Burden of Rare Variants in ALS and Axonal Hereditary Neuropathy Genes Influence Survival in ALS: Insights from a Next Generation Sequencing Study of an Italian ALS Cohort

 ,
, 
Abstract
1. Introduction
2. Results
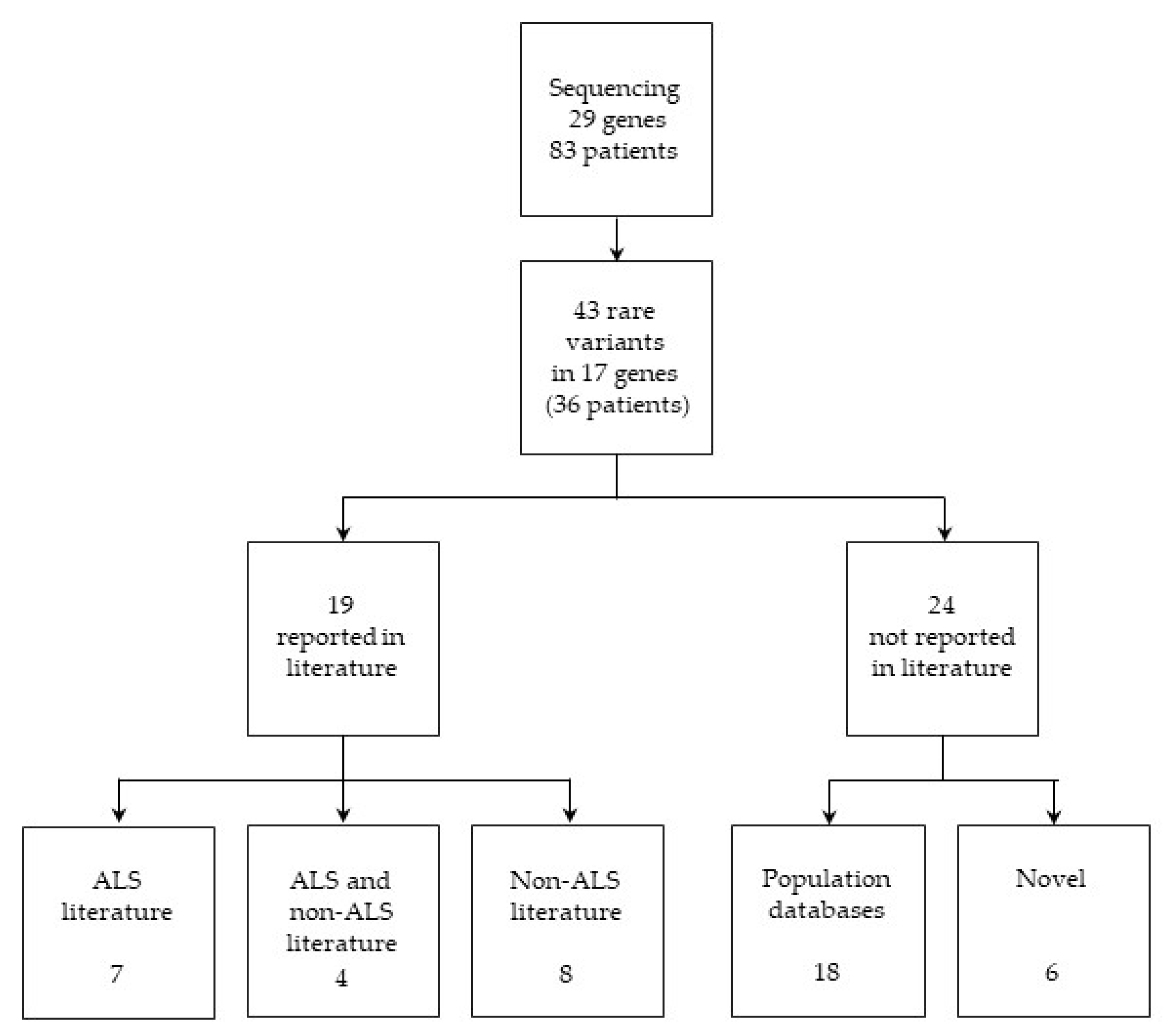
2.1. Variant Identification and Classification
2.1.1. Variants Previously Reported in ALS Literature
2.1.2. Variants Previously Reported in both ALS and Non-ALS Literature
2.1.3. Variants Previously Reported in Non-ALS Literature
2.1.4. Variants Present Only in Population Databases
2.1.5. Novel Candidate ALS-Associated Variants
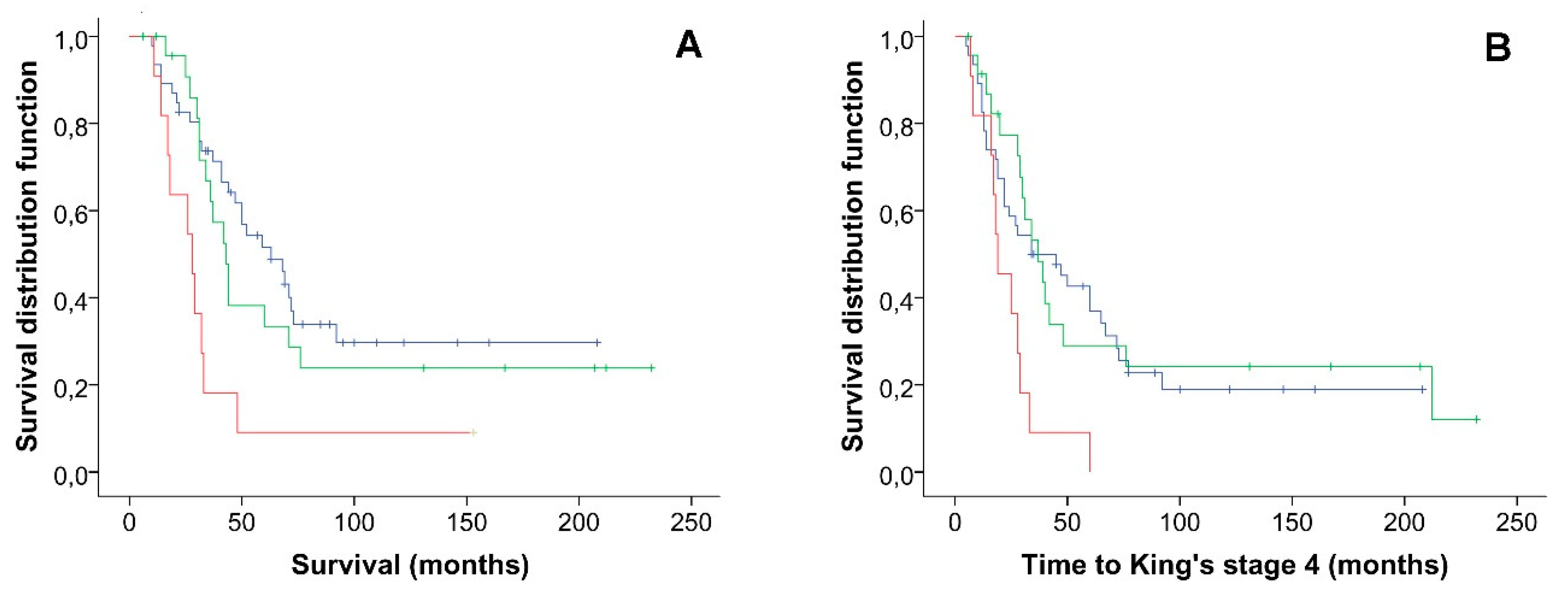
2.2. Co-Occurrence of Variants in ALS Genes Panel and Survival Analysis
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. DNA Preparation
4.3. Targeted Next Generation Sequencing (NGS)
4.4. Filters
4.5. Sanger Sequencing Validation
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| MND | Motor neuron diseases |
| UMN | Upper motor neurons |
| LMN | Lower motor neurons |
| fALS | Familial amyotrophic lateral sclerosis |
| sALS | Sporadic smyotrophic lateral sclerosis |
| SOD1 | Superoxide Dismutase 1 |
| C9orf72 | Chromosome 9 open reading frame 72 |
| TARDBP | TAR DNA binding protein |
| FUS | FUS RNA binding protein |
| CMT | Charcot-Marie-Tooth neuropathy |
| dHMN | Distal hereditary motor neuropathy |
| NGS | Next generation sequencing |
| ALS2 | Alsin Rho guanine nucleotide exchange factor |
| ANG | Angiogenin |
| DCTN1 | Dynactin subunit 1 |
| ERBB4 | Erb-B2 receptor tyrosine kinase 4 |
| FIG4 | FIG4 phosphoinositide 5-phosphatase |
| OPTN | Optineurin |
| PFN1 | Profilin 1 |
| SETX | Senataxin |
| SPAST | Spastin |
| SPG11 | Spatacsin vesicle trafficking associated |
| SQSTM1 | Sequestosome 1 |
| UBQLN2 | Ubiquilin 2 |
| VAPB | VAMP associated protein B and C |
| VCP | Valosin containing protein |
| DYNC1H1 | Dynein cytoplasmic 1 heavy chain 1 |
| GARS | Glycyl-tRNA synthetase |
| PLEKHG5 | Pleckstrin homology and RhoGEF domain containing G5 |
| SPG7 | Paraplegin matrix AAA peptidase subunit |
| BSCL2 | Seipin lipid droplet biogenesis associated |
| HSPB1 | Heat shock protein family B (Small) member 1 |
| HSPB3 | Heat shock protein family B (Small) member 3 |
| HSPB8 | Heat shock protein family B (Small) member 8 |
| MFN2 | Mitofusin 2 |
| TRPV4 | Transient receptor potential cation channel subfamily V member 4 |
| TBK1 | TANK-binding kinase 1 |
| HSP | Hereditary spastic paraplegia |
| dSMA | Distal spinal muscular atrophy |
| POAG | Primary open angle glaucoma |
| AOA2 | Ataxia with oculomotor apraxia type 2 |
| PEO | Progressive external ophthalmoplegia |
| AD | Alzheimer disease |
| FTD | Frontotemporal dementia |
| SMA-LED | Spinal muscular atrophy with predominance of lower extremity involvement |
| PEG | Percutaneous endoscopic gastrostomy |
| NIMV | Non-invasive mechanical ventilation |
References
- Riva, N.; Agosta, F.; Lunetta, C.; Filippi, M.; Quattrini, A. Recent advances in amyotrophic lateral sclerosis. J. Neurol. 2016, 263, 1241–1254. [Google Scholar] [CrossRef] [PubMed]
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chio, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E. Incidence of amyotrophic lateral sclerosis in europe. J. Neurol. Neurosurg. Psychiatry 2010, 81, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Fang, F.; Hanby, M.F.; Leigh, P.N.; Shaw, C.E.; Ye, W.; Rijsdijk, F. An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1324–1326. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Calvo, A.; Chio, A.; Colville, S.; Ellis, C.M.; Hardiman, O.; Heverin, M.; Howard, R.S.; Huisman, M.H.B.; Keren, N.; et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet Neurol. 2014, 13, 1108–1113. [Google Scholar] [CrossRef]
- The scottish motor neuron disease register: A prospective study of adult onset motor neuron disease in scotland. Methodology, demography and clinical features of incident cases in 1989. J. Neurol. Neurosurg. Psychiatry 1992, 55, 536–541.
- Cady, J.; Allred, P.; Bali, T.; Pestronk, A.; Goate, A.; Miller, T.M.; Mitra, R.D.; Ravits, J.; Harms, M.B.; Baloh, R.H. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol. 2015, 77, 100–113. [Google Scholar] [CrossRef]
- Gentile, F.; Scarlino, S.; Falzone, Y.M.; Lunetta, C.; Tremolizzo, L.; Quattrini, A.; Riva, N. The peripheral nervous system in amyotrophic lateral sclerosis: Opportunities for translational research. Front. Neurosci. 2019, 13, 601. [Google Scholar] [CrossRef]
- Riva, N.; Iannaccone, S.; Corbo, M.; Casellato, C.; Sferrazza, B.; Lazzerini, A.; Scarlato, M.; Cerri, F.; Previtali, S.C.; Nobile-Orazio, E.; et al. Motor nerve biopsy: Clinical usefulness and histopathological criteria. Ann. Neurol. 2011, 69, 197–201. [Google Scholar] [CrossRef]
- Agosta, F.; Galantucci, S.; Magnani, G.; Marcone, A.; Martinelli, D.; Antonietta Volonte, M.; Riva, N.; Iannaccone, S.; Ferraro, P.M.; Caso, F.; et al. Mri signatures of the frontotemporal lobar degeneration continuum. Hum. Brain Mapp. 2015, 36, 2602–2614. [Google Scholar] [CrossRef] [PubMed]
- Orrell, R.W.; Marklund, S.L.; deBelleroche, J.S. Familial als is associated with mutations in all exons of sod1: A novel mutation in exon 3 (gly72ser). J. Neurol. Sci. 1997, 153, 46–49. [Google Scholar] [CrossRef]
- Kobayashi, Z.; Tsuchiya, K.; Kubodera, T.; Shibata, N.; Arai, T.; Miura, H.; Ishikawa, C.; Kondo, H.; Ishizu, H.; Akiyama, H.; et al. Fals with gly72ser mutation in sod1 gene: Report of a family including the first autopsy case. J. Neurol. Sci. 2011, 300, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Del Grande, A.; Luigetti, M.; Conte, A.; Mancuso, I.; Lattante, S.; Marangi, G.; Stipa, G.; Zollino, M.; Sabatelli, M. A novel l67p sod1 mutation in an italian als patient. Amyotroph. Lateral Scler. 2011, 12, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Kenna, K.P.; McLaughlin, R.L.; Byrne, S.; Elamin, M.; Heverin, M.; Kenny, E.M.; Cormican, P.; Morris, D.W.; Donaghy, C.G.; Bradley, D.G.; et al. Delineating the genetic heterogeneity of als using targeted high-throughput sequencing. J. Med. Genet. 2013, 50, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, L.; Valenza, F.; Mosca, L.; Dal Mas, A.; Domi, T.; Romano, A.; Tarlarini, C.; Falzone, Y.M.; Tremolizzo, L.; Soraru, G.; et al. Tbk1 mutations in italian patients with amyotrophic lateral sclerosis: Genetic and functional characterisation. J. Neurol. Neurosurg. Psychiatry 2017, 88, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Kruger, S.; Battke, F.; Sprecher, A.; Munz, M.; Synofzik, M.; Schols, L.; Gasser, T.; Grehl, T.; Prudlo, J.; Biskup, S. Rare variants in neurodegeneration associated genes revealed by targeted panel sequencing in a german als cohort. Front. Mol. Neurosci. 2016, 9, 92. [Google Scholar] [CrossRef]
- Morgan, S.; Shoai, M.; Fratta, P.; Sidle, K.; Orrell, R.; Sweeney, M.G.; Shatunov, A.; Sproviero, W.; Jones, A.; Al-Chalabi, A.; et al. Investigation of next-generation sequencing technologies as a diagnostic tool for amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, 1600.e5–1600.e8. [Google Scholar] [CrossRef]
- Bernard, V.; Stricker, S.; Kreuz, F.; Minnerop, M.; Gillessen-Kaesbach, G.; Zuhlke, C. Ataxia with oculomotor apraxia type 2: Novel mutations in six patients with juvenile age of onset and elevated serum alpha-fetoprotein. Neuropediatrics 2008, 39, 347–350. [Google Scholar] [CrossRef]
- Hoyer, H.; Braathen, G.J.; Busk, O.L.; Holla, O.L.; Svendsen, M.; Hilmarsen, H.T.; Strand, L.; Skjelbred, C.F.; Russell, M.B. Genetic diagnosis of charcot-marie-tooth disease in a population by next-generation sequencing. Biomed. Res. Int. 2014, 2014, 210401. [Google Scholar] [CrossRef]
- Arning, L.; Epplen, J.T.; Rahikkala, E.; Hendrich, C.; Ludolph, A.C.; Sperfeld, A.D. The setx missense variation spectrum as evaluated in patients with als4-like motor neuron diseases. Neurogenetics 2013, 14, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Pensato, V.; Castellotti, B.; Gellera, C.; Pareyson, D.; Ciano, C.; Nanetti, L.; Salsano, E.; Piscosquito, G.; Sarto, E.; Eoli, M.; et al. Overlapping phenotypes in complex spastic paraplegias spg11, spg15, spg35 and spg48. Brain 2014, 137, 1907–1920. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.Y.; Zhang, Y.; Dowling, J.J.; Jin, N.; Adamska, M.; Shiga, K.; Szigeti, K.; Shy, M.E.; Li, J.; Zhang, X.; et al. Mutation of fig4 causes neurodegeneration in the pale tremor mouse and patients with cmt4j. Nature 2007, 448, 68–72. [Google Scholar] [CrossRef]
- Nicholson, G.; Lenk, G.M.; Reddel, S.W.; Grant, A.E.; Towne, C.F.; Ferguson, C.J.; Simpson, E.; Scheuerle, A.; Yasick, M.; Hoffman, S.; et al. Distinctive genetic and clinical features of cmt4j: A severe neuropathy caused by mutations in the pi(3,5)p(2) phosphatase fig4. Brain 2011, 134, 1959–1971. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.P.; Waddell, L.; Lenk, G.M.; Kaur, S.; MacArthur, D.G.; Meisler, M.H.; Clarke, N.F. Whole exome sequencing identifies three recessive fig4 mutations in an apparently dominant pedigree with charcot-marie-tooth disease. Neuromuscul. Disord. 2014, 24, 666–670. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lenk, G.M.; Ferguson, C.J.; Chow, C.Y.; Jin, N.; Jones, J.M.; Grant, A.E.; Zolov, S.N.; Winters, J.J.; Giger, R.J.; Dowling, J.J.; et al. Pathogenic mechanism of the fig4 mutation responsible for charcot-marie-tooth disease cmt4j. PLoS Genet. 2011, 7, e1002104. [Google Scholar] [CrossRef]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious variants of fig4, a phosphoinositide phosphatase, in patients with als. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Osmanovic, A.; Rangnau, I.; Kosfeld, A.; Abdulla, S.; Janssen, C.; Auber, B.; Raab, P.; Preller, M.; Petri, S.; Weber, R.G. Fig4 variants in central european patients with amyotrophic lateral sclerosis: A whole-exome and targeted sequencing study. Eur. J. Hum. Genet. 2017, 25, 324–331. [Google Scholar] [CrossRef]
- Takahashi, Y.; Fukuda, Y.; Yoshimura, J.; Toyoda, A.; Kurppa, K.; Moritoyo, H.; Belzil, V.V.; Dion, P.A.; Higasa, K.; Doi, K.; et al. Erbb4 mutations that disrupt the neuregulin-erbb4 pathway cause amyotrophic lateral sclerosis type 19. Am. J. Hum. Genet. 2013, 93, 900–905. [Google Scholar] [CrossRef]
- Rudloff, U.; Samuels, Y. A growing family: Adding mutated erbb4 as a novel cancer target. Cell Cycle 2010, 9, 1487–1503. [Google Scholar] [CrossRef]
- Stevanin, G.; Azzedine, H.; Denora, P.; Boukhris, A.; Tazir, M.; Lossos, A.; Rosa, A.L.; Lerer, I.; Hamri, A.; Alegria, P.; et al. Mutations in spg11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 2008, 131, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Denora, P.S.; Schlesinger, D.; Casali, C.; Kok, F.; Tessa, A.; Boukhris, A.; Azzedine, H.; Dotti, M.T.; Bruno, C.; Truchetto, J.; et al. Screening of arhsp-tcc patients expands the spectrum of spg11 mutations and includes a large scale gene deletion. Hum. Mutat. 2009, 30, E500–E519. [Google Scholar] [CrossRef] [PubMed]
- DiVincenzo, C.; Elzinga, C.D.; Medeiros, A.C.; Karbassi, I.; Jones, J.R.; Evans, M.C.; Braastad, C.D.; Bishop, C.M.; Jaremko, M.; Wang, Z.; et al. The allelic spectrum of charcot-marie-tooth disease in over 17,000 individuals with neuropathy. Mol. Genet. Genom. Med. 2014, 2, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Cuyvers, E.; van der Zee, J.; Bettens, K.; Engelborghs, S.; Vandenbulcke, M.; Robberecht, C.; Dillen, L.; Merlin, C.; Geerts, N.; Graff, C.; et al. Genetic variability in sqstm1 and risk of early-onset alzheimer dementia: A european early-onset dementia consortium study. Neurobiol. Aging 2015, 36, 2005.e15–2005.e22. [Google Scholar] [CrossRef]
- Van der Zee, J.; Van Langenhove, T.; Kovacs, G.G.; Dillen, L.; Deschamps, W.; Engelborghs, S.; Matej, R.; Vandenbulcke, M.; Sieben, A.; Dermaut, B.; et al. Rare mutations in sqstm1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 2014, 128, 397–410. [Google Scholar] [CrossRef]
- Zhang, J.; Grubor, V.; Love, C.L.; Banerjee, A.; Richards, K.L.; Mieczkowski, P.A.; Dunphy, C.; Choi, W.; Au, W.Y.; Srivastava, G.; et al. Genetic heterogeneity of diffuse large b-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1398–1403. [Google Scholar] [CrossRef]
- Giannakis, M.; Hodis, E.; Jasmine Mu, X.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. Rnf43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef]
- Morelli, F.F.; Verbeek, D.S.; Bertacchini, J.; Vinet, J.; Mediani, L.; Marmiroli, S.; Cenacchi, G.; Nasi, M.; De Biasi, S.; Brunsting, J.F.; et al. Aberrant compartment formation by hspb2 mislocalizes lamin a and compromises nuclear integrity and function. Cell Rep. 2017, 20, 2100–2115. [Google Scholar] [CrossRef]
- Morais, S.; Raymond, L.; Mairey, M.; Coutinho, P.; Brandao, E.; Ribeiro, P.; Loureiro, J.L.; Sequeiros, J.; Brice, A.; Alonso, I.; et al. Massive sequencing of 70 genes reveals a myriad of missing genes or mechanisms to be uncovered in hereditary spastic paraplegias. Eur. J. Hum. Genet. 2017, 25, 1217–1228. [Google Scholar] [CrossRef]
- Casasnovas, C.; Banchs, I.; Cassereau, J.; Gueguen, N.; Chevrollier, A.; Martinez-Matos, J.A.; Bonneau, D.; Volpini, V. Phenotypic spectrum of mfn2 mutations in the spanish population. J. Med. Genet. 2010, 47, 249–256. [Google Scholar] [CrossRef]
- Engelfried, K.; Vorgerd, M.; Hagedorn, M.; Haas, G.; Gilles, J.; Epplen, J.T.; Meins, M. Charcot-marie-tooth neuropathy type 2a: Novel mutations in the mitofusin 2 gene (mfn2). BMC Med. Genet. 2006, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- McCorquodale, D.S., 3rd; Montenegro, G.; Peguero, A.; Carlson, N.; Speziani, F.; Price, J.; Taylor, S.W.; Melanson, M.; Vance, J.M.; Zuchner, S. Mutation screening of mitofusin 2 in charcot-marie-tooth disease type 2. J. Neurol. 2011, 258, 1234–1239. [Google Scholar] [CrossRef] [PubMed]
- Cassereau, J.; Casasnovas, C.; Gueguen, N.; Malinge, M.C.; Guillet, V.; Reynier, P.; Bonneau, D.; Amati-Bonneau, P.; Banchs, I.; Volpini, V.; et al. Simultaneous mfn2 and gdap1 mutations cause major mitochondrial defects in a patient with cmt. Neurology 2011, 76, 1524–1526. [Google Scholar] [CrossRef] [PubMed]
- Braathen, G.J.; Sand, J.C.; Lobato, A.; Hoyer, H.; Russell, M.B. Mfn2 point mutations occur in 3.4% of charcot-marie-tooth families. An investigation of 232 norwegian cmt families. BMC Med. Genet. 2010, 11, 48. [Google Scholar] [CrossRef]
- Antoniadi, T.; Buxton, C.; Dennis, G.; Forrester, N.; Smith, D.; Lunt, P.; Burton-Jones, S. Application of targeted multi-gene panel testing for the diagnosis of inherited peripheral neuropathy provides a high diagnostic yield with unexpected phenotype-genotype variability. BMC Med. Genet. 2015, 16, 84. [Google Scholar] [CrossRef]
- Sanchez-Ferrero, E.; Coto, E.; Beetz, C.; Gamez, J.; Corao, A.I.; Diaz, M.; Esteban, J.; del Castillo, E.; Moris, G.; Infante, J.; et al. Spg7 mutational screening in spastic paraplegia patients supports a dominant effect for some mutations and a pathogenic role for p.A510v. Clin. Genet. 2013, 83, 257–262. [Google Scholar] [CrossRef]
- Roxburgh, R.H.; Marquis-Nicholson, R.; Ashton, F.; George, A.M.; Lea, R.A.; Eccles, D.; Mossman, S.; Bird, T.; van Gassen, K.L.; Kamsteeg, E.J.; et al. The p.Ala510val mutation in the spg7 (paraplegin) gene is the most common mutation causing adult onset neurogenetic disease in patients of british ancestry. J. Neurol. 2013, 260, 1286–1294. [Google Scholar] [CrossRef]
- McDermott, C.J.; Dayaratne, R.K.; Tomkins, J.; Lusher, M.E.; Lindsey, J.C.; Johnson, M.A.; Casari, G.; Turnbull, D.M.; Bushby, K.; Shaw, P.J. Paraplegin gene analysis in hereditary spastic paraparesis (hsp) pedigrees in northeast england. Neurology 2001, 56, 467–471. [Google Scholar] [CrossRef]
- Klebe, S.; Depienne, C.; Gerber, S.; Challe, G.; Anheim, M.; Charles, P.; Fedirko, E.; Lejeune, E.; Cottineau, J.; Brusco, A.; et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 2012, 135, 2980–2993. [Google Scholar] [CrossRef]
- Pfeffer, G.; Gorman, G.S.; Griffin, H.; Kurzawa-Akanbi, M.; Blakely, E.L.; Wilson, I.; Sitarz, K.; Moore, D.; Murphy, J.L.; Alston, C.L.; et al. Mutations in the spg7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014, 137, 1323–1336. [Google Scholar] [CrossRef]
- Bonn, F.; Pantakani, K.; Shoukier, M.; Langer, T.; Mannan, A.U. Functional evaluation of paraplegin mutations by a yeast complementation assay. Hum. Mutat. 2010, 31, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Munch, C.; Sedlmeier, R.; Meyer, T.; Homberg, V.; Sperfeld, A.D.; Kurt, A.; Prudlo, J.; Peraus, G.; Hanemann, C.O.; Stumm, G.; et al. Point mutations of the p150 subunit of dynactin (dctn1) gene in als. Neurology 2004, 63, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef] [PubMed]
- Lupo, V.; Aguado, C.; Knecht, E.; Espinos, C. Chaperonopathies: Spotlight on hereditary motor neuropathies. Front. Mol. Biosci. 2016, 3, 81. [Google Scholar] [CrossRef]
- Evgrafov, O.V.; Mersiyanova, I.; Irobi, J.; Van Den Bosch, L.; Dierick, I.; Leung, C.L.; Schagina, O.; Verpoorten, N.; Van Impe, K.; Fedotov, V.; et al. Mutant small heat-shock protein 27 causes axonal charcot-marie-tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004, 36, 602–606. [Google Scholar] [CrossRef]
- Fiorillo, C.; Moro, F.; Yi, J.; Weil, S.; Brisca, G.; Astrea, G.; Severino, M.; Romano, A.; Battini, R.; Rossi, A.; et al. Novel dynein dync1h1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development. Hum. Mutat. 2014, 35, 298–302. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; van Es, M.A.; Hennekam, E.A.; Dooijes, D.; van Rheenen, W.; Medic, J.; Bourque, P.R.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 3776–3784. [Google Scholar] [CrossRef]
- Chornenkyy, Y.; Fardo, D.W.; Nelson, P.T. Tau and tdp-43 proteinopathies: Kindred pathologic cascades and genetic pleiotropy. Lab. Investig. 2019, 99, 993–1007. [Google Scholar] [CrossRef]
- Weedon, M.N.; Hastings, R.; Caswell, R.; Xie, W.; Paszkiewicz, K.; Antoniadi, T.; Williams, M.; King, C.; Greenhalgh, L.; Newbury-Ecob, R.; et al. Exome sequencing identifies a dync1h1 mutation in a large pedigree with dominant axonal charcot-marie-tooth disease. Am. J. Hum. Genet. 2011, 89, 308–312. [Google Scholar] [CrossRef]
- Harms, M.B.; Allred, P.; Gardner, R., Jr.; Fernandes Filho, J.A.; Florence, J.; Pestronk, A.; Al-Lozi, M.; Baloh, R.H. Dominant spinal muscular atrophy with lower extremity predominance: Linkage to 14q32. Neurology 2010, 75, 539–546. [Google Scholar] [CrossRef]
- Beecroft, S.J.; McLean, C.A.; Delatycki, M.B.; Koshy, K.; Yiu, E.; Haliloglu, G.; Orhan, D.; Lamont, P.J.; Davis, M.R.; Laing, N.G.; et al. Expanding the phenotypic spectrum associated with mutations of dync1h1. Neuromuscul. Disord. 2017, 27, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Suzuki, N. Seipinopathy: A novel endoplasmic reticulum stress-associated disease. Brain 2009, 132, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Craveiro Sarmento, A.S.; de Azevedo Medeiros, L.B.; Agnez-Lima, L.F.; Lima, J.G.; de Melo Campos, J.T.A. Exploring seipin: From biochemistry to bioinformatics predictions. Int. J. Cell Biol. 2018, 2018, 5207608. [Google Scholar] [CrossRef] [PubMed]
- Datskevich, P.N.; Nefedova, V.V.; Sudnitsyna, M.V.; Gusev, N.B. Mutations of small heat shock proteins and human congenital diseases. Biochem. Biokhimiia 2012, 77, 1500–1514. [Google Scholar] [CrossRef]
- Asthana, A.; Raman, B.; Ramakrishna, T.; Rao Ch, M. Structural aspects and chaperone activity of human hspb3: Role of the “c-terminal extension”. Cell Biochem. Biophys. 2012, 64, 61–72. [Google Scholar] [CrossRef]
- Corrado, L.; Del Bo, R.; Castellotti, B.; Ratti, A.; Cereda, C.; Penco, S.; Soraru, G.; Carlomagno, Y.; Ghezzi, S.; Pensato, V.; et al. Mutations of fus gene in sporadic amyotrophic lateral sclerosis. J. Med. Genet. 2010, 47, 190–194. [Google Scholar] [CrossRef]
- Kwon, M.J.; Baek, W.; Ki, C.S.; Kim, H.Y.; Koh, S.H.; Kim, J.W.; Kim, S.H. Screening of the sod1, fus, tardbp, ang, and optn mutations in korean patients with familial and sporadic als. Neurobiol. Aging 2012, 33, 1017.e17–1017.e23. [Google Scholar] [CrossRef]
- Van Langenhove, T.; van der Zee, J.; Sleegers, K.; Engelborghs, S.; Vandenberghe, R.; Gijselinck, I.; Van den Broeck, M.; Mattheijssens, M.; Peeters, K.; De Deyn, P.P.; et al. Genetic contribution of fus to frontotemporal lobar degeneration. Neurology 2010, 74, 366–371. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Dogu, O.; Yilmaz, A.; Houlden, H.; Singleton, A. Spg11 mutations are common in familial cases of complicated hereditary spastic paraplegia. Neurology 2008, 70, 1384–1389. [Google Scholar] [CrossRef]
- Orlacchio, A.; Babalini, C.; Borreca, A.; Patrono, C.; Massa, R.; Basaran, S.; Munhoz, R.P.; Rogaeva, E.A.; St George-Hyslop, P.H.; Bernardi, G.; et al. Spatacsin mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010, 133, 591–598. [Google Scholar] [CrossRef]
- Pang, S.Y.; Hsu, J.S.; Teo, K.C.; Li, Y.; Kung, M.H.W.; Cheah, K.S.E.; Chan, D.; Cheung, K.M.C.; Li, M.; Sham, P.C.; et al. Burden of rare variants in als genes influences survival in familial and sporadic als. Neurobiol. Aging 2017, 58, 238.e9–238.e15. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Sone, J.; Atsuta, N.; Tohnai, G.; Watanabe, H.; Yokoi, D.; Nakatochi, M.; Watanabe, H.; Ito, M.; Senda, J.; et al. Next-generation sequencing of 28 als-related genes in a japanese als cohort. Neurobiol. Aging 2016, 39, 219.e1–219.e8. [Google Scholar] [CrossRef] [PubMed]
- Naruse, H.; Ishiura, H.; Mitsui, J.; Takahashi, Y.; Matsukawa, T.; Tanaka, M.; Doi, K.; Yoshimura, J.; Morishita, S.; Goto, J.; et al. Burden of rare variants in causative genes for amyotrophic lateral sclerosis (als) accelerates age at onset of als. J. Neurol. Neurosurg. Psychiatry 2019, 90, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Chio, A.; Mazzini, L.; D’Alfonso, S.; Corrado, L.; Canosa, A.; Moglia, C.; Manera, U.; Bersano, E.; Brunetti, M.; Barberis, M.; et al. The multistep hypothesis of als revisited: The role of genetic mutations. Neurology 2018, 91, e635–e642. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.; Shatunov, A.; Sproviero, W.; Jones, A.R.; Shoai, M.; Hughes, D.; Al Khleifat, A.; Malaspina, A.; Morrison, K.E.; Shaw, P.J.; et al. A comprehensive analysis of rare genetic variation in amyotrophic lateral sclerosis in the uk. Brain 2017, 140, 1611–1618. [Google Scholar] [CrossRef]
- Kim, H.J.; Oh, K.W.; Kwon, M.J.; Oh, S.I.; Park, J.S.; Kim, Y.E.; Choi, B.O.; Lee, S.; Ki, C.S.; Kim, S.H. Identification of mutations in korean patients with amyotrophic lateral sclerosis using multigene panel testing. Neurobiol. Aging 2016, 37, 209.e9–209.e16. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Calvo, A.; Moglia, C.; Lunetta, C.; Marinou, K.; Ticozzi, N.; Ferrante, G.D.; Scialo, C.; Soraru, G.; Trojsi, F.; Conte, A.; et al. Factors predicting survival in als: A multicenter italian study. J. Neurol. 2017, 264, 54–63. [Google Scholar] [CrossRef]
- Westeneng, H.J.; Debray, T.P.A.; Visser, A.E.; van Eijk, R.P.A.; Rooney, J.P.K.; Calvo, A.; Martin, S.; McDermott, C.J.; Thompson, A.G.; Pinto, S.; et al. Prognosis for patients with amyotrophic lateral sclerosis: Development and validation of a personalised prediction model. Lancet Neurol. 2018, 17, 423–433. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Robins, H.; Niedermoser, I.; Wyles, M.; Heath, P.R.; Higginbottom, A.; Walsh, T.; Kazoka, M.; Project Min, E.A.L.S.S.C.; Ince, P.G.; et al. Targeted genetic screen in amyotrophic lateral sclerosis reveals novel genetic variants with synergistic effect on clinical phenotype. Front. Mol. Neurosci. 2017, 10, 370. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [PubMed]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L.; World Federation of Neurology Research Group on Motor Neuron, D. El escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Chio, A.; Calvo, A.; Moglia, C.; Mazzini, L.; Mora, G.; PARALS study group. Phenotypic heterogeneity of amyotrophic lateral sclerosis: A population based study. J. Neurol. Neurosurg. Psychiatry 2011, 82, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (igv): High-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Agosta, F.; Ferraro, P.M.; Riva, N.; Spinelli, E.G.; Domi, T.; Carrera, P.; Copetti, M.; Falzone, Y.; Ferrari, M.; Lunetta, C.; et al. Structural and functional brain signatures of c9orf72 in motor neuron disease. Neurobiol. Aging 2017, 57, 206–219. [Google Scholar] [CrossRef]
- Nishiyama, A.; Niihori, T.; Warita, H.; Izumi, R.; Akiyama, T.; Kato, M.; Suzuki, N.; Aoki, Y.; Aoki, M. Comprehensive targeted next-generation sequencing in japanese familial amyotrophic lateral sclerosis. Neurobiol. Aging 2017, 53, 194.e1–194.e8. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the sift algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Sunyaev, S.; Ramensky, V.; Koch, I.; Lathe, W., 3rd; Kondrashov, A.S.; Bork, P. Prediction of deleterious human alleles. Hum. Mol. Genet. 2001, 10, 591–597. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| PT-ID a | Gene Name | cDNA Change | Protein Change | dbSNP ID b | ACMG Classification | Global MAF c | Population MAF d | SIFT Score | Polyphen Score | Mutation Taster Pred | CADD |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ALS literature | |||||||||||
| ALS_78 | ALS2 | c.1115C > G | p.Pro372Arg | rs190369242 | 3 | 0.00130 | 0.00220 | 0.64 (T) | 0.919 (D) | 0.683 (D) | 19.65 |
| ALS_6 | OPTN | c.941A > T | p.Gln314Leu | rs142812715 | 3 | 0.00017 | 0.00030 | 0.01 (D) | 0.999 (D) | 0.993 (D) | 27.70 |
| ALS_56 | SETX | c.654G > C | p.Lys218Asn | rs117861188 | 3 | 0.00033 | 0.00060 | 0.0 (D) | 0.961 (D) | 0.900 (D) | 23.80 |
| ALS_34 | SOD1 | c.203T > C | p.Leu68Pro | CM110553 | 5 | 0.00000 | 0.00000 | 0.21 (T) | 0.001 (B) | 0.99 (N) | 6.15 |
| ALS_6 | SOD1 | c.217G > A | p.Gly73Ser | rs121912455 | 5 | 0.00000 | 0.00000 | 0.0 (D) | 0.970 (D) | 0.999 (D) | 29.30 |
| ALS_23 | TBK1 | c.1190T > C | p.Ile397Thr | rs755069538 | 5 | 0.00010 | 0.00030 | 0.31 (T) | 0.039 (B) | 0.999 (D) | 22.80 |
| ALS and non-ALS literature | |||||||||||
| ALS_5 | SETX | c.59G > A | p.Arg20His | rs79740039 | 3 | 0.00683 | 0.01051 | 0.25 (T) | 0.001 (B) | 0.999 (N) | 3.43 |
| ALS_18 | SETX | c.59G > A | p.Arg20His | ||||||||
| ALS_60 | SETX | c.59G > A | p.Arg20His | ||||||||
| ALS_4 | SPG11 | c.1348A > G | p.Ile450Val | rs3759873 | 1 | 0.01672 | 0.00450 | 0.78 (T) | 0 (B) | 0.994 (D) | 6.31 |
| ALS_30 | SPG11 | c.3037A > G | p.Lys1013Glu | rs111347025 | 2 | 0.00861 | 0.01448 | 0.73 (T) | 0.002 (B) | 0.963 (D) | 22.70 |
| ALS_43 | SPG11 | c.3037A > G | p.Lys1013Glu | ||||||||
| ALS_32 | SPG11 | c.6224A > G | p.Asn2075Ser | rs140824939 | 3 | 0.00310 | 0.00489 | 0.49 (T) | 0 (B) | 0.999 (N) | 0.28 |
| Non-ALS literature | |||||||||||
| ALS_2 | BSCL2 | c.844G > A | p.Ala282Thr(218)§ | rs190842600 | 3 | 0.00022 | 0.00000 | 0.08 (T) | 1 (D) | 0.999 (D) | 25.90 |
| ALS_53 | BSCL2 | c.1033C > T | p.Arg345Trp(281)§ | rs767820877 | 3 | 0.00000 | 0.00000 | 0.02(D) | 0.987 (D) | 0.999 (D) | 21.40 |
| ALS_41 | HSPB3 | c.347G > C | p.Arg116Pro | rs150931007 | 4 | 0.00011 | 0.00010 | 0 (D) | 1 (D) | 0.999 (D) | 29.70 |
| ALS_1 | MFN2 | c.1574A > G | p.Asn525Ser | rs145654854 | 3 | 0.00017 | 0.00021 | 1 (T) | 0 (B) | 0.753 (N) | 19.62 |
| ALS_6 | SETX | c.4612C > T | p.Arg1538Trp | rs147018359 | 3 | 0.00000 | 0.00000 | 0.21 (T) | 0 (B) | 0.999 (N) | 17.31 |
| ALS_29 | SETX | c.4612C > T | p.Arg1538Trp | ||||||||
| ALS_30 | SPG11 | c.5986_5987insT | p.Cys1996Leufs*4 | rs312262775 | 5 | 0.00002 | 0.00000 | - | - | - | - |
| ALS_7 | SQSTM1 | c.352C > T | p.Pro118Ser | rs200152247 | 3 | 0.00006 | 0.00010 | 0.54 (T) | 0.009 (B) | 0.999 (D) | 20.90 |
| ALS_34 | SQSTM1 | c.802C > G | p.Leu268Val | rs753685955 | 3 | 0.00001 | 0.00000 | 1 (T) | 0.004 (B) | 0.824 (N) | 17.73 |
| Variants present only in population databases | |||||||||||
| ALS_64 | BSCL2 | c.689G > A | p.Ser230Asn (166) § | rs778378228 | 4 | 0.00001 | 0.00000 | 0.11 (T) | 0.820 (P) | 0.824 (N) | 23.40 |
| ALS_67 | BSCL2 | c.785C > T | p.Ala262Val(198) § | rs140896339 | 3 | 0.00028 | 0.00000 | 0.50 (T) | 0.085(B) | 0.999 (N) | 18.85 |
| ALS_7 | BSCL2 | c.1057T > A | p.Ser353Thr(189) § | rs769769807 | 3 | 0.00001 | 0.00000 | 0.17 (T) | 0.006 (B) | 0.999 (N) | 6.03 |
| ALS_69 | BSCL2 | c.1282C > T | p.Pro428Ser(364) § | rs369732238 | 3 | 0.00006 | 0.00010 | 0.1 (T) | 0.573 (P) | 0.905 (N) | 18.59 |
| ALS_43 | BSCL2 | c.1300T > C | p.Ser434Pro(370) § | rs199584887 | 3 | 0.00006 | 0.00000 | 0.44 (T) | 0.001 (B) | 0.999 (N) | 1.58 |
| ALS_52 | DCTN1 | c.1486G > C | p.Val496Leu | rs773897036 | 4 | 0.00002 | 0.00000 | 0.06 (T) | 0.984 (D) | 0.999 (D) | 23.50 |
| ALS_13 | DYNC1H1 | c.265G > A | p.Gly89Ser | rs749973847 | 3 | 0.00002 | 0.00000 | 0.74 (T) | 0.019 (B) | 0.999 (D) | 22.80 |
| ALS_40 | DYNC1H1 | c.3748G > A | p.Val1250Met | rs369914512 | 3 | 0.00006 | 0.00010 | 0.07 (T) | 0.978 (D) | 0.999 (D) | 29.20 |
| ALS_41 | DYNC1H1 | c.12213C > T # | p.Ile4071Ile | rs746950373 | 3 | 0.00002 | 0.00000 | 0.48 (T) | - | 1 (D) | 12.50 |
| ALS_33 | HSPB1 | c.403T > G | p.Ser135Ala | rs766728475 | 4 | 0.00002 | 0.00000 | 0.07 (T) | 0.623 (P) | 0.999 (D) | 25.40 |
| ALS_53 | HSPB3 | c.199G > A | p.Gly67Ser | rs35258119 | 3 | 0.00727 | 0.00000 | 0.2 (T) | 0.036 (B) | 0.999 (N) | 17.28 |
| ALS_4 | HSPB3 | c.346C > T | p.Arg116X | rs757339596 | 4 | 0.00003 | 0.00000 | - | - | 0.999 (D) | 11.58 |
| ALS_58 | PLEKHG5 | c.3049G > A | p.Gly1017Arg | rs755699992 | 3 | 0.00000 | 0.00000 | 0.56 (T) | 0.047 (B) | 0.999 (N) | 0.16 |
| ALS_15 | SETX | c.3182C > T | p.Pro1061Leu | rs12352982 | 3 | 0.01604 | 0.00000 | 1 (T) | 0 (B) | 0.999 (N) | 0.90 |
| ALS_46 | SETX | c.7435A > G | p.Ile2479Val | rs536912256 | 3 | 0.00006 | 0.00000 | 0.43 (T) | 0.001 (B) | 0.999 (N) | 0.76 |
| ALS_73 | SPG11 | c.1675T > A | p.Ser559Thr | rs773680273 | 3 | 0.00001 | 0.00000 | 0.2 (T) | 0.990 (D) | 0.591 (D) | 20.80 |
| ALS_27 | SPG11 | c.2764G > A | p.Val922Ile | rs139399250 | 3 | 0,00006 | 0,00010 | 0.43 (T) | 0.002(B) | 0.999 (N) | 2.25 |
| ALS_67 | SPG11 | c.6201A > T # | p.Gly2067Gly | rs764991726 | 3 | 0,00001 | 0,00000 | 1 (T) | - | 0.987 (D) | 7.48 |
| Novel candidate ALS-associated variants | |||||||||||
| ALS_34 | DYNC1H1 | c.4183A > C | p.Lys1395Gln | - | 4 | - | - | 0.12 (T) | 1.000 (D) | 0.999 (D) | 34.00 |
| ALS_22 | DYNC1H1 | c.5303G > T | p.Ser1768Ile | - | 3 | - | - | 0.1 (T) | 0.028 (B) | 0.999 (N) | 15.71 |
| ALS_27 | FIG4 | c.1030C > A | p.Pro344Thr | - | 4 | - | - | 0.37 (T) | 0.875 (P) | 0.999 (D) | 25.40 |
| ALS_21 | FUS | c.1168 + 7A > G # | - | - | 3 | - | - | - | - | - | - |
| ALS_40 | SETX | c.5852A > T | p.His1951Leu | - | 4 | - | - | 0.13 (T) | 0.961 (D) | 0.974 (D) | 25.80 |
| ALS_70 | SPG11 | c.4826delT | p.Met1609Serfs*31 | - | 4 | - | - | - | - | 1 (D) | 8.99 |
| Pt ID a | Family | Variant 1 (ACMG) | Gene Category | Variant 2 (ACMG) | Gene Category | Variant 3 (ACMG) | Gene Category |
|---|---|---|---|---|---|---|---|
| ALS_34 | fALS | SOD1 p.Leu68Pro (5) | ALS | SQSTM1 p.Leu268Val (3) | ALS | DYNC1H1 p.Lys1395Gln (4) | ALS |
| ALS_6 | sALS | SOD1 p.Gly73Ser (5) | ALS | OPTN p.Gln314Leu (3) | ALS | SETX p.Arg1538Trp (3) | ALS |
| ALS_40 | fALS | C9orf72 expansion (5) | ALS | DYNC1H1 p.Val1250Met (3) | ALS | SETX p.His1951Leu (4) | ALS |
| ALS_52 | fALS | C9orf72 expansion (5) | ALS | DCTN1 p.Val496Leu (4) | ALS | ||
| ALS_22 | sALS | C9orf72 expansion (5) | ALS | DYNC1H1 p.Ser1768Ile (3) | ALS | ||
| ALS_27 | sALS | SPG11 p.Val922Ile (3) | ALS | FIG4 p.Pro344Thr (4) | ALS | ||
| ALS_30 | sALS | SPG11 p.Lys1013Glu (2) | ALS | SPG11 p.Cys1996Leufs*4 (5) | ALS | ||
| ALS_33 | sALS | C9orf72 expansion (5) | ALS | HSPB1 p.Ser135Ala (4) | Other/HMN/CMT2 | ||
| ALS_41 | sALS | DYNC1H1 p.Ile4071Ile (3) | ALS | HSPB3 p.Arg116Pro (4) | Other/HMN/CMT2 | ||
| ALS_4 | sALS | SPG11 p.Ile450Val (1) | ALS | HSPB3 p.Arg116X (4) | Other/HMN/CMT2 | ||
| ALS_43 | fALS | SPG11 p.Lys1013Glu (2) | ALS | BSCL2 p.Ser434Pro (3) | Other/HMN/CMT2 | ||
| ALS_67 | sALS | SPG11 p.Gly2067Gly (3) | ALS | BSCL2 p.Ala262Val (3) | Other/HMN/CMT2 | ||
| ALS_7 | sALS | SQSTM1 p.Pro118Ser (3) | ALS | BSCL2 p.Ser353Thr (3) | Other/HMN/CMT2 | ||
| ALS_53 | sALS | BSCL2 p.Arg345Trp (3) | Other/CMT2/dSMA | HSPB3 p.Gly67Ser (3) | Other/CMT2/dSMA |
| Survival | Time to King Stage 4 | |||
|---|---|---|---|---|
| Factor | HR (95% CI) | p Value | HR (95% CI) | p Value |
| Rare variants (n) | <0.001 | 0.023 | ||
| 0 | 1 | 1 | ||
| 1 | 1.97 (0.98–3.97) | 0.220 | 1.10 (0.56–2.19) | 0.777 |
| ≥2 | 5.61 (2.38–13.22) | <0.001 | 3.09 (1.37–6.97) | 0.007 |
| Site of symptoms onset | ||||
| Spinal | 1 | 0.013 | 1 | 0.042 |
| Bulbar | 2.64 (1.23–5.70) | 2.32 (1.03–5.19) | ||
| ALSFRS-R decline (points/month) | ||||
| ≤0.60 | 1 | 0.003 | 1 | 0.036 |
| >0.60 | 4.38 (1.64–11.69) | 2.83 (1.07–7.49) | ||
| Diagnostic delay (months) | ||||
| ≤10 | ns | ns | ns | Ns |
| >10 | ||||
| Dementia | ||||
| No | ns | ns | ns | Ns |
| Yes | ||||
| Age at the onset (years) | ||||
| <60 | ns | ns | 1 | 0.016 |
| >60 | 2.17 (1.15–4.08) | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarlino, S.; Domi, T.; Pozzi, L.; Romano, A.; Pipitone, G.B.; Falzone, Y.M.; Mosca, L.; Penco, S.; Lunetta, C.; Sansone, V.; et al. Burden of Rare Variants in ALS and Axonal Hereditary Neuropathy Genes Influence Survival in ALS: Insights from a Next Generation Sequencing Study of an Italian ALS Cohort. Int. J. Mol. Sci. 2020, 21, 3346. https://doi.org/10.3390/ijms21093346
Scarlino S, Domi T, Pozzi L, Romano A, Pipitone GB, Falzone YM, Mosca L, Penco S, Lunetta C, Sansone V, et al. Burden of Rare Variants in ALS and Axonal Hereditary Neuropathy Genes Influence Survival in ALS: Insights from a Next Generation Sequencing Study of an Italian ALS Cohort. International Journal of Molecular Sciences. 2020; 21(9):3346. https://doi.org/10.3390/ijms21093346
Chicago/Turabian StyleScarlino, Stefania, Teuta Domi, Laura Pozzi, Alessandro Romano, Giovanni Battista Pipitone, Yuri Matteo Falzone, Lorena Mosca, Silvana Penco, Christian Lunetta, Valeria Sansone, and et al. 2020. "Burden of Rare Variants in ALS and Axonal Hereditary Neuropathy Genes Influence Survival in ALS: Insights from a Next Generation Sequencing Study of an Italian ALS Cohort" International Journal of Molecular Sciences 21, no. 9: 3346. https://doi.org/10.3390/ijms21093346
APA StyleScarlino, S., Domi, T., Pozzi, L., Romano, A., Pipitone, G. B., Falzone, Y. M., Mosca, L., Penco, S., Lunetta, C., Sansone, V., Tremolizzo, L., Fazio, R., Agosta, F., Filippi, M., Carrera, P., Riva, N., & Quattrini, A. (2020). Burden of Rare Variants in ALS and Axonal Hereditary Neuropathy Genes Influence Survival in ALS: Insights from a Next Generation Sequencing Study of an Italian ALS Cohort. International Journal of Molecular Sciences, 21(9), 3346. https://doi.org/10.3390/ijms21093346