Loss of AMPKalpha1 Triggers Centrosome Amplification via PLK4 Upregulation in Mouse Embryonic Fibroblasts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
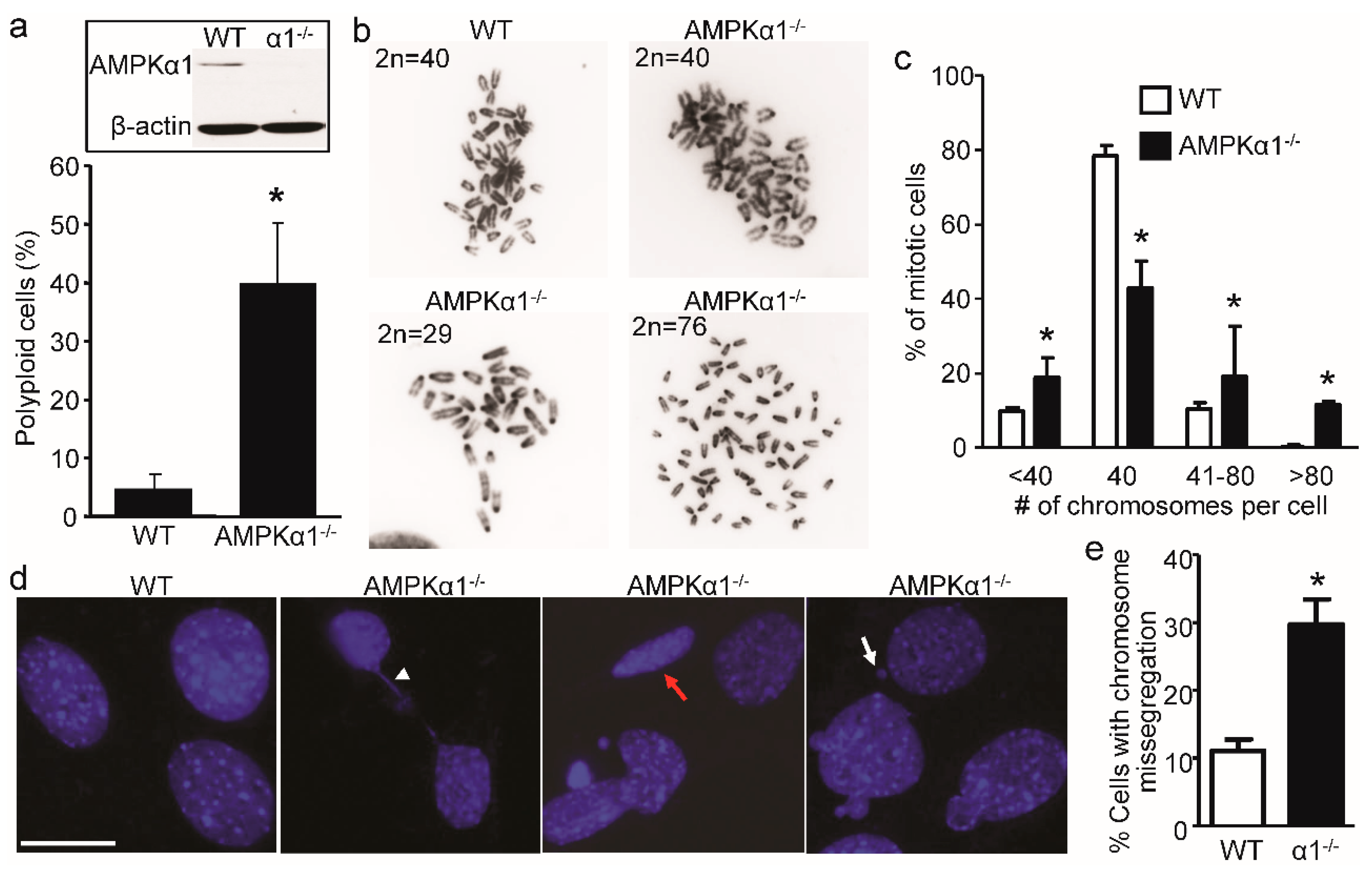
2.1. Loss of AMPKα1 Causes Chromosome Instability and Abnormal Nuclei in Mouse Embryonic Fibroblasts (MEFs)
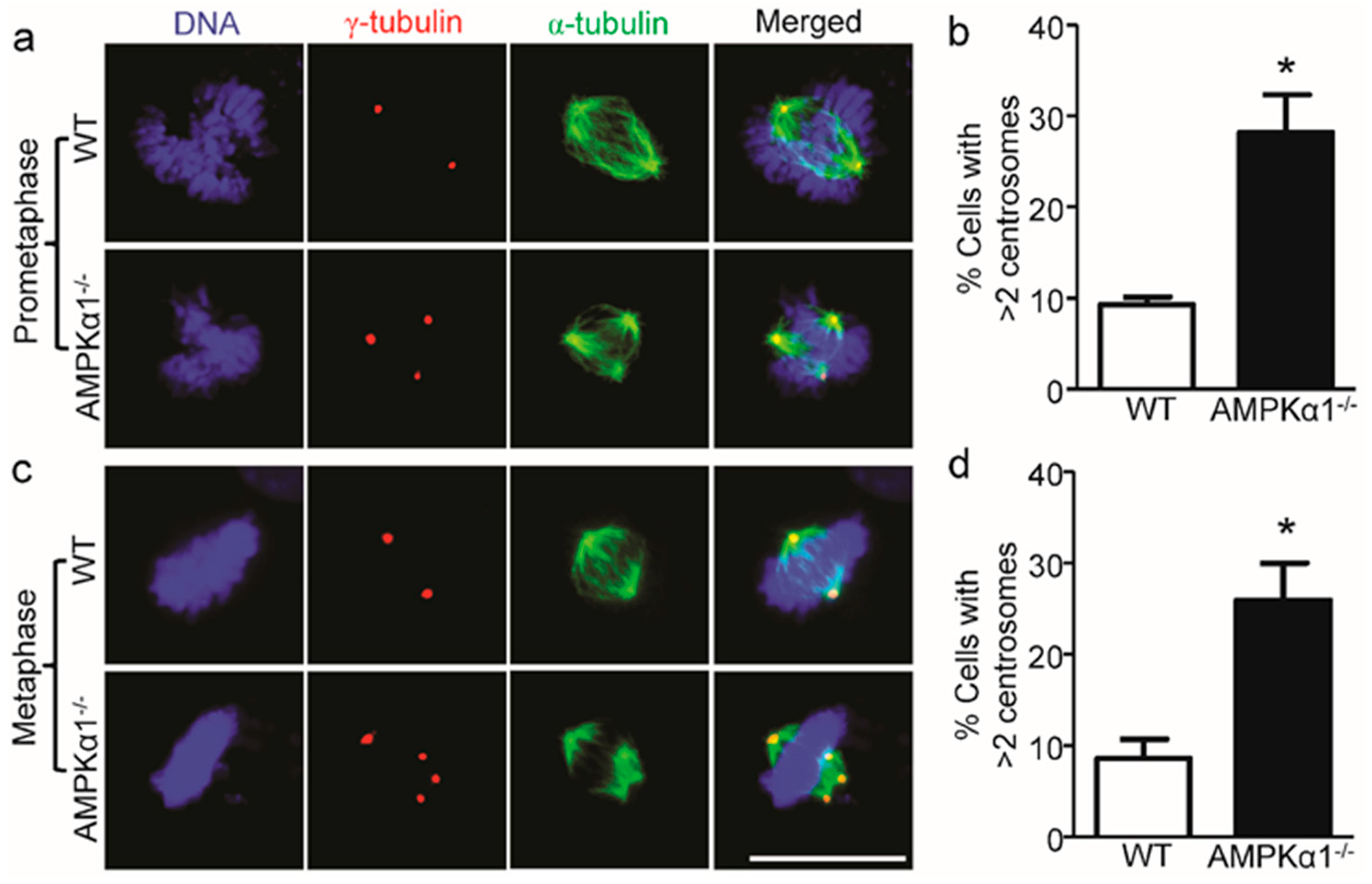
2.2. AMPKα1 Deletion Confers Centrosome Amplification
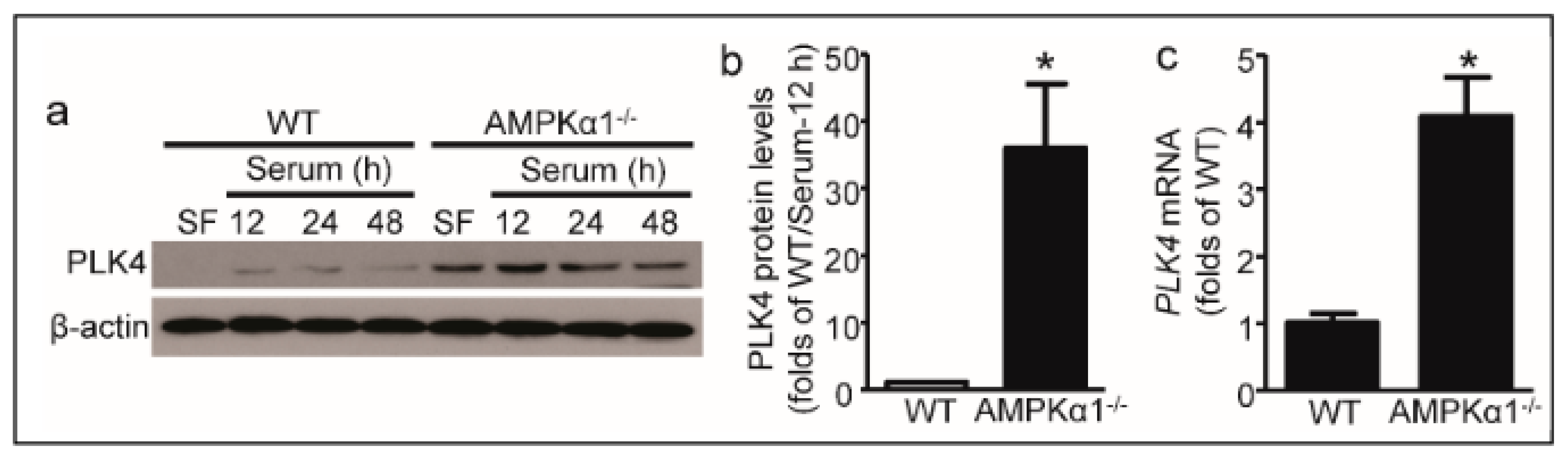
2.3. PLK4 Elevates in AMPKα1-Deleted MEFs
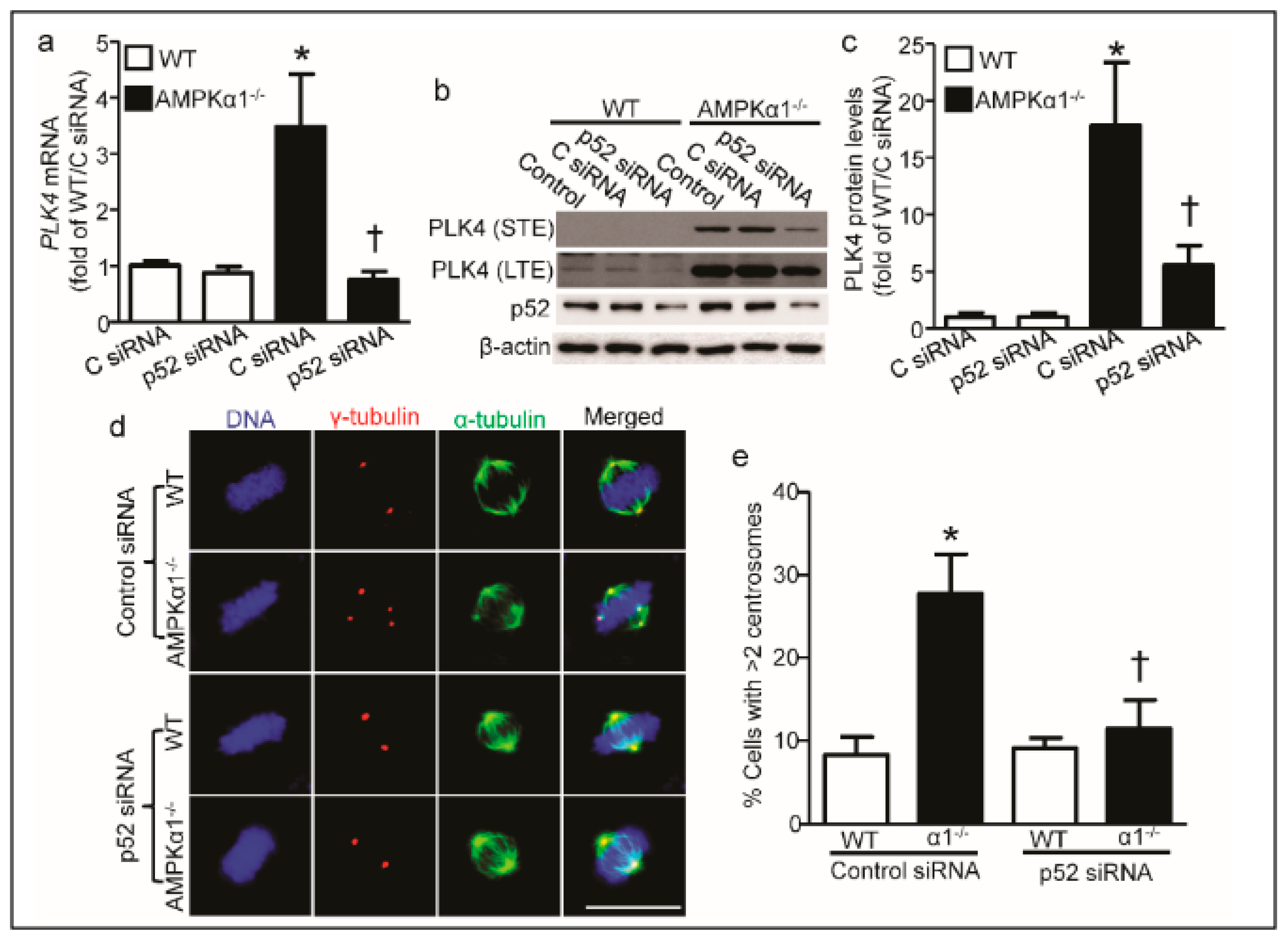
2.4. PLK4 Upregulation in AMPKα1−/− MEFs Is p52-Mediated
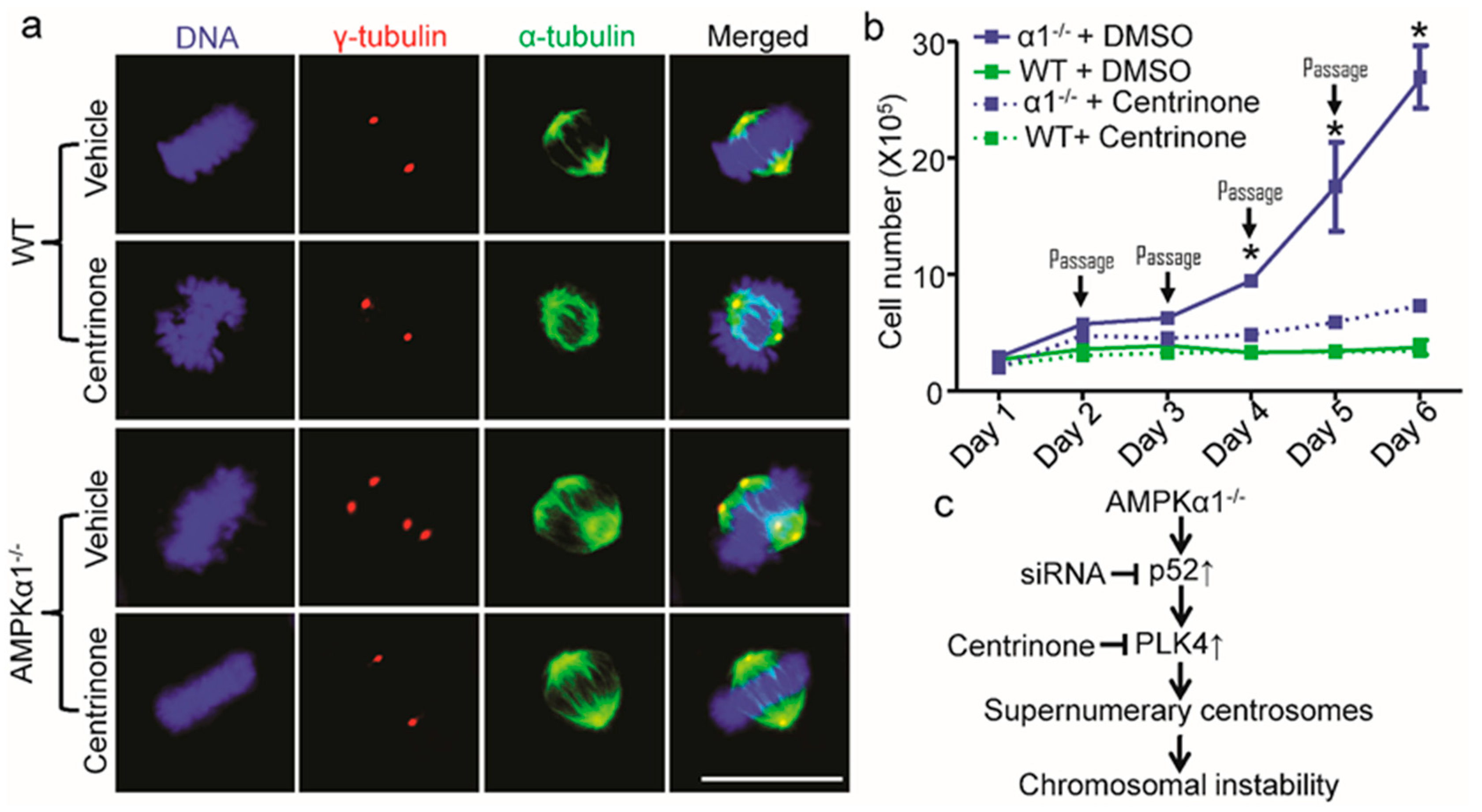
2.5. PLK4 Inhibition Ameliorates Centrosome Amplification in AMPKα1−/− MEFs
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Cell Culture and Transfection
4.3. Indirect Immunofluorescence Analysis
4.4. RNA Extraction, cDNA Synthesis, and Real Time PCR
4.5. Metaphase Chromosome Spreads
4.6. Flow Cytometry Analysis
4.7. Protein Extraction and Western Blotting
4.8. Passaging Assays
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | adenosine monophosphate-activated protein kinase |
| CIN | chromosomal instability |
| MEFs | mouse embryonic fibroblasts |
| PLK4 | Polo-like kinase 4 |
References
- Nigg, E.A.; Holland, A.J. Once and only once: Mechanisms of centriole duplication and their deregulation in disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 297–312. [Google Scholar] [CrossRef]
- Westhorpe, F.G.; Straight, A.F. Functions of the centromere and kinetochore in chromosome segregation. Curr. Opin. Cell Biol. 2013, 25, 334–340. [Google Scholar] [CrossRef]
- Thornton, G.K.; Woods, C.G. Primary microcephaly: Do all roads lead to rome? Trends Genet. 2009, 25, 501–510. [Google Scholar] [CrossRef]
- Dionne, L.K.; Shim, K.; Hoshi, M.; Cheng, T.; Wang, J.; Marthiens, V.; Knoten, A.; Basto, R.; Jain, S.; Mahjoub, M.R. Centrosome amplification disrupts renal development and causes cystogenesis. J. Cell Biol. 2018, 217, 2485–2501. [Google Scholar] [CrossRef]
- Zyss, D.; Gergely, F. Centrosome function in cancer: Guilty or innocent? Trends Cell Biol. 2009, 19, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y. A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci. 2011, 7, 1122–1144. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.M.; Williams, B.R.; Amon, A. Aneuploidy: Cells losing their balance. Genetics 2008, 179, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Lingle, W.L.; Barrett, S.L.; Negron, V.C.; D’Assoro, A.B.; Boeneman, K.; Liu, W.; Whitehead, C.M.; Reynolds, C.; Salisbury, J.L. Centrosome amplification drives chromosomal instability in breast tumor development. Proc. Natl. Acad. Sci. USA 2002, 99, 1978–1983. [Google Scholar] [CrossRef]
- Passerini, V.; Ozeri-Galai, E.; de Pagter, M.S.; Donnelly, N.; Schmalbrock, S.; Kloosterman, W.P.; Kerem, B.; Storchova, Z. The presence of extra chromosomes leads to genomic instability. Nat. Commun. 2016, 7, 10754. [Google Scholar] [CrossRef]
- de Cárcer, G.; Manning, G.; Malumbres, M. From plk1 to plk5. Cell Cycle 2011, 10, 2255–2262. [Google Scholar] [CrossRef]
- Li, S.; Deng, Z.; Fu, J.; Xu, C.; Xin, G.; Wu, Z.; Luo, J.; Wang, G.; Zhang, S.; Zhang, B.; et al. Spatial compartmentalization specializes the function of aurora a and aurora b. J. Biol. Chem. 2015, 290, 17546–17558. [Google Scholar] [CrossRef]
- Habedanck, R.; Stierhof, Y.D.; Wilkinson, C.J.; Nigg, E.A. The polo kinase plk4 functions in centriole duplication. Nat. Cell Biol. 2005, 7, 1140–1146. [Google Scholar] [CrossRef]
- Fournier, M.; Orpinell, M.; Grauffel, C.; Scheer, E.; Garnier, J.M.; Ye, T.; Chavant, V.; Joint, M.; Esashi, F.; Dejaegere, A.; et al. Kat2a/kat2b-targeted acetylome reveals a role for plk4 acetylation in preventing centrosome amplification. Nat. Commun. 2016, 7, 13227. [Google Scholar] [CrossRef]
- Aydogan, M.G.; Wainman, A.; Saurya, S.; Steinacker, T.L.; Caballe, A.; Novak, Z.A.; Baumbach, J.; Muschalik, N.; Raff, J.W. A homeostatic clock sets daughter centriole size in flies. J. Cell Biol. 2018, 217, 1233–1248. [Google Scholar] [CrossRef]
- Coelho, P.A.; Bury, L.; Shahbazi, M.N.; Liakath-Ali, K.; Tate, P.H.; Wormald, S.; Hindley, C.J.; Huch, M.; Archer, J.; Skarnes, W.C.; et al. Over-expression of plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol. 2015, 5, 150209. [Google Scholar] [CrossRef]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef]
- Sercin, O.; Larsimont, J.C.; Karambelas, A.E.; Marthiens, V.; Moers, V.; Boeckx, B.; Le Mercier, M.; Lambrechts, D.; Basto, R.; Blanpain, C. Transient plk4 overexpression accelerates tumorigenesis in p53-deficient epidermis. Nat. Cell Biol. 2016, 18, 100–110. [Google Scholar] [CrossRef]
- Levine, M.S.; Bakker, B.; Boeckx, B.; Moyett, J.; Lu, J.; Vitre, B.; Spierings, D.C.; Lansdorp, P.M.; Cleveland, D.W.; Lambrechts, D.; et al. Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev. Cell 2017, 40, 313–322. [Google Scholar] [CrossRef]
- Wong, Y.L.; Anzola, J.V.; Davis, R.L.; Yoon, M.; Motamedi, A.; Kroll, A.; Seo, C.P.; Hsia, J.E.; Kim, S.K.; Mitchell, J.W.; et al. Cell biology. Reversible centriole depletion with an inhibitor of polo-like kinase 4. Science 2015, 348, 1155–1160. [Google Scholar] [CrossRef]
- Rogers, G.C.; Rusan, N.M.; Roberts, D.M.; Peifer, M.; Rogers, S.L. The scf slimb ubiquitin ligase regulates plk4/sak levels to block centriole reduplication. J. Cell Biol. 2009, 184, 225–239. [Google Scholar] [CrossRef]
- Tategu, M.; Nakagawa, H.; Sasaki, K.; Yamauchi, R.; Sekimachi, S.; Suita, Y.; Watanabe, N.; Yoshid, K. Transcriptional regulation of human polo-like kinases and early mitotic inhibitor. J. Genet. Genom. 2008, 35, 215–224. [Google Scholar] [CrossRef]
- Ledoux, A.C.; Sellier, H.; Gillies, K.; Iannetti, A.; James, J.; Perkins, N.D. Nfkappab regulates expression of polo-like kinase 4. Cell Cycle 2013, 12, 3052–3062. [Google Scholar] [CrossRef]
- Fan, G.; Sun, L.; Shan, P.; Zhang, X.; Huan, J.; Zhang, X.; Li, D.; Wang, T.; Wei, T.; Zhang, X.; et al. Loss of klf14 triggers centrosome amplification and tumorigenesis. Nat. Commun. 2015, 6, 8450. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Ferreira, I.; Rodrigues-Martins, A.; Bento, I.; Riparbelli, M.; Zhang, W.; Laue, E.; Callaini, G.; Glover, D.M.; Bettencourt-Dias, M. The scf/slimb ubiquitin ligase limits centrosome amplification through degradation of sak/plk4. Curr. Biol. 2009, 19, 43–49. [Google Scholar] [CrossRef]
- Brownlee, C.W.; Klebba, J.E.; Buster, D.W.; Rogers, G.C. The protein phosphatase 2a regulatory subunit twins stabilizes plk4 to induce centriole amplification. J. Cell Biol. 2011, 195, 231–243. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. Ampk: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zou, M.H. Regulation of nad(p)h oxidases by ampk in cardiovascular systems. Free Radic. Biol. Med. 2012, 52, 1607–1619. [Google Scholar] [CrossRef] [PubMed]
- Banko, M.R.; Allen, J.J.; Schaffer, B.E.; Wilker, E.W.; Tsou, P.; White, J.L.; Villen, J.; Wang, B.; Kim, S.R.; Sakamoto, K.; et al. Chemical genetic screen for ampkalpha2 substrates uncovers a network of proteins involved in mitosis. Mol. Cell 2011, 44, 878–892. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Lopez-Bonet, E.; Oliveras-Ferraros, C.; Perez-Martinez, M.C.; Bernado, L.; Menendez, J.A. Mitotic kinase dynamics of the active form of ampk (phospho-ampkalphathr172) in human cancer cells. Cell Cycle 2009, 8, 788–791. [Google Scholar] [CrossRef]
- Thaiparambil, J.T.; Eggers, C.M.; Marcus, A.I. Ampk regulates mitotic spindle orientation through phosphorylation of myosin regulatory light chain. Mol. Cell Biol. 2012, 32, 3203–3217. [Google Scholar] [CrossRef]
- Lee, J.H.; Koh, H.; Kim, M.; Kim, Y.; Lee, S.Y.; Karess, R.E.; Lee, S.H.; Shong, M.; Kim, J.M.; Kim, J.; et al. Energy-dependent regulation of cell structure by amp-activated protein kinase. Nature 2007, 447, 1017–1020. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. The active form of the metabolic sensor: Amp-activated protein kinase (ampk) directly binds the mitotic apparatus and travels from centrosomes to the spindle midzone during mitosis and cytokinesis. Cell Cycle 2009, 8, 2385–2398. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Cufi, S.; Menendez, J.A. Polo-like kinase 1 regulates activation of amp-activated protein kinase (ampk) at the mitotic apparatus. Cell Cycle 2011, 10, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Huang, W.; Tao, R.; Ibaragi, S.; Lan, F.; Ido, Y.; Wu, X.; Alekseyev, Y.O.; Lenburg, M.E.; Hu, G.F.; et al. Inactivation of ampk alters gene expression and promotes growth of prostate cancer cells. Oncogene 2009, 28, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, H.; Ding, Y.; Lu, Q.; Zou, M.H.; Song, P. Ampkalpha1 deletion in fibroblasts promotes tumorigenesis in athymic nude mice by p52-mediated elevation of erythropoietin and cdk2. Oncotarget 2016, 7, 53654–53667. [Google Scholar] [PubMed]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. Ampk is a negative regulator of the warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Pineda, C.T.; Ramanathan, S.; Fon Tacer, K.; Weon, J.L.; Potts, M.B.; Ou, Y.H.; White, M.A.; Potts, P.R. Degradation of ampk by a cancer-specific ubiquitin ligase. Cell 2015, 160, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Vila, I.K.; Yao, Y.; Kim, G.; Xia, W.; Kim, H.; Kim, S.J.; Park, M.K.; Hwang, J.P.; Gonzalez-Billalabeitia, E.; Hung, M.C.; et al. A ube2o-ampkalpha2 axis that promotes tumor initiation and progression offers opportunities for therapy. Cancer Cell 2017, 31, 208–224. [Google Scholar] [CrossRef]
- Wu, D.; Hu, D.; Chen, H.; Shi, G.; Fetahu, I.S.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-regulated phosphorylation of tet2 by ampk reveals a pathway linking diabetes to cancer. Nature 2018, 559, 637–641. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, Y.; Coughlan, K.A.; Ding, Y.; Wang, S.; Wu, Y.; Song, P.; Zou, M.H. Ampkalpha1 deficiency promotes cellular proliferation and DNA damage via p21 reduction in mouse embryonic fibroblasts. Biochim. Biophys. Acta 2015, 1853, 65–73. [Google Scholar] [CrossRef]
- Igata, M.; Motoshima, H.; Tsuruzoe, K.; Kojima, K.; Matsumura, T.; Kondo, T.; Taguchi, T.; Nakamaru, K.; Yano, M.; Kukidome, D.; et al. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ. Res. 2005, 97, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Wang, S.; He, C.; Liang, B.; Viollet, B.; Zou, M.H. Ampkalpha2 deletion exacerbates neointima formation by upregulating skp2 in vascular smooth muscle cells. Circ. Res. 2011, 109, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Humbert, N.; Navaratnam, N.; Augert, A.; Da Costa, M.; Martien, S.; Wang, J.; Martinez, D.; Abbadie, C.; Carling, D.; de Launoit, Y.; et al. Regulation of ploidy and senescence by the ampk-related kinase nuak1. EMBO J. 2010, 29, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Merlen, G.; Gentric, G.; Celton-Morizur, S.; Foretz, M.; Guidotti, J.E.; Fauveau, V.; Leclerc, J.; Viollet, B.; Desdouets, C. Ampkα1 controls hepatocyte proliferation independently of energy balance by regulating cyclin a2 expression. J. Hepatol. 2014, 60, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Li, N.; Guo, Y.; Xu, X.; Gao, L.; Xu, Y.; Zhou, L.; Liu, W. Ampk phosphorylates gbf1 for mitotic golgi disassembly. J. Cell Sci. 2013, 126, 1498–1505. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Sullivan, B.A.; Trazzi, S.; Della Valle, G.; Robertson, K.D. Dnmt3b interacts with constitutive centromere protein cenp-c to modulate DNA methylation and the histone code at centromeric regions. Hum. Mol. Genet. 2009, 18, 3178–3193. [Google Scholar] [CrossRef]
- Hinchcliffe, E.H.; Day, C.A.; Karanjeet, K.B.; Fadness, S.; Langfald, A.; Vaughan, K.T.; Dong, Z. Chromosome missegregation during anaphase triggers p53 cell cycle arrest through histone h3.3 ser31 phosphorylation. Nat. Cell Biol. 2016, 18, 668–675. [Google Scholar] [CrossRef]
- Fischer, M.; Quaas, M.; Steiner, L.; Engeland, K. The p53-p21-dream-cde/chr pathway regulates g2/m cell cycle genes. Nucleic Acids Res. 2016, 44, 164–174. [Google Scholar] [CrossRef]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Lambrus, B.G.; Daggubati, V.; Uetake, Y.; Scott, P.M.; Clutario, K.M.; Sluder, G.; Holland, A.J. A usp28-53bp1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J. Cell Biol. 2016, 214, 143–153. [Google Scholar] [CrossRef]
- Li, S.; Lavagnino, Z.; Lemacon, D.; Kong, L.; Ustione, A.; Ng, X.; Zhang, Y.; Wang, Y.; Zheng, B.; Piwnica-Worms, H.; et al. Ca(2+)-stimulated ampk-dependent phosphorylation of exo1 protects stressed replication forks from aberrant resection. Mol. Cell 2019, 74, 1123–1137.e1126. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, J.; Song, P.; Viollet, B.; Zou, M.H. In vivo activation of amp-activated protein kinase attenuates diabetes-enhanced degradation of gtp cyclohydrolase i. Diabetes 2009, 58, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.C.; Williams, B.R.; Siegel, J.J.; Amon, A. Identification of aneuploidy-selective antiproliferation compounds. Cell 2011, 144, 499–512. [Google Scholar] [CrossRef]
- Jorgensen, S.B.; Viollet, B.; Andreelli, F.; Frosig, C.; Birk, J.B.; Schjerling, P.; Vaulont, S.; Richter, E.A.; Wojtaszewski, J.F. Knockout of the alpha2 but not alpha1 5’-amp-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J. Biol. Chem. 2004, 279, 1070–1079. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Ding, Y.; Zhang, M.; Lu, Q.; Wu, S.; Zhu, H.; Song, P.; Zou, M.H. Ablation of adenosine monophosphate-activated protein kinase alpha1 in vascular smooth muscle cells promotes diet-induced atherosclerotic calcification in vivo. Circ. Res. 2016, 119, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, P.; Zou, M.H. Inhibition of amp-activated protein kinase alpha (ampkalpha) by doxorubicin accentuates genotoxic stress and cell death in mouse embryonic fibroblasts and cardiomyocytes: Role of p53 and sirt1. J. Biol Chem. 2012, 287, 8001–8012. [Google Scholar] [CrossRef] [PubMed]
- Todaro, G.J.; Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 1963, 17, 299–313. [Google Scholar] [CrossRef]
- Deng, W.; Tsao, S.W.; Lucas, J.N.; Leung, C.S.; Cheung, A.L. A new method for improving metaphase chromosome spreading. Cytometry A 2003, 51, 46–51. [Google Scholar] [CrossRef]
- Ding, Y.; Han, Y.; Lu, Q.; An, J.; Zhu, H.; Xie, Z.; Song, P.; Zou, M.H. Peroxynitrite-mediated sirt (sirtuin)-1 inactivation contributes to nicotine-induced arterial stiffness in mice. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1419–1431. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Q.; Coughlan, K.A.; Zou, M.-H.; Song, P. Loss of AMPKalpha1 Triggers Centrosome Amplification via PLK4 Upregulation in Mouse Embryonic Fibroblasts. Int. J. Mol. Sci. 2020, 21, 2772. https://doi.org/10.3390/ijms21082772
Zhao Q, Coughlan KA, Zou M-H, Song P. Loss of AMPKalpha1 Triggers Centrosome Amplification via PLK4 Upregulation in Mouse Embryonic Fibroblasts. International Journal of Molecular Sciences. 2020; 21(8):2772. https://doi.org/10.3390/ijms21082772
Chicago/Turabian StyleZhao, Qiang, Kathleen A Coughlan, Ming-Hui Zou, and Ping Song. 2020. "Loss of AMPKalpha1 Triggers Centrosome Amplification via PLK4 Upregulation in Mouse Embryonic Fibroblasts" International Journal of Molecular Sciences 21, no. 8: 2772. https://doi.org/10.3390/ijms21082772
APA StyleZhao, Q., Coughlan, K. A., Zou, M.-H., & Song, P. (2020). Loss of AMPKalpha1 Triggers Centrosome Amplification via PLK4 Upregulation in Mouse Embryonic Fibroblasts. International Journal of Molecular Sciences, 21(8), 2772. https://doi.org/10.3390/ijms21082772