Journal of Molecular Sciences 22, no. 12: 6255. https://doi.org/10.3390/ijms22126255
APA StyleStainer, A., Faverio, P., Busnelli, S., Catalano, M., Della Zoppa, M., Marruchella, A., Pesci, A., & Luppi, F. (2021). Molecular Biomarkers in Idiopathic Pulmonary Fibrosis: State of the Art and Future Directions. International Journal of Molecular Sciences, 22(12), 6255. https://doi.org/10.3390/ijms22126255