Whole-Genome Deep Sequencing Reveals Host-Driven in-planta Evolution of Columnea Latent Viroid (CLVd) Quasi-Species Populations

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
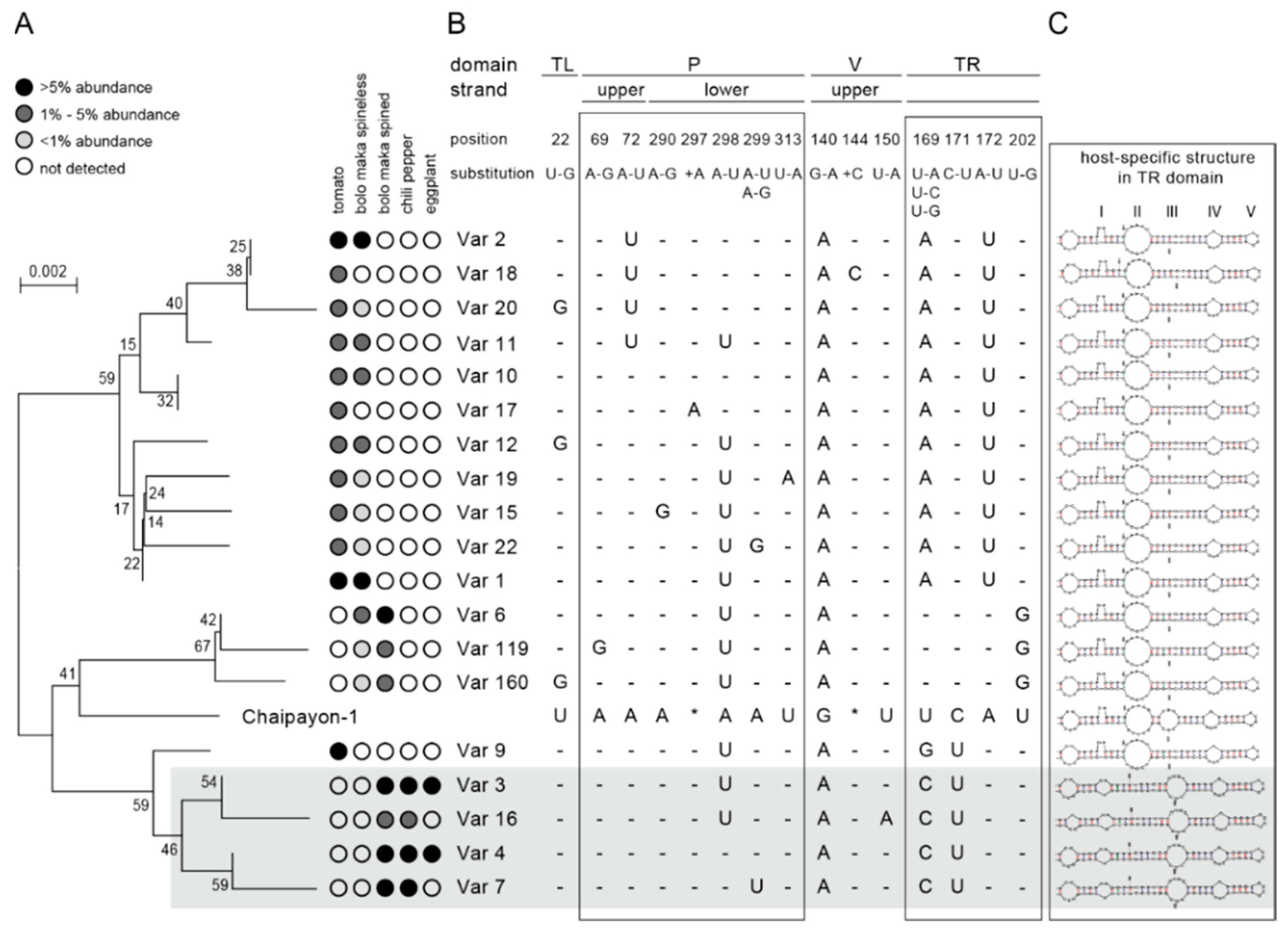
2. Results
2.1. Infectivity of a dsDNA CLVd-Chaipayon-1 Founder Isolate
2.2. Whole-Genome Deep Amplicon Sequencing of Quasi-Species Populations
2.3. Identification of True Variants and Analysis of Variant Frequency Distribution
2.3.1. Reproducibility and Quasi-Species Composition
2.3.2. Variant Distribution
2.3.3. Phylogenetic Analysis
2.4. Characteristics of the CLVd Progeny Variants
2.4.1. Detailed Description of the Major SNPs and Their Effect on the Secondary Structure
TL Domain
P Domain
V Domain
TR Domain
2.4.2. Secondary Structure Prediction of Major Variants
3. Discussion
3.1. Infectivity of Infectious dsDNA CLVd Clones
3.2. Mutation Distribution in CLVd Chaipayon-1 Progeny Population in Different Host Species
3.3. Host-Driven Adaptation
4. Materials and Methods
4.1. Preparation of Infectious dsDNA CLVd Chaipayon-1
4.2. Plant Inoculation and Maintenance
4.3. RNA Extraction, Library Preparation and Amplicon Sequencing
4.4. Read Processing and Downstream Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ding, B.; Itaya, A. Viroid: A useful model for studying the basic principles of infection and RNA biology. Mol. Plant Microbe. 2007, 20, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Navarro, B.; Gisel, A.; Rodio, M.E.; Delgado, S.; Flores, R.; Di Serio, F. Viroids: How to infect a host and cause disease without encoding proteins. Biochimie 2012, 94, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Gago-Zachert, S. Viroids, infectious long non-coding RNAs with autonomous replication. Virus Res. 2016, 212, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Dingley, A.J.; Steger, G.; Esters, B.; Riesner, D.; Grzesiek, S. Structural characterization of the 69 nucleotide potato spindle tuber viroid left-terminal domain by NMR and thermodynamic analysis. J. Mol. Biol. 2003, 334, 751–767. [Google Scholar] [CrossRef] [PubMed]
- Palukaitis, P. What has been happening with viroids? Virus Genes 2014, 49, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.A.; Smith, D.R.; Diener, T.O. Measurement of viroid sequence homology by hybridization with complementary DNA prepared in vitro. Virology 1978, 89, 388–394. [Google Scholar] [CrossRef]
- Matsushita, Y.; Tsuda, S. Seed transmission of potato spindle tuber viroid, tomato chlorotic dwarf viroid, tomato apical stunt viroid, and Columnea latent viroid in horticultural plants. Eur. J. Plant Pathol. 2016, 145, 1007–1011. [Google Scholar] [CrossRef]
- Tangkanchanapas, P.; Reanwarakorn, K.; Kirdpipat, W. The new strain of Columnea latent viroid (CLVd) causes severe symptoms on bolo maka (Solanum stramonifolium). Thai Agric. Res. J. 2013, 31, 53–68. [Google Scholar]
- Tansuwan, K.; Reanwarakorn, K. Seed transmission of Columnea latent viroid in cucumber. Thai Agric. Res. J. 2018, 36, 130–140. [Google Scholar]
- Verhoeven, J.T.J.; Jansen, C.C.C.; Willemen, T.M.; Kox, L.F.F.; Owens, R.A.; Roenhorst, J.W. Natural infections of tomato by Citrus exocortis viroid, Columnea latent viroid, Potato spindle tuber viroid and Tomato chlorotic dwarf viroid. Eur. J. Plant Pathol. 2004, 110, 823–831. [Google Scholar] [CrossRef]
- Marach, S. Infectious clones of Columnea latent viroid and its effects on commercial Tomato Varieties. Master’s Thesis, Kasetsart University, Bangkok, Thailand, 2008. [Google Scholar]
- Hammond, R.; Smith, D.R.; Diener, T.O. Nucleotide sequence and proposed secondary structure of Columnea latent viroid: A natural mosaic of viroid sequences. Nucleic Acids Res. 1989, 17, 10083–10094. [Google Scholar] [CrossRef] [PubMed]
- Constable, F.; Chambers, G.; Penrose, L.; Daly, A.; Mackie, J.; Davis, K.; Rodoni, B.; Gibbs, M. Viroid-infected tomato and capsicum seed shipments to Australia. Viruses 2019, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Tangkanchanapas, P. Viroid detection in tomato (Lycopersicon esculentum Mill.) seed production plantation in northeast of Thailand. Master’s Thesis, Kasetsart University, Bangkok, Thailand, 2005. [Google Scholar]
- Morris, C.S.J. Food and Environment Research Agency (Fera) Pest Risk Analysis for Columnea latent viroid, 3rd ed.; Food and Environment Research Agency: Sand Hutton, York, UK, 2010. [Google Scholar]
- Tangkanchanapas, P.; Reanwarakorn, K.; Kirdpipat, W. Detection of Columnea latent viroid (CLVd) and Pepper chat fruit viroid (PCFVd). Thai Agric. Res. J. 2013, 31, 107–122. [Google Scholar]
- Flores, R.; Grubb, D.; Elleuch, A.; Nohales, M.A.; Delgado, S.; Gago, S. Rolling-circle replication of viroids, viroid-like satellite RNAs and hepatitis delta virus: Variations on a theme. RNA Biol. 2011, 8, 200–206. [Google Scholar] [CrossRef]
- Gago, S.; Elena, S.F.; Flores, R.; Sanjuan, R. Extremely high mutation rate of a hammerhead viroid. Science 2009. [Google Scholar] [CrossRef]
- Brass, J.R.; Owens, R.A.; Matousek, J.; Steger, G. Viroid quasispecies revealed by deep sequencing. RNA Biol. 2017, 14, 317–325. [Google Scholar] [CrossRef]
- Lopez-Carrasco, A.; Ballesteros, C.; Sentandreu, V.; Delgado, S.; Gago-Zachert, S.; Flores, R.; Sanjuan, R. Different rates of spontaneous mutation of chloroplastic and nuclear viroids as determined by high-fidelity ultra-deep sequencing. PLoS Pathog. 2017. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Bull, J.J.; Meyers, L.A.; Lachmann, M. Quasispecies made simple. PLoS Comput. Biol. 2005, 1, 450–460. [Google Scholar] [CrossRef]
- Eigen, M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar] [CrossRef] [PubMed]
- Gora-Sochacka, A.; Kierzek, A.; Candresse, T.; Zagorski, W. The genetic stability of potato spindle tuber viroid (PSTVd) molecular variants. RNA 1997, 3, 68–74. [Google Scholar] [PubMed]
- Nie, X. Analysis of sequence polymorphism and population structure of tomato chlorotic dwarf viroid and potato spindle tuber viroid in viroid-infected tomato plants. Viruses 2012, 4, 940–953. [Google Scholar] [CrossRef]
- Podstolski, W.; Gora-Sochacka, A.; Zagorski, W. Co-inoculation with two non-infectious cDNA copies of potato spindle tuber viroid (PSTVd) leads to the appearance of novel fully infectious variants. Acta Biochim. Pol. 2005, 52, 87–98. [Google Scholar] [CrossRef]
- Bernad, L.; Duran-Vila, N.; Elena, S.F. Effect of citrus hosts on the generation, maintenance and evolutionary fate of genetic variability of citrus exocortis viroid. J. Gen. Virol. 2009, 90, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Bernad, L.; Gandía, M.; Duran-Vila, N. Host effect on the genetic variability of Citrus exocortis viroid (CEVd). In Proceedings of the Sixteenth IOCV Conference, Valencia, Spain, 2005. [Google Scholar]
- Gandia, M.; Bernad, L.; Rubio, L.; Duran-Vila, N. Host effect on the molecular and biological properties of a Citrus exocortis viroid isolate from Vicia faba. Phytopathology 2007, 97, 1004–1010. [Google Scholar] [CrossRef]
- Hajeri, S.; Ramadugu, C.; Manjunath, K.; Ng, J.; Lee, R.; Vidalakis, G. In vivo generated Citrus exocortis viroid progeny variants display a range of phenotypes with altered levels of replication, systemic accumulation and pathogenicity. Virology 2011, 417, 400–409. [Google Scholar] [CrossRef]
- Semancik, J.S.; Szychowski, J.A.; Rakowski, A.G.; Symons, R.H. Isolates of Citrus exocortis viroid recovered by host and tissue selection. J. Gen. Virol. 1993, 74, 2427–2436. [Google Scholar] [CrossRef]
- Choi, H.; Jo, Y.; Yoon, J.Y.; Choi, S.K.; Cho, W.K. Sequence variability of Chrysanthemum stunt viroid in different chrysanthemum cultivars. PEERJ 2017. [Google Scholar] [CrossRef]
- Jiang, D.M.; Gao, R.; Qin, L.; Wu, Z.J.; Xie, L.H.; Hou, W.Y.; Li, S.F. Infectious cDNA clones of four viroids in Coleus blumei and molecular characterization of their progeny. Virus Res. 2014, 180, 97–101. [Google Scholar] [CrossRef]
- Hamdi, I.; Elleuch, A.; Bessaies, N.; Fakhfakh, H. Insights on genetic diversity and phylogenetic analysis of Hop stunt viroid (HSVd) population from symptomatic citrus tree in Tunisia. Afr. J. Microbiol. Res. 2011, 5, 3422–3431. [Google Scholar] [CrossRef]
- Vidalakis, G.; Davis, J.Z.; Semancik, J.S. Intra-population diversity between citrus viroid II variants described as agents of cachexia disease. Ann. Appl. Biol. 2005, 146, 449–458. [Google Scholar] [CrossRef]
- Gandia, M.; Duran-Vila, N. Variability of the progeny of a sequence variant Citrus bent leaf viroid (CBLVd). Arch. Virol. 2004, 149, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Hong, N.; Wang, G.; Fan, X. Population structure and genetic diversity within Peach latent mosaic viroid field isolates from peach showing three symptoms. J. Phytopathol. 2008, 156, 565–572. [Google Scholar] [CrossRef]
- Glouzon, J.P.S.; Bolduc, F.; Wang, S.; Najmanovich, R.J.; Perreault, J.P. Deep-Sequencing of the Peach latent mosaic viroid reveals new aspects of population heterogeneity. PLoS ONE 2014, 9, e87297. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Gao, S.; Hernandez, A.G.; Wechter, W.P.; Fei, Z.; Ling, K.S. Deep sequencing of small RNAs in tomato for virus and viroid identification and strain differentiation. PLoS ONE 2012, 7, e37127. [Google Scholar] [CrossRef]
- Seguin, J.; Rajeswaran, R.; Malpica-Lopez, N.; Martin, R.R.; Kasschau, K.; Dolja, V.V.; Otten, P.; Farinelli, L.; Pooggin, M.M. De novo reconstruction of consensus master genomes of plant RNA and DNA viruses from siRNAs. PLoS ONE 2014, 9, e88513. [Google Scholar] [CrossRef]
- Zhang, Z.; Qi, S.; Tang, N.; Zhang, X.; Chen, S.; Zhu, P.; Ma, L.; Cheng, J.; Xu, Y.; Lu, M.; et al. Discovery of replicating circular RNAs by RNA-seq and computational algorithms. PLoS Pathog. 2014, 10, e1004553. [Google Scholar] [CrossRef]
- Hammond, R.W.; Owens, R.A. Mutational analysis of Potato spindle tuber viroid reveals complex relationships between structure and infectivity. Proc. Natl. Acad. Sci. USA 1987, 84, 3967–3971. [Google Scholar] [CrossRef]
- Hu, Y.; Feldstein, P.A.; Bottino, P.J.; Owens, R.A. Role of the variable domain in modulating potato spindle tuber viroid replication. Virology 1996, 219, 45–56. [Google Scholar] [CrossRef]
- Loss, P.; Schmitz, M.; Steger, G.; Riesner, D. Formation of a thermodynamically metastable structure containing hairpin II is critical for infectivity of potato spindle tuber viroid RNA. EMBO J. 1991, 10, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.A.; Chen, W.; Hu, Y.; Hsu, Y.H. Suppression of Potato spindle tuber viroid replication and symptom expression by mutations which stabilize the pathogenicity domain. Virology 1995, 208, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Owens, R.A.; Thompson, S.M.; Steger, G. Effects of random mutagenesis upon Potato Spindle Tuber Viroid replication and symptom expression. Virology 1991, 185, 18–31. [Google Scholar] [CrossRef]
- Qu, F.; Heinrich, C.; Loss, P.; Steger, G.; Tien, P.; Riesner, D. Multiple pathways of reversion in viroids for conservation of structural elements. EMBO J. 1993, 12, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Wassenegger, M.; Spieker, R.L.; Thalmeir, S.; Gast, F.U.; Riedel, L.; Sanger, H.L. A single nucleotide substitution converts potato spindle tuber viroid (PSTVd) from a noninfectious to an infectious RNA for nicotiana tabacum. Virology 1996, 226, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Serra, P.; Minoia, S.; Di Serio, F.; Navarro, B. Viroids: From genotype to phenotype just relying on RNA sequence and structural motifs. Front. Microbiol. 2012, 3, 217. [Google Scholar] [CrossRef]
- Gozmanova, M.; Denti, M.A.; Minkov, I.N.; Tsagris, M.; Tabler, M. Characterization of the RNA motif responsible for the specific interaction of potato spindle tuber viroid RNA (PSTVd) and the tomato protein Virp1. Nucleic Acids Res. 2003, 31, 5534–5543. [Google Scholar] [CrossRef]
- Kalantidis, K.; Denti, M.A.; Tzortzakaki, S.; Marinou, E.; Tabler, M.; Tsagris, M. Virp1 is a host protein with a major role in Potato Spindle Tuber Viroid infection in Nicotiana plants. J. Virol. 2007, 81, 12872–12880. [Google Scholar] [CrossRef]
- Steger, G. Modelling the three-dimensional structure of the right-terminal domain of pospiviroids. Sci. Rep. 2017, 7, 711. [Google Scholar] [CrossRef]
- Maniataki, E.; De Alba, A.E.M.; Sagesser, R.; Tabler, M.; Tsagris, M. Viroid RNA systemic spread may depend on the interaction of a 71-nucleotide bulged hairpin with the host protein VirP1. RNA 2003, 9, 346–354. [Google Scholar] [CrossRef]
- Zhong, X.H.; Archual, A.J.; Amin, A.A.; Ding, B. A genomic map of viroid RNA motifs critical for replication and systemic trafficking. Plant Cell 2008, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Biebricher, C.K.; Eigen, M. The error threshold. Virus Res. 2005, 107, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. Error catastrophe and antiviral strategy. Proc. Natl. Acad. Sci. USA. 2002, 99, 13374–13376. [Google Scholar] [CrossRef] [PubMed]
- Tangkanchanapas, P.; Reanwarakorn, K.; Sermsiri, C.; Ratchanee, H. An RT-PCR primer pair for the detection of six Pospiviroid in tomato plants. Thai Phytopathol. 2005, 19, 13–21. [Google Scholar]
- Self-Circularization-How to Prevent Other Unwanted Ligations. Available online: http://www.protocol-online.org/biology-forums-2/posts/20611.html. (accessed on 8 September 2017).
- 3 more DNA Ligation Tips. Available online: https://bitesizebio.com/10263/3-more-dna-ligation-tips/. (accessed on 8 September 2017).
- Nunez, A.N.; Kavlick, M.F.; Robertson, J.M.; Budowle, B. Application of Circular Ligase to Provide Template for Rolling Circle Amplification of Low Amounts of Fragmented DNA. Available online: https://pdfs.semanticscholar.org/607e/5beef92ea8bf3718e80fb1ec7bd0163d22f6.pdf?_ga=2.162454140.1710484673.1585829258-1238950593.1585829258. (accessed on 8 September 2017).
- Self-Circularization of Linear DNA. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0013192_Selof_Linear_DNA_UG.pdf. (accessed on 8 September 2017).
- T4 DNA Ligase. Available online: https://www.nhm.ac.uk/content/dam/nhmwww/our-science/dpts-facilities-staff/Coreresearchlabs/t4-dna-ligase_aug12.pdf. (accessed on 8 September 2017).
- Zhang, J.J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from High-Throughput sequencing reads. Embnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PEERJ 2016. [Google Scholar] [CrossRef]
- Boyer, F.; Mercier, C.; Bonin, A.; Le Bras, Y.; Taberlet, P.; Coissac, E. OBITOOLS: A UNIX-inspired software package for DNA metabarcoding. Mol. Ecol. Resour. 2016, 16, 176–182. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tangkanchanapas, P.; Haegeman, A.; Ruttink, T.; Höfte, M.; De Jonghe, K. Whole-Genome Deep Sequencing Reveals Host-Driven in-planta Evolution of Columnea Latent Viroid (CLVd) Quasi-Species Populations. Int. J. Mol. Sci. 2020, 21, 3262. https://doi.org/10.3390/ijms21093262
Tangkanchanapas P, Haegeman A, Ruttink T, Höfte M, De Jonghe K. Whole-Genome Deep Sequencing Reveals Host-Driven in-planta Evolution of Columnea Latent Viroid (CLVd) Quasi-Species Populations. International Journal of Molecular Sciences. 2020; 21(9):3262. https://doi.org/10.3390/ijms21093262
Chicago/Turabian StyleTangkanchanapas, Parichate, Annelies Haegeman, Tom Ruttink, Monica Höfte, and Kris De Jonghe. 2020. "Whole-Genome Deep Sequencing Reveals Host-Driven in-planta Evolution of Columnea Latent Viroid (CLVd) Quasi-Species Populations" International Journal of Molecular Sciences 21, no. 9: 3262. https://doi.org/10.3390/ijms21093262
APA StyleTangkanchanapas, P., Haegeman, A., Ruttink, T., Höfte, M., & De Jonghe, K. (2020). Whole-Genome Deep Sequencing Reveals Host-Driven in-planta Evolution of Columnea Latent Viroid (CLVd) Quasi-Species Populations. International Journal of Molecular Sciences, 21(9), 3262. https://doi.org/10.3390/ijms21093262