Multiple Roles of Transforming Growth Factor Beta in Amyotrophic Lateral Sclerosis

 ,
, 
 ,
,  ,
, 

Abstract
1. Introduction
2. Amyotrophic Lateral Sclerosis as a Non-Cell-Autonomous Disease
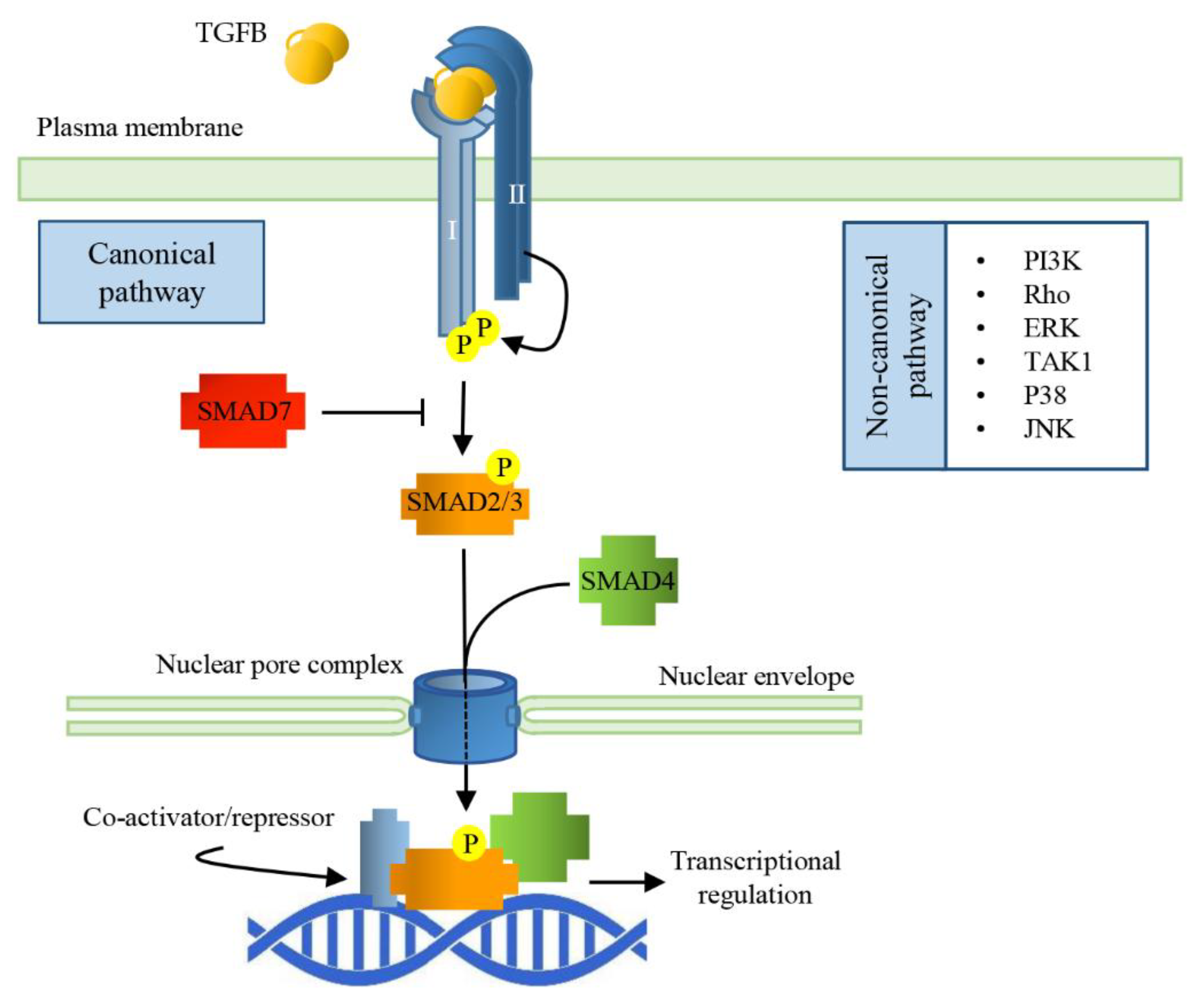
3. Transforming Growth Factor Beta
4. TGFB Plasma Levels in ALS Patients
5. TGFB and ALS-Nervous System
6. TGFB Pathway in ALS Skeletal Muscle
7. TGFB and NMJ in ALS
8. TGFB: A Target for ALS Treatment
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| SOD1 | superoxide dismutase type 1 |
| C9orf72 | chromosome 9 open reading frame 72 |
| TDP-43 | TAR DNA-binding protein 43 |
| CNS | central nervous system |
| PNS | peripheral nervous system |
| TGFB | transforming growth factor beta |
| TGFBRI | TGFB type I receptor |
| TGFBRII | TGFB type II receptor |
| SMAD | small mother against decapentaplegic |
| SBE | SMAD binding element |
References
- Marin, B.; Boumediene, F.; Logroscino, G.; Couratier, P.; Babron, M.C.; Leutenegger, A.L.; Copetti, M.; Preux, P.M.; Beghi, E. Variation in worldwide incidence of amyotrophic lateral sclerosis: A meta-analysis. Int. J. Epidemiol. 2017, 46, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Vegeto, E.; Villa, A.; Della Torre, S.; Crippa, V.; Rusmini, P.; Cristofani, R.; Galbiati, M.; Maggi, A.; Poletti, A. The Role of sex and sex hormones in neurodegenerative diseases. Endocr. Rev. 2020, 41, 273–319. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.L.; Dejesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W., III; Rademakers, R.; et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Lashley, T.; Hardy, J.; Isaacs, A.M. RANTing about C9orf72. Neuron 2013, 77, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Boillee, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillee, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef]
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38. [Google Scholar] [CrossRef]
- Yamanaka, K.; Yamashita, H. [ALS and microglia--a player for non-cell-autonomous neuron death]. Brain Nerve = Shinkei Kenkyu Shinpo 2007, 59, 1163–1170. [Google Scholar]
- Endo, F.; Komine, O.; Fujimori-Tonou, N.; Katsuno, M.; Jin, S.; Watanabe, S.; Sobue, G.; Dezawa, M.; Wyss-Coray, T.; Yamanaka, K. Astrocyte-derived TGF-beta1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 2015, 11, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Higginbottom, A.; Heath, P.R.; Barber, S.; Greenald, D.; Kirby, J.; Shaw, P.J. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 2011, 134, 2627–2641. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, L.T.; Trotti, D. EAAT2 and the molecular signature of amyotrophic lateral sclerosis. Adv. Neurobiol. 2017, 16, 117–136. [Google Scholar] [PubMed]
- Alexianu, M.E.; Kozovska, M.; Appel, S.H. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 2001, 57, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hao, W.; Dawson, A.; Liu, S.; Fassbender, K. Expression of amyotrophic lateral sclerosis-linked SOD1 mutant increases the neurotoxic potential of microglia via TLR2. J. Biol. Chem. 2009, 284, 3691–3699. [Google Scholar] [CrossRef]
- Boillee, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Gerber, Y.N.; Sabourin, J.C.; Rabano, M.; Vivanco, M.; Perrin, F.E. Early functional deficit and microglial disturbances in a mouse model of amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e36000. [Google Scholar] [CrossRef]
- Nonneman, A.; Robberecht, W.; Van Den Bosch, L. The role of oligodendroglial dysfunction in amyotrophic lateral sclerosis. Neurodegener. Dis. Manag. 2014, 4, 223–239. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Meyer, K.; Sherwood, T.W.; Vick, J.; Likhite, S.; Frakes, A.; Miranda, C.J.; Braun, L.; Heath, P.R.; Pineda, R.; et al. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, E6496–E6505. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Boncompagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Giacinti, C.; Pelosi, L.; Nicoletti, C.; Winn, N.; Barberi, L.; Molinaro, M.; Rosenthal, N.; Musaro, A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 2005, 168, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet 2010, 19, 2284–2302. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Manzano, R.; Toivonen, J.M.; Olivan, S.; Calvo, A.C.; Moreno-Igoa, M.; Munoz, M.J.; Zaragoza, P.; Garcia-Redondo, A.; Osta, R. Altered expression of myogenic regulatory factors in the mouse model of amyotrophic lateral sclerosis. Neurodegener. Dis. 2011, 8, 386–396. [Google Scholar] [CrossRef]
- Pradat, P.F.; Barani, A.; Wanschitz, J.; Dubourg, O.; Lombes, A.; Bigot, A.; Mouly, V.; Bruneteau, G.; Salachas, F.; Lenglet, T.; et al. Abnormalities of satellite cells function in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2011, 12, 264–271. [Google Scholar] [CrossRef]
- Crippa, V.; Boncoraglio, A.; Galbiati, M.; Aggarwal, T.; Rusmini, P.; Giorgetti, E.; Cristofani, R.; Carra, S.; Pennuto, M.; Poletti, A. Differential autophagy power in the spinal cord and muscle of transgenic ALS mice. Front. Cell. Neurosci. 2013, 7, 234. [Google Scholar] [CrossRef]
- Galbiati, M.; Onesto, E.; Zito, A.; Crippa, V.; Rusmini, P.; Mariotti, R.; Bentivoglio, M.; Bendotti, C.; Poletti, A. The anabolic/androgenic steroid nandrolone exacerbates gene expression modifications induced by mutant SOD1 in muscles of mice models of amyotrophic lateral sclerosis. Pharmacol. Res. 2012, 65, 221–230. [Google Scholar] [CrossRef]
- Meroni, M.; Crippa, V.; Cristofani, R.; Rusmini, P.; Cicardi, M.E.; Messi, E.; Piccolella, M.; Tedesco, B.; Ferrari, V.; Soraru, G.; et al. Transforming growth factor beta 1 signaling is altered in the spinal cord and muscle of amyotrophic lateral sclerosis mice and patients. Neurobiol. Aging 2019, 82, 48–59. [Google Scholar] [CrossRef]
- Hernandez Lain, A.; Millecamps, S.; Dubourg, O.; Salachas, F.; Bruneteau, G.; Lacomblez, L.; LeGuern, E.; Seilhean, D.; Duyckaerts, C.; Meininger, V.; et al. Abnormal TDP-43 and FUS proteins in muscles of sporadic IBM: Similarities in a TARDBP-linked ALS patient. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1414–1416. [Google Scholar] [CrossRef]
- Soraru, G.; Orsetti, V.; Buratti, E.; Baralle, F.; Cima, V.; Volpe, M.; D’Ascenzo, C.; Palmieri, A.; Koutsikos, K.; Pegoraro, E.; et al. TDP-43 in skeletal muscle of patients affected with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 11, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Cykowski, M.D.; Dickson, D.W.; Powell, S.Z.; Arumanayagam, A.S.; Rivera, A.L.; Appel, S.H. Dipeptide repeat (DPR) pathology in the skeletal muscle of ALS patients with C9ORF72 repeat expansion. Acta Neuropathol. 2019, 138, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Cicardi, M.E.; Cristofani, R.; Rusmini, P.; Meroni, M.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Piccolella, M.; Messi, E.; Galbiati, M.; et al. Tdp-25 Routing to autophagy and proteasome ameliorates its aggregation in amyotrophic lateral sclerosis target cells. Sci. Rep. 2018, 8, 12390. [Google Scholar] [CrossRef]
- Crippa, V.; Galbiati, M.; Boncoraglio, A.; Rusmini, P.; Onesto, E.; Giorgetti, E.; Cristofani, R.; Zito, A.; Poletti, A. Motoneuronal and muscle-selective removal of ALS-related misfolded proteins. Biochem. Soc. Trans. 2013, 41, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
- Onesto, E.; Rusmini, P.; Crippa, V.; Ferri, N.; Zito, A.; Galbiati, M.; Poletti, A. Muscle cells and motoneurons differentially remove mutant SOD1 causing familial amyotrophic lateral sclerosis. J. Neurochem. 2011, 118, 266–280. [Google Scholar] [CrossRef]
- Massague, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef]
- Mu, Y.; Gudey, S.K.; Landstrom, M. Non-Smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef]
- Weiss, A.; Attisano, L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef]
- Izzi, L.; Attisano, L. Regulation of the TGFbeta signalling pathway by ubiquitin-mediated degradation. Oncogene 2004, 23, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad signaling pathways of the TGF-beta family. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Abrigo, J.; Campos, F.; Simon, F.; Riedel, C.; Cabrera, D.; Vilos, C.; Cabello-Verrugio, C. TGF-beta requires the activation of canonical and non-canonical signalling pathways to induce skeletal muscle atrophy. Biol. Chem. 2018, 399, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Ala, T.A.; Hu, S.; Crossley, K.B.; Sherman, R.E.; Peterson, P.K.; Frey, W.H. 2nd, Serum cytokine levels in patients with Alzheimer’s disease. Clin. Diagn. Lab. Immunol. 1994, 1, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Kondo, T.; Narabayashi, H.; Riederer, P.; Nagatsu, T. Transforming growth factor-beta 1 levels are elevated in the striatum and in ventricular cerebrospinal fluid in Parkinson’s disease. Neurosci. Lett. 1995, 193, 129–132. [Google Scholar] [CrossRef]
- Houi, K.; Kobayashi, T.; Kato, S.; Mochio, S.; Inoue, K. Increased plasma TGF-beta1 in patients with amyotrophic lateral sclerosis. Acta Neurol. Scand. 2002, 106, 299–301. [Google Scholar] [CrossRef]
- Ilzecka, J.; Stelmasiak, Z.; Dobosz, B. Transforming growth factor-Beta 1 (tgf-Beta 1) in patients with amyotrophic lateral sclerosis. Cytokine 2002, 20, 239–243. [Google Scholar] [CrossRef]
- Duque, T.; Gromicho, M.; Pronto-Laborinho, A.C.; de Carvalho, M. Transforming growth factor-beta plasma levels and its role in amyotrophic lateral sclerosis. Med. Hypotheses 2020, 139, 109632. [Google Scholar] [CrossRef]
- Zubiri, I.; Lombardi, V.; Bremang, M.; Mitra, V.; Nardo, G.; Adiutori, R.; Lu, C.H.; Leoni, E.; Yip, P.; Yildiz, O.; et al. Tissue-enhanced plasma proteomic analysis for disease stratification in amyotrophic lateral sclerosis. Mol. Neurodegener. 2018, 13, 60. [Google Scholar] [CrossRef]
- Masuda, T.; Itoh, J.; Koide, T.; Tomidokoro, Y.; Takei, Y.; Ishii, K.; Tamaoka, A. Transforming growth factor-beta1 in the cerebrospinal fluid of patients with distinct neurodegenerative diseases. J. Clin. Neurosci. 2017, 35, 47–49. [Google Scholar] [CrossRef][Green Version]
- Iida, A.; Takahashi, A.; Kubo, M.; Saito, S.; Hosono, N.; Ohnishi, Y.; Kiyotani, K.; Mushiroda, T.; Nakajima, M.; Ozaki, K.; et al. A functional variant in ZNF512B is associated with susceptibility to amyotrophic lateral sclerosis in Japanese. Hum. Mol. Genet 2011, 20, 3684–3692. [Google Scholar] [CrossRef] [PubMed]
- Tetsuka, S.; Morita, M.; Iida, A.; Uehara, R.; Ikegawa, S.; Nakano, I. ZNF512B gene is a prognostic factor in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2013, 324, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Komuta, Y.; Teng, X.; Yanagisawa, H.; Sango, K.; Kawamura, K.; Kawano, H. Expression of transforming growth factor-beta receptors in meningeal fibroblasts of the injured mouse brain. Cell. Mol. Neurobiol. 2010, 30, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Unsicker, K.; Flanders, K.C.; Cissel, D.S.; Lafyatis, R.; Sporn, M.B. Transforming growth factor beta isoforms in the adult rat central and peripheral nervous system. Neuroscience 1991, 44, 613–625. [Google Scholar] [CrossRef]
- Vincze, C.; Pal, G.; Wappler, E.A.; Szabo, E.R.; Nagy, Z.G.; Lovas, G.; Dobolyi, A. Distribution of mRNAs encoding transforming growth factors-beta1, -2, and -3 in the intact rat brain and after experimentally induced focal ischemia. J. Comp. Neurol. 2010, 518, 3752–3770. [Google Scholar] [CrossRef]
- Bottner, M.; Krieglstein, K.; Unsicker, K. The transforming growth factor-betas: Structure, signaling, and roles in nervous system development and functions. J. Neurochem. 2000, 75, 2227–2240. [Google Scholar] [CrossRef]
- Diniz, L.P.; Matias, I.; Siqueira, M.; Stipursky, J.; Gomes, F.C.A. Astrocytes and the TGF-beta1 Pathway in the Healthy and Diseased Brain: A Double-Edged Sword. Mol. Neurobiol. 2019, 56, 4653–4679. [Google Scholar] [CrossRef]
- Fukushima, T.; Liu, R.Y.; Byrne, J.H. Transforming growth factor-beta2 modulates synaptic efficacy and plasticity and induces phosphorylation of CREB in hippocampal neurons. Hippocampus 2007, 17, 5–9. [Google Scholar] [CrossRef]
- Poon, V.Y.; Choi, S.; Park, M. Growth factors in synaptic function. Front. Synaptic Neurosci. 2013, 5, 6. [Google Scholar] [CrossRef]
- Miller, M.W. Expression of transforming growth factor-beta in developing rat cerebral cortex: Effects of prenatal exposure to ethanol. J. Comp. Neurol. 2003, 460, 410–424. [Google Scholar] [CrossRef]
- Siqueira, M.; Francis, D.; Gisbert, D.; Gomes, F.C.A.; Stipursky, J. Radial glia cells control angiogenesis in the developing cerebral cortex through TGF-beta1 signaling. Mol. Neurobiol. 2018, 55, 3660–3675. [Google Scholar] [PubMed]
- Koeglsperger, T.; Li, S.; Brenneis, C.; Saulnier, J.L.; Mayo, L.; Carrier, Y.; Selkoe, D.J.; Weiner, H.L. Impaired glutamate recycling and GluN2B-mediated neuronal calcium overload in mice lacking TGF-beta1 in the CNS. Glia 2013, 61, 985–1002. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; McLennan, I.S.; Koishi, K.; Hendry, I.A. Transforming growth factor-beta 2 is anterogradely and retrogradely transported in motoneurons and up-regulated after nerve injury. Neuroscience 2000, 97, 735–742. [Google Scholar] [CrossRef]
- Oppenheim, R.W.; Prevette, D.; Haverkamp, L.J.; Houenou, L.; Yin, Q.W.; McManaman, J. Biological studies of a putative avian muscle-derived neurotrophic factor that prevents naturally occurring motoneuron death In Vivo. J. Neurobiol. 1993, 24, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Prehn, J.H.; Peruche, B.; Unsicker, K.; Krieglstein, J. Isoform-specific effects of transforming growth factors-beta on degeneration of primary neuronal cultures induced by cytotoxic hypoxia or glutamate. J. Neurochem. 1993, 60, 1665–1672. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Shiojima, T.; Tagaya, N.; Kobayashi, T.; Kinoshita, M. Effect of transforming growth factor beta 1 on spinal motor neurons after axotomy. J. Neurol. Sci. 1997, 147, 9–12. [Google Scholar] [CrossRef]
- Murakami, N.; McLennan, I.S.; Nonaka, I.; Koishi, K.; Baker, C.; Hammond-Tooke, G. Transforming growth factor-beta2 is elevated in skeletal muscle disorders. Muscle Nerve 1999, 22, 889–898. [Google Scholar] [CrossRef]
- Wong, C.O.; Venkatachalam, K. Motor neurons from ALS patients with mutations in C9ORF72 and SOD1 exhibit distinct transcriptional landscapes. Hum. Mol. Genet 2019, 28, 2799–2810. [Google Scholar] [CrossRef]
- Nakamura, M.; Ito, H.; Wate, R.; Nakano, S.; Hirano, A.; Kusaka, H. Phosphorylated Smad2/3 immunoreactivity in sporadic and familial amyotrophic lateral sclerosis and its mouse model. Acta Neuropathol. 2008, 115, 327–334. [Google Scholar] [CrossRef]
- Nakamura, M.; Kaneko, S.; Wate, R.; Asayama, S.; Nakamura, Y.; Fujita, K.; Ito, H.; Kusaka, H. Regionally different immunoreactivity for Smurf2 and pSmad2/3 in TDP-43-positive inclusions of amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2013, 39, 144–156. [Google Scholar] [CrossRef]
- Grunseich, C.; Wang, I.X.; Watts, J.A.; Burdick, J.T.; Guber, R.D.; Zhu, Z.; Bruzel, A.; Lanman, T.; Chen, K.; Schindler, A.B.; et al. Senataxin mutation reveals how R-Loops Promote transcription by blocking DNA methylation at gene promoters. Mol. Cell 2018, 69, 426–437 e7. [Google Scholar] [CrossRef] [PubMed]
- Henrich-Noack, P.; Prehn, J.H.; Krieglstein, J. Neuroprotective effects of TGF-beta 1. J. Neural Transm. Suppl. 1994, 43, 33–45. [Google Scholar] [PubMed]
- Phatnani, H.P.; Guarnieri, P.; Friedman, B.A.; Carrasco, M.A.; Muratet, M.; O’Keeffe, S.; Nwakeze, C.; Pauli-Behn, F.; Newberry, K.M.; Meadows, S.K.; et al. Intricate interplay between astrocytes and motor neurons in ALS. Proc. Natl. Acad. Sci. USA 2013, 110, E756–E765. [Google Scholar] [CrossRef] [PubMed]
- D’Arrigo, A.; Colavito, D.; Pena-Altamira, E.; Fabris, M.; Dam, M.; Contestabile, A.; Leon, A. Transcriptional profiling in the lumbar spinal cord of a mouse model of amyotrophic lateral sclerosis: A role for wild-type superoxide dismutase 1 in sporadic disease? J. Mol. Neurosci. 2010, 41, 404–415. [Google Scholar] [CrossRef]
- Kirby, J.; Ning, K.; Ferraiuolo, L.; Heath, P.R.; Ismail, A.; Kuo, S.W.; Valori, C.F.; Cox, L.; Sharrack, B.; Wharton, S.B.; et al. Phosphatase and tensin homologue/protein kinase B pathway linked to motor neuron survival in human superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 2011, 134, 506–517. [Google Scholar] [CrossRef]
- Zhang, J.; Ito, H.; Wate, R.; Ohnishi, S.; Nakano, S.; Kusaka, H. Altered distributions of nucleocytoplasmic transport-related proteins in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. 2006, 112, 673–680. [Google Scholar] [CrossRef]
- Jiang, Y.M.; Yamamoto, M.; Kobayashi, Y.; Yoshihara, T.; Liang, Y.; Terao, S.; Takeuchi, H.; Ishigaki, S.; Katsuno, M.; Adachi, H.; et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 2005, 57, 236–251. [Google Scholar] [CrossRef]
- Kim, M.S.; Hwang, N.S.; Lee, J.; Kim, T.K.; Leong, K.; Shamblott, M.J.; Gearhart, J.; Elisseeff, J. Musculoskeletal differentiation of cells derived from human embryonic germ cells. Stem Cells 2005, 23, 113–123. [Google Scholar] [CrossRef]
- Gumucio, J.P.; Sugg, K.B.; Mendias, C.L. TGF-beta superfamily signaling in muscle and tendon adaptation to resistance exercise. Exerc. Sport Sci. Rev. 2015, 43, 93–99. [Google Scholar] [CrossRef]
- Liu, D.; Black, B.L.; Derynck, R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 2001, 15, 2950–2966. [Google Scholar] [CrossRef]
- Mendias, C.L.; Gumucio, J.P.; Davis, M.E.; Bromley, C.W.; Davis, C.S.; Brooks, S.V. Transforming growth factor-beta induces skeletal muscle atrophy and fibrosis through the induction of atrogin-1 and scleraxis. Muscle Nerve 2012, 45, 55–59. [Google Scholar] [CrossRef]
- Narola, J.; Pandey, S.N.; Glick, A.; Chen, Y.W. Conditional expression of TGF-beta1 in skeletal muscles causes endomysial fibrosis and myofibers atrophy. PLoS ONE 2013, 8, e79356. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Colgan, T.D.; Walton, K.L.; Gregorevic, P.; Harrison, C.A. The TGF-beta signalling network in muscle development, adaptation and disease. Adv. Exp. Med. Biol. 2016, 900, 97–131. [Google Scholar] [PubMed]
- Katsuno, M.; Adachi, H.; Minamiyama, M.; Waza, M.; Doi, H.; Kondo, N.; Mizoguchi, H.; Nitta, A.; Yamada, K.; Banno, H.; et al. Disrupted transforming growth Factor-{beta} signaling in spinal and bulbar muscular atrophy. J. Neurosci. 2010, 30, 5702–5712. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Polanco, M.J.; Cristofani, R.; Cicardi, M.E.; Meroni, M.; Galbiati, M.; Piccolella, M.; Messi, E.; Giorgetti, E.; Lieberman, A.P.; et al. Aberrant autophagic response in the muscle of a knock-in mouse model of spinal and bulbar muscular atrophy. Sci. Rep. 2015, 5, 15174. [Google Scholar] [CrossRef]
- Burks, T.N.; Cohn, R.D. Role of TGF-beta signaling in inherited and acquired myopathies. Skelet. Muscle 2011, 1, 19. [Google Scholar] [CrossRef]
- Cohn, R.D.; van Erp, C.; Habashi, J.P.; Soleimani, A.A.; Klein, E.C.; Lisi, M.T.; Gamradt, M.; Ap Rhys, C.M.; Holm, T.M.; Loeys, B.L.; et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 2007, 13, 204–210. [Google Scholar] [CrossRef]
- Aggarwal, T.; Polanco, M.J.; Scaramuzzino, C.; Rocchi, A.; Milioto, C.; Emionite, L.; Ognio, E.; Sambataro, F.; Galbiati, M.; Poletti, A.; et al. Androgens affect muscle, motor neuron, and survival in a mouse model of SOD1-related amyotrophic lateral sclerosis. Neurobiol. Aging 2014, 35, 1929–1938. [Google Scholar] [CrossRef]
- Pradat, P.F.; Dubourg, O.; de Tapia, M.; di Scala, F.; Dupuis, L.; Lenglet, T.; Bruneteau, G.; Salachas, F.; Lacomblez, L.; Corvol, J.C.; et al. Muscle gene expression is a marker of amyotrophic lateral sclerosis severity. Neurodegener. Dis. 2012, 9, 38–52. [Google Scholar] [CrossRef]
- Si, Y.; Kim, S.; Cui, X.; Zheng, L.; Oh, S.J.; Anderson, T.; AlSharabati, M.; Kazamel, M.; Volpicelli-Daley, L.; Bamman, M.M.; et al. Transforming growth factor beta (TGF-beta) is a muscle biomarker of disease progression in ALS and correlates with smad expression. PLoS ONE 2015, 10, e0138425. [Google Scholar] [CrossRef]
- Si, Y.; Cui, X.; Kim, S.; Wians, R.; Sorge, R.; Oh, S.J.; Kwan, T.; AlSharabati, M.; Lu, L.; Claussen, G.; et al. Smads as muscle biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2014, 1, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Zitzelsperger, E.; Kuespert, S.; Iberl, S.; Heydn, R.; Johannesen, S.; Petri, S.; Aigner, L.; Thal, D.R.; Hermann, A.; et al. The TGF-beta System As a Potential Pathogenic Player in Disease Modulation of Amyotrophic Lateral Sclerosis. Front. Neurol. 2017, 8, 669. [Google Scholar] [CrossRef] [PubMed]
- Saris, C.G.; Groen, E.J.; van Vught, P.W.; van Es, M.A.; Blauw, H.M.; Veldink, J.H.; van den Berg, L.H. Gene expression profile of SOD1-G93A mouse spinal cord, blood and muscle. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Ferri, A.; Carri, M.T. Amyotrophic lateral sclerosis: From current developments in the laboratory to clinical implications. Antioxid Redox Signal 2008, 10, 405–443. [Google Scholar] [CrossRef]
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I.; et al. Mutation of SOD1 in ALS: A gain of a loss of function. Hum. Mol. Genet 2007, 16, 1604–1618. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar]
- Gonzalez, D.; Contreras, O.; Rebolledo, D.L.; Espinoza, J.P.; van Zundert, B.; Brandan, E. ALS skeletal muscle shows enhanced TGF-beta signaling, fibrosis and induction of fibro/adipogenic progenitor markers. PLoS ONE 2017, 12, e0177649. [Google Scholar] [CrossRef]
- Tsujihata, M.; Hazama, R.; Yoshimura, T.; Satoh, A.; Mori, M.; Nagataki, S. The motor end-plate fine structure and ultrastructural localization of acetylcholine receptors in amyotrophic lateral sclerosis. Muscle Nerve 1984, 7, 243–249. [Google Scholar] [CrossRef]
- Moloney, E.B.; de Winter, F.; Verhaagen, J. ALS as a distal axonopathy: Molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 2014, 8, 252. [Google Scholar] [CrossRef]
- Cappello, V.; Francolini, M. Neuromuscular junction dismantling in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2017, 18, 2092. [Google Scholar] [CrossRef]
- Feng, Z.; Ko, C.P. Schwann cells promote synaptogenesis at the neuromuscular junction via transforming growth factor-beta1. J. Neurosci. 2008, 28, 9599–9609. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.I.; Seburn, K.L.; Pinter, M.J. Altered terminal Schwann cell morphology precedes denervation in SOD1 mice. Exp. Neurol. 2016, 275, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Taetzsch, T.; Tenga, M.J.; Valdez, G. Muscle fibers secrete FGFBP1 to slow degeneration of neuromuscular synapses during aging and progression of ALS. J. Neurosci. 2017, 37, 70–82. [Google Scholar] [CrossRef]
- Pennetta, G.; Hiesinger, P.R.; Fabian-Fine, R.; Meinertzhagen, I.A.; Bellen, H.J. Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron 2002, 35, 291–306. [Google Scholar] [CrossRef]
- Ratnaparkhi, A.; Lawless, G.M.; Schweizer, F.E.; Golshani, P.; Jackson, G.R. A Drosophila model of ALS: Human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS ONE 2008, 3, e2334. [Google Scholar] [CrossRef]
- Day, W.A.; Koishi, K.; Nukuda, H.; McLennan, I.S. Transforming growth factor-beta 2 causes an acute improvement in the motor performance of transgenic ALS mice. Neurobiol. Dis. 2005, 19, 323–330. [Google Scholar] [CrossRef]
- Holzbaur, E.L.; Howland, D.S.; Weber, N.; Wallace, K.; She, Y.; Kwak, S.; Tchistiakova, L.A.; Murphy, E.; Hinson, J.; Karim, R.; et al. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 23, 697–707. [Google Scholar] [CrossRef]
- Morrison, B.M.; Lachey, J.L.; Warsing, L.C.; Ting, B.L.; Pullen, A.E.; Underwood, K.W.; Kumar, R.; Sako, D.; Grinberg, A.; Wong, V.; et al. A soluble activin type IIB receptor improves function in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2009, 217, 258–268. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| TGFB Super Family | Family | Family Members | Type I Receptor | Type II Receptor | R-SMAD | I-SMAD |
| TGFB | TGFB 1–5 | TGFBR1 | TGFBR2 | SMAD2/3 | SMAD7 | |
| ACTIVINS/INHIBIN | ACTIVIN A, B | ACVR1B, ACVR1C | ACVR2, ACVR2B | SMAD2/3 | SMAD7 | |
| INHIBIN A, B | / | ACVR2 | / | / | ||
| LEFTTY A, B | / | / | / | / | ||
| NODAL | / | ACVR2, ACVR2B | SMAD2/3 | SMAD6/7 | ||
| BMP | BMP 2, 4 | BMPR1A, BMPR1B | ACVR2, ACVR2B, BMPR2 | SMAD1/5 | SMAD6/7 | |
| BMP 3 | / | ACVR2B | / | SMAD6/7 | ||
| BMP 5–8 | ACVR1A, BMPR1A, BMPR1B | ACVR2, ACVR2B, BMPR2 | SMAD1/5 | SMAD6/7 | ||
| BMP 9, 10 | ALK1 | ACVR2, BMPR2 | SMAD1/5 | SMAD6/7 | ||
| BMP 15 | BMPR1B | BMPR2 | SMAD1/5 | SMAD6/7 | ||
| AMH | ACVR1A, BMPR1A | AMHR2 | SMAD1/5 | SMAD6/7 | ||
| GDF | GDF 1, 3 | ACVR1B, ACVR1C | ACVR2, ACVR2B | SMAD2/3 | SMAD7 | |
| GDF 8 (MYOSTATIN) | ACVR1B, TGFBR1 | ACVR2 | SMAD2/3 | SMAD7 | ||
| GDF 9 | ACVR1B | BMPR2 | SMAD2/3 | SMAD7 | ||
| GDF 11 | ACVR1B | ACVR2, ACVR2B | SMAD2/3 | SMAD7 | ||
| GDF 5–7 | BMPR1A, BMPR1B | ACVR2, ACVR2B, BMPR2 | SMAD1/5 | SMAD7 | ||
| GDF 15 | GFRAL | / | / | / |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galbiati, M.; Crippa, V.; Rusmini, P.; Cristofani, R.; Messi, E.; Piccolella, M.; Tedesco, B.; Ferrari, V.; Casarotto, E.; Chierichetti, M.; et al. Multiple Roles of Transforming Growth Factor Beta in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 4291. https://doi.org/10.3390/ijms21124291
Galbiati M, Crippa V, Rusmini P, Cristofani R, Messi E, Piccolella M, Tedesco B, Ferrari V, Casarotto E, Chierichetti M, et al. Multiple Roles of Transforming Growth Factor Beta in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2020; 21(12):4291. https://doi.org/10.3390/ijms21124291
Chicago/Turabian StyleGalbiati, Mariarita, Valeria Crippa, Paola Rusmini, Riccardo Cristofani, Elio Messi, Margherita Piccolella, Barbara Tedesco, Veronica Ferrari, Elena Casarotto, Marta Chierichetti, and et al. 2020. "Multiple Roles of Transforming Growth Factor Beta in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 21, no. 12: 4291. https://doi.org/10.3390/ijms21124291
APA StyleGalbiati, M., Crippa, V., Rusmini, P., Cristofani, R., Messi, E., Piccolella, M., Tedesco, B., Ferrari, V., Casarotto, E., Chierichetti, M., & Poletti, A. (2020). Multiple Roles of Transforming Growth Factor Beta in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 21(12), 4291. https://doi.org/10.3390/ijms21124291