The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction: The Metformin Mechanism and AMPK Activation
2. AMPK Activation
2.1. Control by Adenine Nucleotides, Calcium and F1,6P2
2.2. Changes Linked to Nutritional State or the Gluconeogenic/Glycolytic Poise
2.3. Activation by Biguanides and Mitochondrial Inhibitors (The Canonical Pathway)
2.4. Metformin Activation of Lysosomal AMPK (Non-Canonical Pathways)
2.5. AMPK Activation by A-769662, 991 and AMP Analogues Does Not Mimic Metformin on Gluconeogenesis
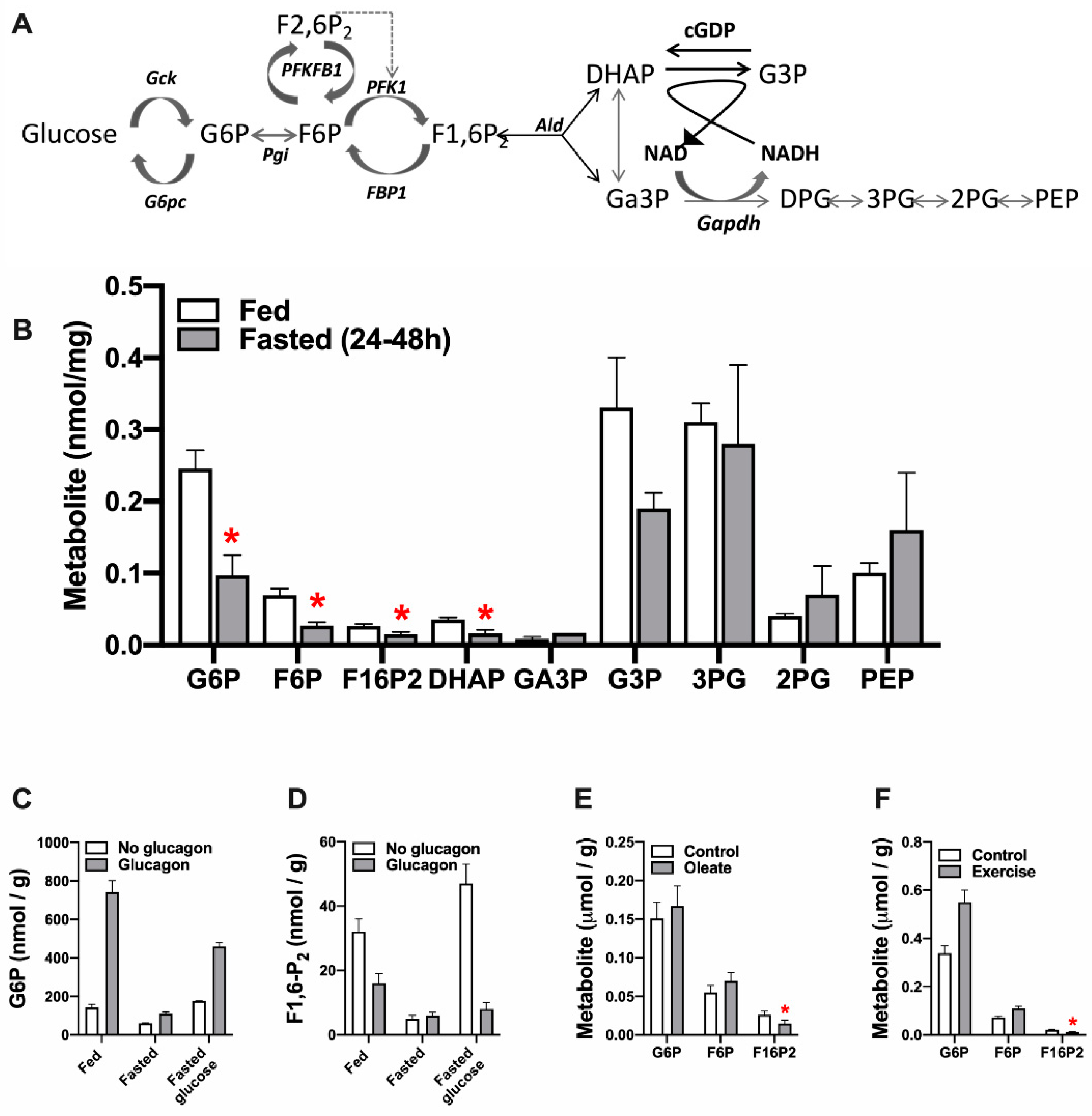
3. Adaptive Changes in Hepatic Intermediates of Glycolysis and Gluconeogenesis
3.1. Effects of Nutritional State, Glucagon, Fatty Acids and Exercise on F1,6-P2
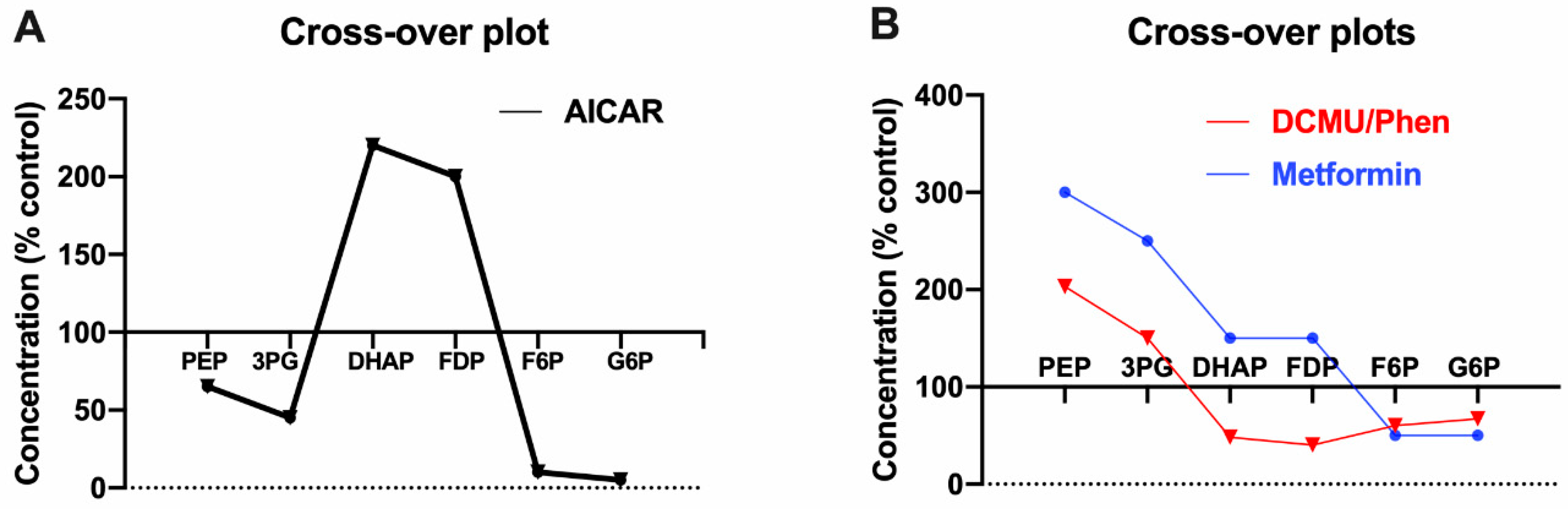
3.2. AICAR Has a Unique Effect on F1,6P2
3.3. Similarities in Cross-Over Plots for Biguanides and Respiratory Chain Inhibitors

3.4. Metformin Has a Biphasic Effect on the Mitochondrial NADH/NAD Redox State
3.5. Candidate Mechanisms for the Oxidized Mitochondrial NADH/NAD State
3.6. Metformin Effects on the Cytoplasmic NAD/NADH State and Glycerol 3-P (G3P)
3.7. The Metformin Mechanism on Gluconeogenic Flux: Deductions from Cell Metabolites
4. Control of Gene Expression of Gluconeogenic Enzymes by Metformin and AMPK Activation
4.1. Interactions between AMPK Signalling and PKA Signalling in the Metformin Mechanism
4.2. Metformin Counter-Regulation of Glucose Induction of G6pc
5. Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACC | acetyl-CoA carboxylase |
| AICAR | 5-amino-4-imidazole carboxamide riboside |
| AMPK | AMP-activated protein kinase |
| cGPD | cytoplasmic glycerophosphate dehydrogenase |
| ChREBP | Carbohydrate response element binding protein |
| DHA | dihydroxyacetone |
| DHAP | dihydroxyaceyone phopsphate |
| FBP1 | fructose 1,6-bisphosphatase-1 |
| F6P | fructose 6-phosphate |
| F1,6P2 | fructose 1,6-bisphosphate |
| F2,6P2 | fructose 2,6-bisphosphate |
| G6P | glucose 6-phosphate |
| Ga3P | glyceraldehyde 3-phosphate |
| G3P | glycerol 3-phosphate |
| LKB1 | liver kinase B1 |
| mGPD | mitochondrial glycerophosphate dehydrogenase |
| MFF | mitochondrial fission factor |
| PEP | phosphoenolpyruvate |
| PFK1 | phosphofructokinase-1 |
| 2PG | 2-phosphoglycerate |
| 3PG | 3-phosphoglycerate |
| TP | triose phosphates |
| ZMP | AICA-ribotide |
References
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Natali, A.; Ferrannini, E. Effects of metformin and thiazolidinediones on suppression of hepatic glucose production and stimulation of glucose uptake in type 2 diabetes: A systematic review. Diabetologia 2006, 49, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef]
- Carling, D.; Clarke, P.R.; Zammit, V.A.; Hardie, D.G. Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur. J. Biochem. 1989, 186, 129–136. [Google Scholar] [CrossRef]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/ AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, C.S.; Zong, Y.; Feng, J.W.; Ma, T.; Hu, M.; Lin, Z.; Li, X.; Xie, C.; Wu, Y.; et al. Transient Receptor Potential V Channels Are Essential for Glucose Sensing by Aldolase and AMPK. Cell Metab. 2019, 30, 508–524. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Zhang, C.S.; Li, M.; Wang, W.; Wang, Z.; Hawley, S.A.; Ma, T.; Feng, J.W.; Tian, X.; Qi, Q.; et al. Hierarchical activation of compartmentalized pools of AMPK depends on severity of nutrient or energy stress. Cell Res. 2019, 29, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef]
- Göransson, O.; McBride, A.; Hawley, S.A.; Ross, F.A.; Shpiro, N.; Foretz, M.; Viollet, B.; Hardie, D.G.; Sakamoto, K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J. Biol. Chem. 2007, 282, 32549–32560. [Google Scholar] [CrossRef]
- Ubl, J.J.; Chen, S.; Stucki, J.W. Anti-diabetic biguanides inhibit hormone-induced intracellular Ca2+ concentration oscillations in rat hepatocytes. Biochem. J. 1994, 304, 561–567. [Google Scholar] [CrossRef]
- Neumann, D. Is TAK1 a Direct Upstream Kinase of AMPK? Int. J. Mol. Sci. 2018, 19, 2412. [Google Scholar] [CrossRef]
- Jia, J.; Abudu, Y.P.; Claude-Taupin, A.; Gu, Y.; Kumar, S.; Choi, S.W.; Peters, R.; Mudd, M.H.; Allers, L.; Salemi, M.; et al. Galectins Control mTOR in Response to Endomembrane Damage. Mol. Cell 2018, 70, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Bissa, B.; Brecht, L.; Allers, L.; Choi, S.W.; Gu, Y.; Zbinden, M.; Burge, M.R.; Timmins, G.; Hallows, K.; et al. AMPK, a Regulator of Metabolism and Autophagy, Is Activated by Lysosomal Damage via a Novel Galectin-Directed Ubiquitin Signal Transduction System. Mol. Cell 2020, 77, 951–969. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi-Shimizu, S.; Park, E.J.; Roh, Y.S.; Yang, L.; Zhang, B.; Song, J.; Liang, S.; Pimienta, M.; Taniguchi, K.; Wu, X.; et al. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J. Clin. Investig. 2014, 124, 3566–3578. [Google Scholar] [CrossRef] [PubMed]
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef]
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem. J. 2016, 473, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Salt, I.; Celler, J.W.; Hawley, S.A.; Prescott, A.; Woods, A.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem. J. 1998, 334, 177–187. [Google Scholar] [CrossRef]
- Carroll, B.; Dunlop, E.A. The lysosome: A crucial hub for AMPK and mTORC1 signalling. Biochem. J. 2017, 474, 1453–1466. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Ford, R.J.; Fullerton, M.D.; Pinkosky, S.L.; Day, E.A.; Scott, J.W.; Oakhill, J.S.; Bujak, A.L.; Smith, B.K.; Crane, J.D.; Blümer, R.M.; et al. Metformin and salicylate synergistically activate liver AMPK, inhibit lipogenesis and improve insulin sensitivity. Biochem. J. 2015, 468, 125–132. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Assifi, M.M.; Suchankova, G.; Constant, S.; Prentki, M.; Saha, A.K.; Ruderman, N.B. AMP-activated protein kinase and coordination of hepatic fatty acid metabolism of starved/carbohydrate-refed rats. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E794–E800. [Google Scholar] [CrossRef]
- Huet, C.; Boudaba, N.; Guigas, B.; Viollet, B.; Foretz, M. Glucose availability but not changes in pancreatic hormones sensitizes hepatic AMPK activity during nutritional transition in rodents. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- You, M.; Matsumoto, M.; Pacold, C.M.; Cho, W.K.; Crabb, D.W. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004, 127, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Liangpunsakul, S.; Sozio, M.S.; Shin, E.; Zhao, Z.; Xu, Y.; Ross, R.A.; Zeng, Y.; Crabb, D.W. Inhibitory effect of ethanol on AMPK phosphorylation is mediated in part through elevated ceramide levels. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G1004–G1012. [Google Scholar] [CrossRef]
- Agius, L. Hormonal and Metabolite Regulation of Hepatic Glucokinase. Annu. Rev. Nutr. 2016, 36, 389–415. [Google Scholar] [CrossRef] [PubMed]
- Droppelmann, C.A.; Sáez, D.E.; Asenjo, J.L.; Yáñez, A.J.; García-Rocha, M.; Concha, I.I.; Grez, M.; Guinovart, J.J.; Slebe, J.C. A new level of regulation in gluconeogenesis: Metabolic state modulates the intracellular localization of aldolase B and its interaction with liver fructose-1,6-bisphosphatase. Biochem. J. 2015, 472, 225–237. [Google Scholar] [CrossRef]
- Stephenne, X.; Foretz, M.; Taleux, N.; van der Zon, G.C.; Sokal, E.; Hue, L.; Viollet, B.; Guigas, B. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia 2011, 54, 3101–3110. [Google Scholar] [CrossRef]
- Hou, W.L.; Yin, J.; Alimujiang, M.; Yu, X.Y.; Ai, L.G.; Bao, Y.Q.; Liu, F.; Jia, W.P. Inhibition of mitochondrial complex I improves glucose metabolism independently of AMPKactivation. J. Cell. Mol. Med. 2018, 22, 1316–1328. [Google Scholar]
- Ota, S.; Horigome, K.; Ishii, T.; Nakai, M.; Hayashi, K.; Kawamura, T.; Kishino, A.; Taiji, M.; Kimura, T. Metformin suppresses glucose-6-phosphatase expression by a complex I inhibition and AMPK activation-independent mechanism. Biochem. Biophys. Res. Commun. 2009, 388, 311–316. [Google Scholar] [CrossRef]
- Bridges, H.R.; Sirviö, V.A.; Agip, A.N.; Hirst, J. Molecular features of biguanides required for targeting of mitochondrial respiratory complex I and activation of AMP-kinase. BMC Biol. 2016, 14, 65. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.B.; Wang, J.; Flotte, T.R.; Yagi, T.; Matsuno-Yagi, A. Use of the NADH-quinone oxidoreductase (NDI1) gene of Saccharomyces cerevisiae as a possible cure for complex I defects in human cells. J. Biol. Chem. 2000, 275, 37774–37778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, C.; Wyre, N.D.; Bailey, C.J. Subcellular distribution of metformin in rat liver. J. Pharm. Pharmacol. 1991, 43, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, D.; Raymer, M.L.; Lockwood, T.D. Antidiabetic and antimalarial biguanide drugs are metal-interactive antiproteolytic agents. Biochem. Pharmacol. 2003, 66, 663–677. [Google Scholar] [CrossRef]
- Xiao, B.; Sanders, M.J.; Carmena, D.; Bright, N.J.; Haire, L.F.; Underwood, E.; Patel, B.R.; Heath, R.B.; Walker, P.A.; Hallen, S.; et al. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 2013, 4, 3017. [Google Scholar] [CrossRef]
- Rajamohan, F.; Reyes, A.R.; Frisbie, R.K.; Hoth, L.R.; Sahasrabudhe, P.; Magyar, R.; Landro, J.A.; Withka, J.M.; Caspers, N.L.; Calabrese, M.F.; et al. Probing the enzyme kinetics, allosteric modulation and activation of α1- and α2-subunit-containing AMP-activated protein kinase (AMPK) heterotrimeric complexes by pharmacological and physiological activators. Biochem. J. 2016, 473, 581–592. [Google Scholar] [CrossRef]
- Hunter, R.W.; Foretz, M.; Bultot, L.; Fullerton, M.D.; Deak, M.; Ross, F.A.; Hawley, S.A.; Shpiro, N.; Viollet, B.; Barron, D.; et al. Mechanism of action of compound-13: An α1-selective small molecule activator of AMPK. Chem. Biol. 2014, 21, 866–879. [Google Scholar] [CrossRef]
- Moonira, T.; Chachra, S.S.; Ford, B.E.; Marin, S.; Alshawi, A.; Adam-Primus, N.S.; Arden, C.; Al-Oanzi, Z.H.; Foretz, M.; Viollet, B.; et al. Metformin lowers glucose 6-phosphate in hepatocytes by activation of glycolysis downstream of glucose phosphorylation. J Biol Chem. 2020, 295, 3330–3346. [Google Scholar] [CrossRef]
- Alshawi, A.; Agius, L. Low metformin causes a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibits gluconeogenesis by a redox-independent mechanism. J. Biol. Chem. 2019, 294, 2839–2853. [Google Scholar] [CrossRef]
- Agius, L. Dietary carbohydrate and control of hepatic gene expression: Mechanistic links from ATP and phosphate ester homeostasis to the carbohydrate-response element-binding protein. Proc. Nutr. Soc. 2016, 75, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Bergmeyer, H.U. Methods in Enzymatic Analysis, Concentrations of Metabolites in Animal Tissues; Verla Chemie: Weinheim, Germany, 1974; Volume 4, pp. 2267–2279. [Google Scholar]
- Van Schaftingen, E.; Hue, L.; Hers, H.G. Study of the fructose 6-phosphate/fructose 1,6-bi-phosphate cycle in the liver in vivo. Biochem. J. 1980, 192, 263–271. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bartrons, R.; Hue, L.; Van Schaftingen, E.; Hers, H.G. Hormonal control of fructose 2,6-bisphosphate concentration in isolated rat hepatocytes. Biochem. J. 1983, 214, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Ochs, R.S.; Harris, R.A. Mechanism for the oleate stimulation of gluconeogenesis from dihydroxyacetone by hepatocytes from fasted rats. Biochim. Biophys. Acta 1986, 886, 40–47. [Google Scholar] [CrossRef]
- Jenkins, C.M.; Yang, J.; Sims, H.F.; Gross, R.W. Reversible high affinity inhibition of phosphofructokinase-1 by acyl-CoA: A mechanism integrating glycolytic flux with lipid metabolism. J. Biol. Chem. 2011, 286, 11937–11950. [Google Scholar] [CrossRef]
- Dohm, G.L.; Newsholme, E.A. Metabolic control of hepatic gluconeogenesis during exercise. Biochem. J. 1983, 212, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Hers, H.G.; Hue, L. Gluconeogenesis and related aspects of glycolysis. Annu. Rev. Biochem. 1983, 52, 617–653. [Google Scholar] [CrossRef]
- Hue, L. Role of fructose 2,6-bisphosphate in the stimulation of glycolysis by anoxia in isolated hepatocytes. Biochem. J. 1982, 206, 359–365. [Google Scholar] [CrossRef]
- Al-Oanzi, Z.H.; Fountana, S.; Moonira, T.; Tudhope, S.J.; Petrie, J.L.; Alshawi, A.; Patman, G.; Arden, C.; Reeves, H.L.; Agius, L. Opposite effects of a glucokinase activator and metformin on glucose-regulated gene expression in hepatocytes. Diabetes Obes. Metab. 2017, 19, 1078–1087. [Google Scholar] [CrossRef]
- Vincent, M.F.; Marangos, P.J.; Gruber, H.E.; Van den Berghe, G. Inhibition by AICA riboside of gluconeogenesis in isolated rat hepatocytes. Diabetes 1991, 40, 1259–1266. [Google Scholar] [CrossRef]
- Hue, L.; Bartrons, R. Role of fructose 2,6-bisphosphate in the control by glucagon of gluconeogenesis from various precursors in isolated rat hepatocytes. Biochem. J. 1984, 218, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.F.; Bontemps, F.; Van den Berghe, G. Inhibition of glycolysis by 5-amino-4-imidazolecarboxamide riboside in isolated rat hepatocytes. Biochem. J. 1992, 281, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Javaux, F.; Vincent, M.F.; Wagner, D.R.; van den Berghe, G. Cell-type specificity of inhibition of glycolysis by 5-amino-4-imidazolecarboxamide riboside. Lack of effect in rabbit cardiomyocytes and human erythrocytes, and inhibition in FTO-2B rat hepatoma cells. Biochem. J. 1995, 305, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Hasenour, C.M.; Ridley, D.E.; Hughey, C.C.; James, F.D.; Donahue, E.P.; Shearer, J.; Viollet, B.; Foretz, M.; Wasserman, D.H. 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) effect on glucose production, but not energy metabolism, is independent of hepatic AMPK in vivo. J. Biol. Chem. 2014, 289, 5950–5959. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.B. The effect of alkylguanidines on mitochondrial metabolism. J. Biol. Chem. 1963, 238, 410–417. [Google Scholar] [PubMed]
- Davidoff, F. Effects of guanidine derivatives on mitochondrial function. 3. The mechanism of phenethylbiguanide accumulation and its relationship to in vitro respiratory inhibition. J. Biol. Chem. 1971, 246, 4017–4027. [Google Scholar]
- Pryor, H.J.; Smyth, J.E.; Quinlan, P.T.; Halestrap, A.P. Evidence that the flux control coefficient of the respiratory chain is high during gluconeogenesis from lactate in hepatocytes from starved rats. Implications for the hormonal control of gluconeogenesis and action of hypoglycaemic agents. Biochem. J. 1987, 247, 449–457. [Google Scholar] [CrossRef]
- Owen, M.R.; Halestrap, A.P. The mechanisms by which mild respiratory chain inhibitors inhibit hepatic gluconeogenesis. Biochim. Biophys. Acta 1993, 1142, 11–22. [Google Scholar] [CrossRef]
- Cook, D.E.; Blair, J.B.; Gilfillan, C.; Lardy, H.A. Mode of action of hypoglycemic agents. IV. Control of the hypoglycemic activity of phenethylbiguanide in rats and guinea-pigs. Biochem. Pharmacol. 1973, 22, 2121–2128. [Google Scholar] [CrossRef]
- Ballard, F.J. The development of gluconeogenesis in rat liver. Controlling factors in the newborn. Biochem. J. 1971, 124, 265–274. [Google Scholar] [CrossRef]
- Hunter, R.W.; Hughey, C.C.; Lantier, L.; Sundelin, E.I.; Peggie, M.; Zeqiraj, E.; Sicheri, F.; Jessen, N.; Wasserman, D.H.; Sakamoto, K. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat. Med. 2018, 24, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.H.; Lund, P.; Krebs, H.A. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J. 1967, 103, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Fulgencio, J.P.; Kohl, C.; Girard, J.; Pégorier, J.P. Effect of metformin on fatty acid and glucose metabolism in freshly isolated hepatocytes and on specific gene expression in cultured hepatocytes. Biochem. Pharmacol. 2001, 62, 439–446. [Google Scholar] [CrossRef]
- Gouaref, I.; Detaille, D.; Wiernsperger, N.; Khan, N.A.; Leverve, X.; Koceir, E.A. The desert gerbil Psammomys obesus as a model for metformin-sensitive nutritional type 2 diabetes to protect hepatocellular metabolic damage: Impact of mitochondrial redox state. PLoS ONE 2017, 12, e0172053. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, C.; Bailey, C.J. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; An, H.; Liu, T.; Qin, C.; Sesaki, H.; Guo, S.; Radovick, S.; Hussain, M.; Maheshwari, A.; Wondisford, F.E.; et al. Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 2019, 29, 1511–1523. [Google Scholar] [CrossRef]
- Kanamura, S.; Kanai, K.; Oka, M.; Watanabe, J.; Asada-Kubota, M. Development of morphologic heterogeneity of hepatocyte mitochondria in the mouse. Anat. Rec. 1984, 210, 315–325. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.M.; Zhang, D.; Camporez, J.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef]
- Qi, H.; Nielsen, P.M.; Schroeder, M.; Bertelsen, L.B.; Palm, F.; Laustsen, C. Acute renal metabolic effect of metformin assessed with hyperpolarised MRI in rats. Diabetologia 2018, 61, 445–454. [Google Scholar] [CrossRef]
- von Morze, C.; Ohliger, M.A.; Marco-Rius, I.; Wilson, D.M.; Flavell, R.R.; Pearce, D.; Vigneron, D.B.; Kurhanewicz, J.; Wang, Z.J. Direct assessment of renal mitochondrial redox state using hyperpolarized (13) C-acetoacetate. Magn. Reson. Med. 2018, 79, 1862–1869. [Google Scholar] [CrossRef] [PubMed]
- Mráček, T.; Drahota, Z.; Houštěk, J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 2013, 1827, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Pecinova, A.; Drahota, Z.; Kovalcikova, J.; Kovarova, N.; Pecina, P.; Alan, L.; Zima, M.; Houstek, J.; Mracek, T. Pleiotropic Effects of Biguanides on Mitochondrial Reactive Oxygen Species Production. Oxid. Med. Cell. Longev. 2017, 2017, 7038603. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, M.J.; Israr-ul, A.; Longacre, M.; Stoker, S. If Metformin Inhibited the Mitochondrial Glycerol Phosphate Dehydrogenase It Might Not Benefit Diabetes. BioRxiv 2020. [Google Scholar] [CrossRef]
- Cameron, A.R.; Logie, L.; Patel, K.; Erhardt, S.; Bacon, S.; Middleton, P.; Harthill, J.; Forteath, C.; Coats, J.T.; Kerr, C.; et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol. 2018, 14, 187–197. [Google Scholar] [CrossRef]
- Batandier, C.; Guigas, B.; Detaille, D.; El-Mir, M.Y.; Fontaine, E.; Rigoulet, M.; Leverve, X.M. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J. Bioenerg. Biomembr. 2006, 38, 33–42. [Google Scholar] [CrossRef]
- Kane, D.A.; Anderson, E.J.; Price, J.W., 3rd; Woodlief, T.L.; Lin, C.T.; Bikman, B.T.; Cortright, R.N.; Neufer, P.D. Metformin selectively attenuates mitochondrial H2O2 emission without affecting respiratory capacity in skeletal muscle of obese rats. Free Radic. Biol. Med. 2010, 49, 1082–1087. [Google Scholar] [CrossRef]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef]
- Lewis, A.J.; Miller, J.J.; McCallum, C.; Rider, O.J.; Neubauer, S.; Heather, L.C.; Tyler, D.J. Assessment of Metformin-Induced Changes in Cardiac and Hepatic Redox State Using Hyperpolarized[1-13C]Pyruvate. Diabetes 2016, 65, 3544–3551. [Google Scholar] [CrossRef]
- Baur, J.A.; Birnbaum, M.J. Control of gluconeogenesis by metformin: Does redox trump energy charge? Cell Metab. 2014, 20, 197–199. [Google Scholar] [CrossRef]
- Saheki, T.; Iijima, M.; Li, M.X.; Kobayashi, K.; Horiuchi, M.; Ushikai, M.; Okumura, F.; Meng, X.J.; Inoue, I.; Tajima, A.; et al. Citrin/mitochondrial glycerol-3-phosphate dehydrogenase double knock-out mice recapitulate features of human citrin deficiency. J. Biol. Chem. 2007, 282, 25041–25052. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.J.; Bremer, J.; Akerman, K.E. Thermodynamic aspects of translocation of reducing equivalents by mitochondria. J. Biol. Chem. 1980, 255, 2277–2283. [Google Scholar] [PubMed]
- Sibille, B.; Keriel, C.; Fontaine, E.; Catelloni, F.; Rigoulet, M.; Leverve, X.M. Octanoate affects 2,4-dinitrophenol uncoupling in intact isolated rat hepatocytes. Eur. J. Biochem. 1995, 231, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Dykens, J.A.; Jamieson, J.; Marroquin, L.; Nadanaciva, S.; Billis, P.A.; Will, Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol. Appl. Pharmacol. 2008, 233, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Argaud, D.; Roth, H.; Wiernsperger, N.; Leverve, X.M. Metformin decreases gluconeogenesis by enhancing the pyruvate kinase flux in isolated rat hepatocytes. Eur. J. Biochem. 1993, 213, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Van Schaftingen, E.; Bartrons, R.; Hers, H.G. The mechanism by which ethanol decreases the concentration of fructose 2,6-bisphosphate in the liver. Biochem. J. 1984, 222, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Woods, H.F.; Eggleston, L.V.; Krebs, H.A. The cause of hepatic accumulation of fructose 1-phosphate on fructose loading. Biochem. J. 1970, 119, 501–510. [Google Scholar] [CrossRef]
- Woods, H.F.; Krebs, H.A. Xylitol metabolism in the isolated perfused rat liver. Biochem. J. 1973, 134, 437–443. [Google Scholar] [CrossRef]
- Calza, G.; Nyberg, E.; Mäkinen, M.; Soliymani, R.; Cascone, A.; Lindholm, D.; Barborini, E.; Baumann, M.; Lalowski, M.; Eriksson, O. Lactate-Induced Glucose Output Is Unchanged by Metformin at a Therapeutic Concentration—A Mass Spectrometry Imaging Study of the Perfused Rat Liver. Front. Pharmacol. 2018, 9, 141. [Google Scholar] [CrossRef]
- Glossmann, H.H.; Lutz, O.M.D. Commentary: Lactate-Induced Glucose Output Is Unchanged by Metformin at a Therapeutic Concentration-A Mass Spectrometry Imaging Study of the Perfused Rat Liver. Front. Pharmacol. 2019, 10, 90. [Google Scholar] [CrossRef]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Sum, C.F.; Webster, J.M.; Johnson, A.B.; Catalano, C.; Cooper, B.G.; Taylor, R. The effect of intravenous metformin on glucose metabolism during hyperglycaemia in type 2 diabetes. Diabet. Med. 1992, 9, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Heishi, M.; Ichihara, J.; Teramoto, R.; Itakura, Y.; Hayashi, K.; Ishikawa, H.; Gomi, H.; Sakai, J.; Kanaoka, M.; Taiji, M.; et al. Global gene expression analysis in liver of obese diabetic db/db mice treated with metformin. Diabetologia 2006, 49, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activatedprotein kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Mosquera, A.; Muntané, J.; Ferrandiz, F.; Rodriguez-Mañas, L.; de Pablo, F.; González-Canudas, J.; Valverde, Á.M. Differential effects of metformin glycinate and hydrochloride in glucose production, AMPK phosphorylation and insulin sensitivity in hepatocytes from non-diabetic and diabetic mice. Food Chem. Toxicol. 2019, 123, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Williams, J.R.; Muckett, P.J.; Mayer, F.V.; Liljevald, M.; Bohlooly-Y, M.; Carling, D. Liver-Specific Activation of AMPK Prevents Steatosis on a High-Fructose Diet. Cell Rep. 2017, 18, 3043–3051. [Google Scholar] [CrossRef] [PubMed]
- Johanns, M.; Lai, Y.C.; Hsu, M.F.; Jacobs, R.; Vertommen, D.; Van Sande, J.; Dumont, J.E.; Woods, A.; Carling, D.; Hue, L.; et al. AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nat. Commun. 2016, 7, 10856. [Google Scholar] [CrossRef]
- Ma, L.; Robinson, L.N.; Towle, H.C. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem. 2006, 281, 28721–28730. [Google Scholar] [CrossRef]
- Arden, C.; Petrie, J.L.; Tudhope, S.J.; Al-Oanzi, Z.; Claydon, A.J.; Beynon, R.J.; Towle, H.C.; Agius, L. Elevated glucose represses liver glucokinase and induces its regulatory protein to safeguard hepatic phosphate homeostasis. Diabetes. 2011, 60, 3110–3120. [Google Scholar] [CrossRef]
- Arden, C.; Tudhope, S.J.; Petrie, J.L.; Al-Oanzi, Z.H.; Cullen, K.S.; Lange, A.J.; Towle, H.C.; Agius, L. Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochem. J. 2012, 443, 111–123. [Google Scholar] [CrossRef]
- Mattila, J.; Havula, E.; Suominen, E.; Teesalu, M.; Surakka, I.; Hynynen, R.; Kilpinen, H.; Väänänen, J.; Hovatta, I.; Käkelä, R.; et al. Mondo-Mlx Mediates Organismal Sugar Sensing through the Gli-Similar Transcription Factor Sugarbabe. Cell Rep. 2015, 13, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Jung, H.; Nakagawa, T.; Pawlosky, R.; Takeshima, T.; Lee, W.R.; Sakiyama, H.; Laxman, S.; Wynn, R.M.; Tu, B.P.; et al. Metabolite Regulation of Nuclear Localization of Carbohydrate-responseElement-binding Protein (ChREBP): ROLE OF AMP AS AN ALLOSTERIC INHIBITOR. J. Biol. Chem. 2016, 291, 10515–10527. [Google Scholar] [CrossRef] [PubMed]
- Grefhorst, A.; Schreurs, M.; Oosterveer, M.H.; Cortés, V.A.; Havinga, R.; Herling, A.W.; Reijngoud, D.J.; Groen, A.K.; Kuipers, F. Carbohydrate-response-element-binding protein (ChREBP) and not the liver X receptor α (LXRα) mediates elevated hepatic lipogenic gene expression in a mouse model of glycogen storage disease type 1. Biochem. J. 2010, 432, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Kalemba, K.M.; Wang, Y.; Xu, H.; Chiles, E.; McMillin, S.M.; Kwon, H.; Su, X.; Wondisford, F.E. Glycerol induces G6pc in primary mouse hepatocytes and is the preferred substrate for gluconeogenesis both in vitro and in vivo. J. Biol. Chem. 2019, 294, 18017–18028. [Google Scholar] [CrossRef] [PubMed]
- McCreight, L.J.; Mari, A.; Coppin, L.; Jackson, N.; Umpleby, A.M.; Pearson, E.R. Metformin increases fasting glucose clearance and endogenous glucose production in non-diabetic individuals. Diabetologia 2020, 63, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Gormsen, L.C.; Søndergaard, E.; Christensen, N.L.; Brøsen, K.; Jessen, N.; Nielsen, S. Metformin increases endogenous glucose production in non-diabetic individuals and individuals with recent-onset type 2 diabetes. Diabetologia 2019, 62, 1251–1256. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agius, L.; Ford, B.E.; Chachra, S.S. The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective. Int. J. Mol. Sci. 2020, 21, 3240. https://doi.org/10.3390/ijms21093240
Agius L, Ford BE, Chachra SS. The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective. International Journal of Molecular Sciences. 2020; 21(9):3240. https://doi.org/10.3390/ijms21093240
Chicago/Turabian StyleAgius, Loranne, Brian E. Ford, and Shruti S. Chachra. 2020. "The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective" International Journal of Molecular Sciences 21, no. 9: 3240. https://doi.org/10.3390/ijms21093240
APA StyleAgius, L., Ford, B. E., & Chachra, S. S. (2020). The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective. International Journal of Molecular Sciences, 21(9), 3240. https://doi.org/10.3390/ijms21093240