The Sophisticated Transcriptional Response Governed by Transposable Elements in Human Health and Disease

 , , , ,
, , , ,
Abstract
1. Introduction
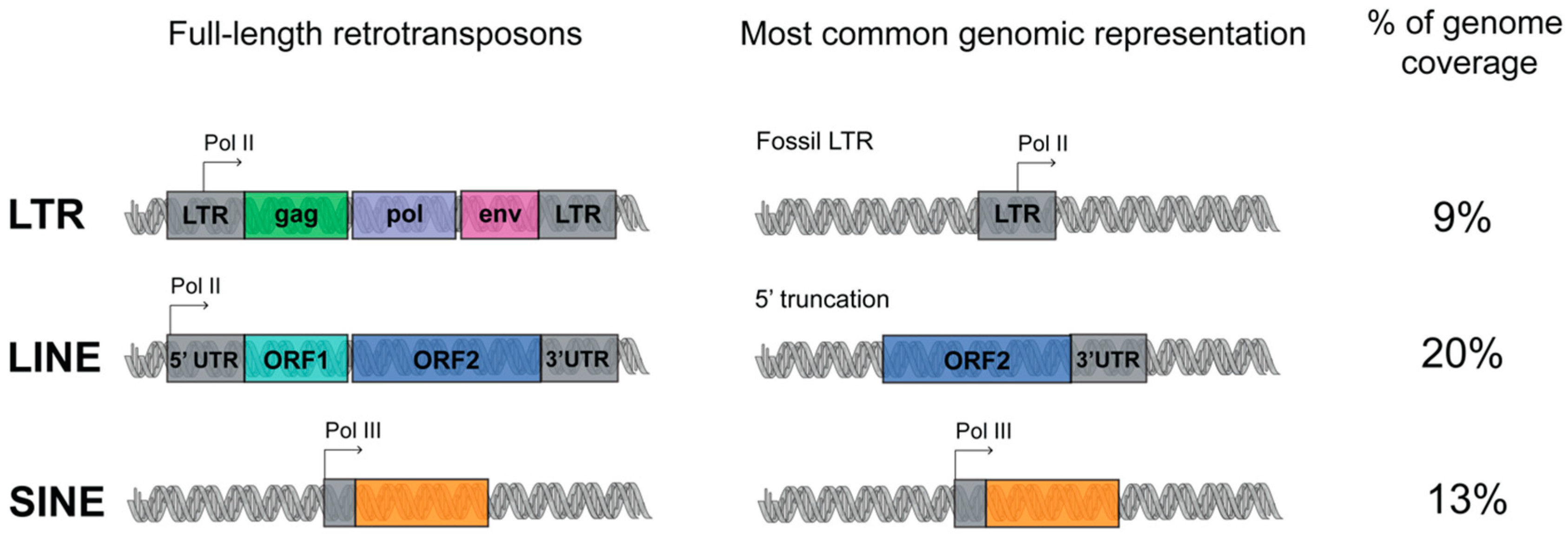
1.1. Transposable Elements (TEs) Account for Genome Evolution and Inter-Individual Genetic Variability
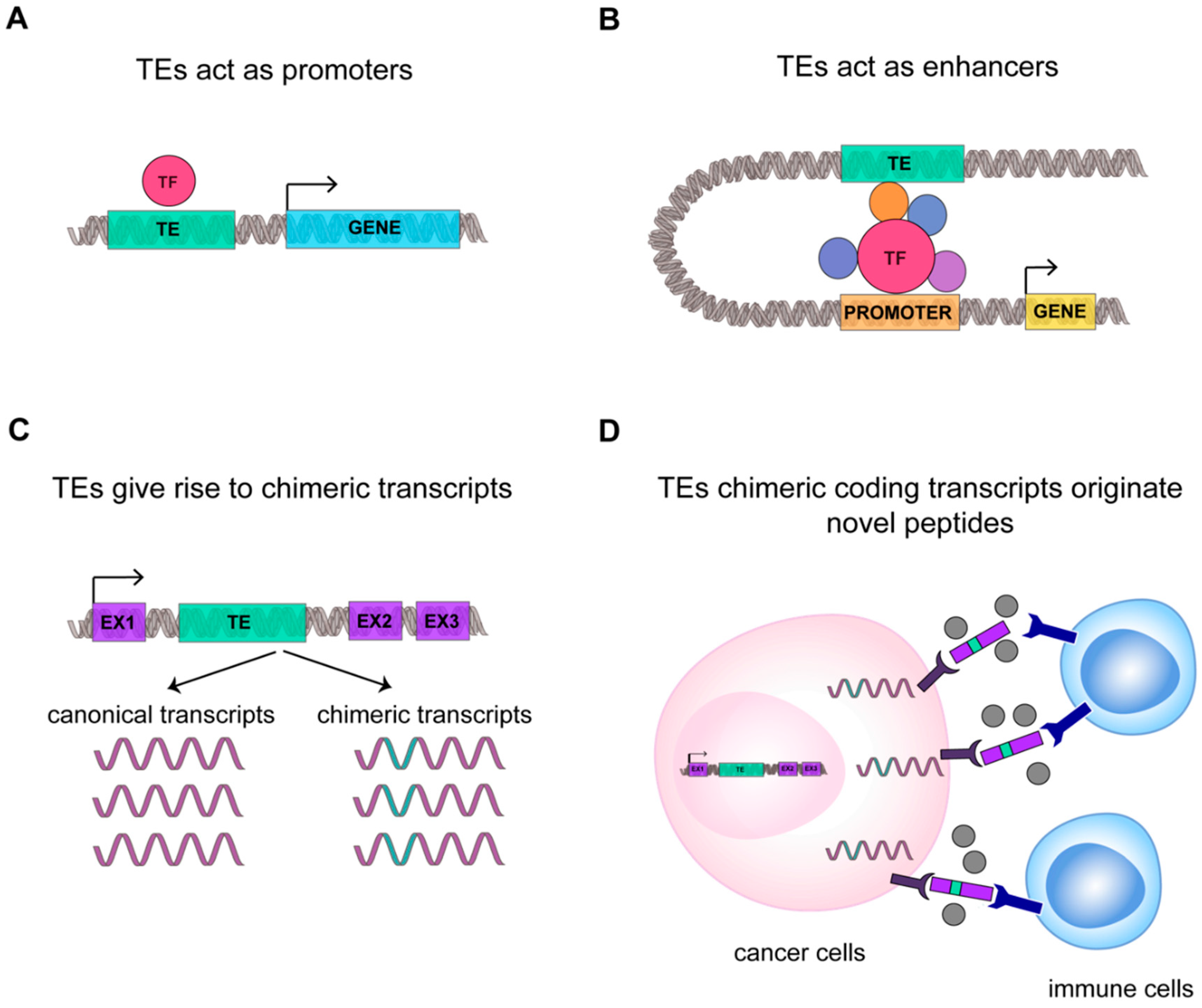
1.2. Not Just Transposition: TEs RNAs Are a Prolific Source for Novel Regulatory Functions
2. The Transcriptional Role of TEs in Shaping the Innate and Adaptive Immune Response
2.1. TEs RNAs Boost Innate and Adaptive Immune Response
2.2. Deregulation of the Expression Levels of TEs is Implicated in Autoimmunity and Inflammation
2.3. TEs RNAs are Novel Players in Cancer Immunity
3. TEs Transcriptional Landscape in Cancer Tissue
3.1. The Expression of TEs is Widely Dysregulated in Cancer Tissue
3.2. TEs RNAs Improve Cancer Specific Transcriptional Complexity and Plasticity
3.3. TEs Regulate Cancer Tumorigenicity and Progression
4. Next-Generation Sequencing (NGS) Approaches for the Analysis of TEs
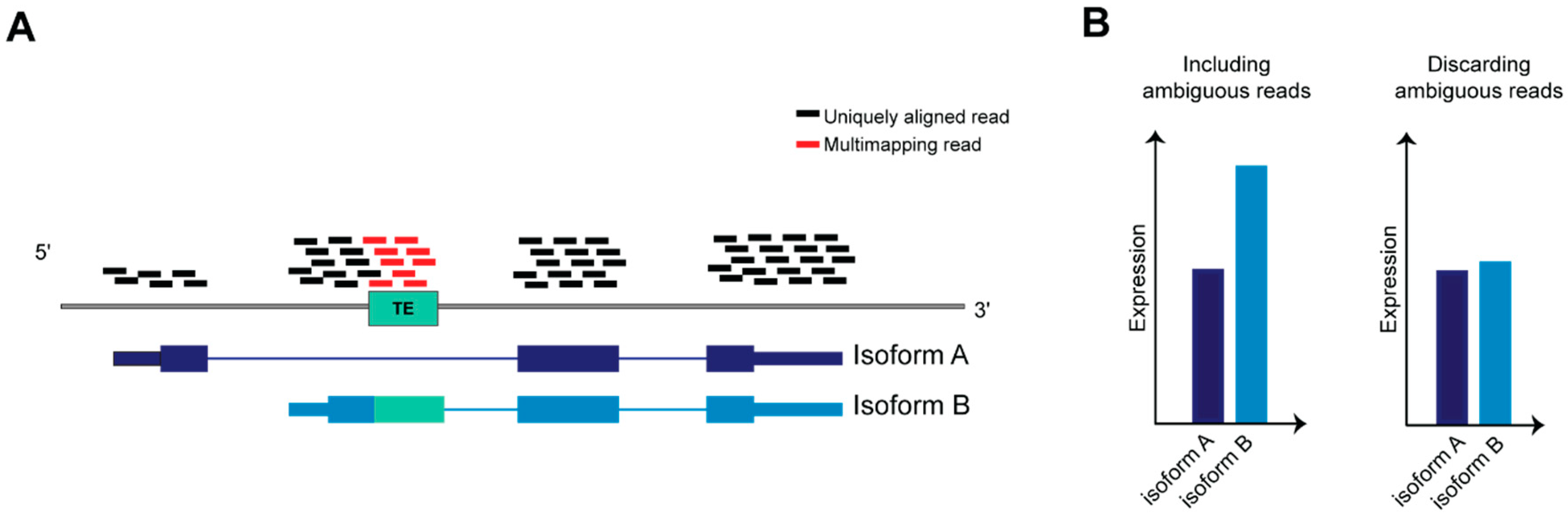
4.1. Dealing with Ambiguity in RNA-Seq Reads Alignment: A Challenge to Resolve TEs Expression Quantification
4.2. Current Computational Methods for TEs Transcriptome Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| LINE | Long interspersed element |
| L1 | Long interspersed element 1 |
| SINE | Short interspersed element |
| LTR | Long terminal repeat |
| TE | Transposable element |
| ERV | Endogenous retrovirus |
| NGS | Next generation sequence |
| IFN | Interferon |
| NK | Natural killer |
| DC | Dendritic cell |
| NF-κB | Nuclear factor kappa B |
| AP-1 | Activator protein-1 |
| IRF | Interferon response factor |
| PRR | Pattern recognition receptor |
| PAMP | Pathogens associated molecular patterns |
| RLR | retinoic acid-inducible gene I-like receptors |
| RIG-I | Retinoic acid inducible gene-I |
| MDA5 | Melanoma differentiation-associated gene 5 |
| TLR | Toll like receptor |
| cGAS | GMP-AMP synthase |
| AID | Activation-induced cytidine deaminase |
| ss | Single strand |
| ds | Double strand |
| lnc | Long non coding |
| CRC | Colon rectal cancer |
| AGS | Aicardi-Goutières Syndrome |
| TREX1 | DNA sensing pathways 3′repair exonuclease |
| ADAR1 | Adenosine deaminase acting on RNA |
| DNMTi | DNA methyltransferase inhibitors |
| AML | Acute myeloid leukemia |
| CDK4/6 | Cyclin dependent kinase 4/6 |
| SLE | Systemic lupus erythematosus |
| ALS | Amyothropic latelar sclerosis |
| CIC | Cancer initiating cell |
| MSI | Microsatellite instable |
| MSS | Microsatellite stable |
| HL | Hodgkin Lymphoma |
| GAGE6 | protooncogene G antigen 6 |
References
- De Koning, A.P.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Percharde, M.; Sultana, T.; Ramalho-Santos, M. What Doesn’t Kill You Makes You Stronger: Transposons as Dual Players in Chromatin Regulation and Genomic Variation. Bioessays 2020, 42, e1900232. [Google Scholar] [CrossRef] [PubMed]
- Kazazian, H.H., Jr.; Moran, J.V. The impact of L1 retrotransposons on the human genome. Nat. Genet. 1998, 19, 19–24. [Google Scholar] [CrossRef]
- Viollet, S.; Monot, C.; Cristofari, G. L1 retrotransposition: The snap-velcro model and its consequences. Mob. Genet. Elements 2014, 4, e28907. [Google Scholar] [CrossRef][Green Version]
- Okada, N.; Hamada, M.; Ogiwara, I.; Ohshima, K. SINEs and LINEs share common 3’ sequences: A review. Gene 1997, 205, 229–243. [Google Scholar] [CrossRef]
- Malik, H.S.; Henikoff, S.; Eickbush, T.H. Poised for contagion: Evolutionary origins of the infectious abilities of invertebrate retroviruses. Genome Res. 2000, 10, 1307–1318. [Google Scholar] [CrossRef]
- Canapa, A.; Barucca, M.; Biscotti, M.A.; Forconi, M.; Olmo, E. Transposons, Genome Size, and Evolutionary Insights in Animals. Cytogenet Genome Res. 2015, 147, 217–239. [Google Scholar] [CrossRef]
- Kazazian, H.H., Jr. Mobile elements: Drivers of genome evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Belancio, V.P.; Hedges, D.J.; Deininger, P. Mammalian non-LTR retrotransposons: For better or worse, in sickness and in health. Genome Res. 2008, 18, 343–358. [Google Scholar] [CrossRef]
- Muotri, A.R.; Chu, V.T.; Marchetto, M.C.; Deng, W.; Moran, J.V.; Gage, F.H. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005, 435, 903–910. [Google Scholar] [CrossRef]
- Beck, C.R.; Collier, P.; Macfarlane, C.; Malig, M.; Kidd, J.M.; Eichler, E.E.; Badge, R.M.; Moran, J.V. LINE-1 retrotransposition activity in human genomes. Cell 2010, 141, 1159–1170. [Google Scholar] [CrossRef]
- Ewing, A.D.; Kazazian, H.H., Jr. High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res. 2010, 20, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Iskow, R.C.; McCabe, M.T.; Mills, R.E.; Torene, S.; Pittard, W.S.; Neuwald, A.F.; Van Meir, E.G.; Vertino, P.M.; Devine, S.E. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell 2010, 141, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Baillie, J.K.; Barnett, M.W.; Upton, K.R.; Gerhardt, D.J.; Richmond, T.A.; De Sapio, F.; Brennan, P.M.; Rizzu, P.; Smith, S.; Fell, M.; et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 2011, 479, 534–537. [Google Scholar] [CrossRef]
- Perrat, P.N.; DasGupta, S.; Wang, J.; Theurkauf, W.; Weng, Z.; Rosbash, M.; Waddell, S. Transposition-driven genomic heterogeneity in the Drosophila brain. Science 2013, 340, 91–95. [Google Scholar] [CrossRef]
- Upton, K.R.; Gerhardt, D.J.; Jesuadian, J.S.; Richardson, S.R.; Sanchez-Luque, F.J.; Bodea, G.O.; Ewing, A.D.; Salvador-Palomeque, C.; van der Knaap, M.S.; Brennan, P.M.; et al. Ubiquitous L1 mosaicism in hippocampal neurons. Cell 2015, 161, 228–239. [Google Scholar] [CrossRef]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Yeo, G.W.; Mu, Y.; Lovci, M.T.; Morell, M.; O’Shea, K.S.; Moran, J.V.; Gage, F.H. L1 retrotransposition in human neural progenitor cells. Nature 2009, 460, 1127–1131. [Google Scholar] [CrossRef]
- Reilly, M.T.; Faulkner, G.J.; Dubnau, J.; Ponomarev, I.; Gage, F.H. The role of transposable elements in health and diseases of the central nervous system. J. Neurosci 2013, 33, 17577–17586. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Macia, A.; Muotri, A.R. Transposable Elements, Inflammation, and Neurological Disease. Front. Neurol 2019, 10, 894. [Google Scholar] [CrossRef] [PubMed]
- Payer, L.M.; Burns, K.H. Transposable elements in human genetic disease. Nat. Rev. Genet. 2019, 20, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Deniz, O.; Frost, J.M.; Branco, M.R. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 2019, 20, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Mc, C.B. The origin and behavior of mutable loci in maize. Proc. Natl Acad Sci USA 1950, 36, 344–355. [Google Scholar] [CrossRef]
- McClintock, B. Controlling elements and the gene. Cold Spring Harb Symp Quant. Biol 1956, 21, 197–216. [Google Scholar] [CrossRef]
- Sundaram, V.; Cheng, Y.; Ma, Z.; Li, D.; Xing, X.; Edge, P.; Snyder, M.P.; Wang, T. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014, 24, 1963–1976. [Google Scholar] [CrossRef]
- Bodega, B.; Orlando, V. Repetitive elements dynamics in cell identity programming, maintenance and disease. Curr. Opin. Cell Biol. 2014, 31, 67–73. [Google Scholar] [CrossRef]
- Pennisi, E. Genomics. ENCODE project writes eulogy for junk DNA. Science 2012, 337, 1159–1161. [Google Scholar] [CrossRef]
- Bourque, G.; Leong, B.; Vega, V.B.; Chen, X.; Lee, Y.L.; Srinivasan, K.G.; Chew, J.L.; Ruan, Y.; Wei, C.L.; Ng, H.H.; et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 2008, 18, 1752–1762. [Google Scholar] [CrossRef]
- Imbeault, M.; Helleboid, P.Y.; Trono, D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017, 543, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.D.; Sutherland, H.G.; Martin, D.I.; Whitelaw, E. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 1999, 23, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; de Llobet Cucalon, L.I.; Di Vona, C.; Le Dilly, F.; Vidal, E.; Lioutas, A.; Oliete, J.Q.; Jochem, L.; Cutts, E.; Dieci, G.; et al. TFIIIC Binding to Alu Elements Controls Gene Expression via Chromatin Looping and Histone Acetylation. Mol. Cell 2020, 77, 475–487 e411. [Google Scholar] [CrossRef]
- Schmidt, D.; Schwalie, P.C.; Wilson, M.D.; Ballester, B.; Goncalves, A.; Kutter, C.; Brown, G.D.; Marshall, A.; Flicek, P.; Odom, D.T. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 2012, 148, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, T.; Preissl, S.; Amaral, M.L.; Grinstein, J.D.; Farah, E.N.; Destici, E.; Qiu, Y.; Hu, R.; Lee, A.Y.; et al. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat. Genet. 2019, 51, 1380–1388. [Google Scholar] [CrossRef]
- Faulkner, G.J.; Kimura, Y.; Daub, C.O.; Wani, S.; Plessy, C.; Irvine, K.M.; Schroder, K.; Cloonan, N.; Steptoe, A.L.; Lassmann, T.; et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009, 41, 563–571. [Google Scholar] [CrossRef]
- Rodriguez-Terrones, D.; Hartleben, G.; Gaume, X.; Eid, A.; Guthmann, M.; Iturbide, A.; Torres-Padilla, M.E. A distinct metabolic state arises during the emergence of 2-cell-like cells. EMBO Rep. 2020, 21, e48354. [Google Scholar] [CrossRef]
- Lu, J.Y.; Shao, W.; Chang, L.; Yin, Y.; Li, T.; Zhang, H.; Hong, Y.; Percharde, M.; Guo, L.; Wu, Z.; et al. Genomic Repeats Categorize Genes with Distinct Functions for Orchestrated Regulation. Cell Rep. 2020, 30, 3296–3311 e3295. [Google Scholar] [CrossRef]
- Attig, J.; Agostini, F.; Gooding, C.; Chakrabarti, A.M.; Singh, A.; Haberman, N.; Zagalak, J.A.; Emmett, W.; Smith, C.W.J.; Luscombe, N.M.; et al. Heteromeric RNP Assembly at LINEs Controls Lineage-Specific RNA Processing. Cell 2018, 174, 1067–1081 e1017. [Google Scholar] [CrossRef]
- Nekrutenko, A.; Li, W.H. Transposable elements are found in a large number of human protein-coding genes. Trends Genet. 2001, 17, 619–621. [Google Scholar] [CrossRef]
- Perepelitsa-Belancio, V.; Deininger, P. RNA truncation by premature polyadenylation attenuates human mobile element activity. Nat. Genet. 2003, 35, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Roy-Engel, A.M.; El-Sawy, M.; Farooq, L.; Odom, G.L.; Perepelitsa-Belancio, V.; Bruch, H.; Oyeniran, O.O.; Deininger, P.L. Human retroelements may introduce intragenic polyadenylation signals. Cytogenet Genome Res. 2005, 110, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3’ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Kronenberg, Z.; Lynch, V.J.; Zhuo, X.; Ramsay, L.; Bourque, G.; Yandell, M.; Feschotte, C. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013, 9, e1003470. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.; Rinn, J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012, 13, R107. [Google Scholar] [CrossRef]
- Fort, A.; Hashimoto, K.; Yamada, D.; Salimullah, M.; Keya, C.A.; Saxena, A.; Bonetti, A.; Voineagu, I.; Bertin, N.; Kratz, A.; et al. Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat. Genet. 2014, 46, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Sachs, F.; Ramsay, L.; Jacques, P.E.; Goke, J.; Bourque, G.; Ng, H.H. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat. Struct. Mol. Biol. 2014, 21, 423–425. [Google Scholar] [CrossRef]
- Hall, L.L.; Carone, D.M.; Gomez, A.V.; Kolpa, H.J.; Byron, M.; Mehta, N.; Fackelmayer, F.O.; Lawrence, J.B. Stable C0T-1 repeat RNA is abundant and is associated with euchromatic interphase chromosomes. Cell 2014, 156, 907–919. [Google Scholar] [CrossRef]
- Jachowicz, J.W.; Bing, X.; Pontabry, J.; Boskovic, A.; Rando, O.J.; Torres-Padilla, M.E. LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nat. Genet. 2017, 49, 1502–1510. [Google Scholar] [CrossRef]
- Fadloun, A.; Le Gras, S.; Jost, B.; Ziegler-Birling, C.; Takahashi, H.; Gorab, E.; Carninci, P.; Torres-Padilla, M.E. Chromatin signatures and retrotransposon profiling in mouse embryos reveal regulation of LINE-1 by RNA. Nat. Struct Mol. Biol. 2013, 20, 332–338. [Google Scholar] [CrossRef]
- Percharde, M.; Lin, C.J.; Yin, Y.; Guan, J.; Peixoto, G.A.; Bulut-Karslioglu, A.; Biechele, S.; Huang, B.; Shen, X.; Ramalho-Santos, M. A LINE1-Nucleolin Partnership Regulates Early Development and ESC Identity. Cell 2018, 174, 391–405 e319. [Google Scholar] [CrossRef] [PubMed]
- Hoebe, K.; Janssen, E.; Beutler, B. The interface between innate and adaptive immunity. Nat. Immunol. 2004, 5, 971–974. [Google Scholar] [CrossRef]
- Broecker, F.; Moelling, K. Evolution of Immune Systems From Viruses and Transposable Elements. Front. Microbiol. 2019, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Grandi, N.; Tramontano, E. Human Endogenous Retroviruses Are Ancient Acquired Elements Still Shaping Innate Immune Responses. Front. Immunol. 2018, 9, 2039. [Google Scholar] [CrossRef] [PubMed]
- Kassiotis, G.; Stoye, J.P. Immune responses to endogenous retroelements: Taking the bad with the good. Nat. Rev. Immunol. 2016, 16, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, E.; Thanos, D. Virus Infection Induces NF-kappaB-dependent interchromosomal associations mediating monoallelic IFN-beta gene expression. Cell 2008, 134, 85–96. [Google Scholar] [CrossRef]
- Thomson, S.J.; Goh, F.G.; Banks, H.; Krausgruber, T.; Kotenko, S.V.; Foxwell, B.M.; Udalova, I.A. The role of transposable elements in the regulation of IFN-lambda1 gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11564–11569. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef]
- Lee, H.C.; Chathuranga, K.; Lee, J.S. Intracellular sensing of viral genomes and viral evasion. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Hurst, T.P.; Magiorkinis, G. Activation of the innate immune response by endogenous retroviruses. J. Gen. Virol. 2015, 96, 1207–1218. [Google Scholar] [CrossRef]
- Rolland, A.; Jouvin-Marche, E.; Viret, C.; Faure, M.; Perron, H.; Marche, P.N. The envelope protein of a human endogenous retrovirus-W family activates innate immunity through CD14/TLR4 and promotes Th1-like responses. J. Immunol. 2006, 176, 7636–7644. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Du, J.; Peng, Y.; Li, P.; Wang, S.; Wang, Y.; Hou, J.; Kang, J.; Zheng, W.; Hua, S.; et al. LINE1 contributes to autoimmunity through both RIG-I- and MDA5-mediated RNA sensing pathways. J. Autoimmun 2018, 90, 105–115. [Google Scholar] [CrossRef]
- Williams, B.R. PKR; a sentinel kinase for cellular stress. Oncogene 1999, 18, 6112–6120. [Google Scholar] [CrossRef]
- Chu, W.M.; Ballard, R.; Carpick, B.W.; Williams, B.R.; Schmid, C.W. Potential Alu function: Regulation of the activity of double-stranded RNA-activated kinase PKR. Mol. Cell Biol. 1998, 18, 58–68. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef]
- Zeng, M.; Hu, Z.; Shi, X.; Li, X.; Zhan, X.; Li, X.D.; Wang, J.; Choi, J.H.; Wang, K.W.; Purrington, T.; et al. MAVS, cGAS, and endogenous retroviruses in T-independent B cell responses. Science 2014, 346, 1486–1492. [Google Scholar] [CrossRef] [PubMed]
- Metzner, M.; Jack, H.M.; Wabl, M. LINE-1 retroelements complexed and inhibited by activation induced cytidine deaminase. PLoS ONE 2012, 7, e49358. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Manghera, M.; Ferguson-Parry, J.; Lin, R.; Douville, R.N. NF-kappaB and IRF1 Induce Endogenous Retrovirus K Expression via Interferon-Stimulated Response Elements in Its 5’ Long Terminal Repeat. J. Virol. 2016, 90, 9338–9349. [Google Scholar] [CrossRef] [PubMed]
- Manghera, M.; Douville, R.N. Endogenous retrovirus-K promoter: A landing strip for inflammatory transcription factors? Retrovirology 2013, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, M.H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; von Geldern, G.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Pratt, G.A.; Sundararaman, B.; Townsend, M.J.; Chaivorapol, C.; Bhangale, T.; Graham, R.R.; Ortmann, W.; Criswell, L.A.; Yeo, G.W.; et al. The Ro60 autoantigen binds endogenous retroelements and regulates inflammatory gene expression. Science 2015, 350, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.H.; Gordon, T.P. Autoimmunity: Ro60-associated RNA takes its toll on disease pathogenesis. Nat. Rev. Rheumatol. 2016, 12, 136–138. [Google Scholar] [CrossRef]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef]
- Thomas, C.A.; Tejwani, L.; Trujillo, C.A.; Negraes, P.D.; Herai, R.H.; Mesci, P.; Macia, A.; Crow, Y.J.; Muotri, A.R. Modeling of TREX1-Dependent Autoimmune Disease using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell 2017, 21, 319–331 e318. [Google Scholar] [CrossRef]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997 e924. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef]
- Robbez-Masson, L.; Tie, C.H.C.; Rowe, H.M. Cancer cells, on your histone marks, get SETDB1, silence retrotransposons, and go! J. Cell Biol. 2017, 216, 3429–3431. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef]
- Cuellar, T.L.; Herzner, A.M.; Zhang, X.; Goyal, Y.; Watanabe, C.; Friedman, B.A.; Janakiraman, V.; Durinck, S.; Stinson, J.; Arnott, D.; et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol. 2017, 216, 3535–3549. [Google Scholar] [CrossRef]
- Canadas, I.; Thummalapalli, R.; Kim, J.W.; Kitajima, S.; Jenkins, R.W.; Christensen, C.L.; Campisi, M.; Kuang, Y.; Zhang, Y.; Gjini, E.; et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat. Med. 2018, 24, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; De Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Investig. 2018, 128, 4804–4820. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Solovyov, A.; Vabret, N.; Arora, K.S.; Snyder, A.; Funt, S.A.; Bajorin, D.F.; Rosenberg, J.E.; Bhardwaj, N.; Ting, D.T.; Greenbaum, B.D. Global Cancer Transcriptome Quantifies Repeat Element Polarization between Immunotherapy Responsive and T Cell Suppressive Classes. Cell Rep. 2018, 23, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.J.; Blanchette, C.; Albert, M.L.; et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Lopez, M.; Macia, A.; Garcia-Canadas, M.; Badge, R.M.; Garcia-Perez, J.L. An epi [c] genetic battle: LINE-1 retrotransposons and intragenomic conflict in humans. Mob. Genet. Elements 2011, 1, 122–127. [Google Scholar] [CrossRef]
- Scott, E.C.; Gardner, E.J.; Masood, A.; Chuang, N.T.; Vertino, P.M.; Devine, S.E. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Res. 2016, 26, 745–755. [Google Scholar] [CrossRef]
- Krug, B.; De Jay, N.; Harutyunyan, A.S.; Deshmukh, S.; Marchione, D.M.; Guilhamon, P.; Bertrand, K.C.; Mikael, L.G.; McConechy, M.K.; Chen, C.C.L.; et al. Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27M Gliomas. Cancer Cell 2019, 35, 782–797 e788. [Google Scholar] [CrossRef]
- Jiang, J.C.; Upton, K.R. Human transposons are an abundant supply of transcription factor binding sites and promoter activities in breast cancer cell lines. Mob. DNA 2019, 10, 16. [Google Scholar] [CrossRef]
- Choi, S.H.; Worswick, S.; Byun, H.M.; Shear, T.; Soussa, J.C.; Wolff, E.M.; Douer, D.; Garcia-Manero, G.; Liang, G.; Yang, A.S. Changes in DNA methylation of tandem DNA repeats are different from interspersed repeats in cancer. Int. J. Cancer 2009, 125, 723–729. [Google Scholar] [CrossRef]
- Alves, P.M.; Levy, N.; Stevenson, B.J.; Bouzourene, H.; Theiler, G.; Bricard, G.; Viatte, S.; Ayyoub, M.; Vuilleumier, H.; Givel, J.C.; et al. Identification of tumor-associated antigens by large-scale analysis of genes expressed in human colorectal cancer. Cancer Immun. 2008, 8, 11. [Google Scholar]
- Buscher, K.; Trefzer, U.; Hofmann, M.; Sterry, W.; Kurth, R.; Denner, J. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 2005, 65, 4172–4180. [Google Scholar] [CrossRef] [PubMed]
- Stengel, S.; Fiebig, U.; Kurth, R.; Denner, J. Regulation of human endogenous retrovirus-K expression in melanomas by CpG methylation. Genes Chromosomes Cancer 2010, 49, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Siebenthall, K.T.; Miller, C.P.; Vierstra, J.D.; Mathieu, J.; Tretiakova, M.; Reynolds, A.; Sandstrom, R.; Rynes, E.; Haugen, E.; Johnson, A.; et al. Integrated epigenomic profiling reveals endogenous retrovirus reactivation in renal cell carcinoma. EBioMedicine 2019, 41, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Radvanyi, L.; Yin, B.; Rycaj, K.; Li, J.; Chivukula, R.; Lin, K.; Lu, Y.; Shen, J.; Chang, D.Z.; et al. Downregulation of Human Endogenous Retrovirus Type K (HERV-K) Viral env RNA in Pancreatic Cancer Cells Decreases Cell Proliferation and Tumor Growth. Clin. Cancer Res. 2017, 23, 5892–5911. [Google Scholar] [CrossRef]
- Yandim, C.; Karakulah, G. Dysregulated expression of repetitive DNA in ER+/HER2- breast cancer. Cancer Genet. 2019, 239, 36–45. [Google Scholar] [CrossRef]
- Wang-Johanning, F.; Liu, J.; Rycaj, K.; Huang, M.; Tsai, K.; Rosen, D.G.; Chen, D.T.; Lu, D.W.; Barnhart, K.F.; Johanning, G.L. Expression of multiple human endogenous retrovirus surface envelope proteins in ovarian cancer. Int. J. Cancer 2007, 120, 81–90. [Google Scholar] [CrossRef]
- Clayton, E.A.; Wang, L.; Rishishwar, L.; Wang, J.; McDonald, J.F.; Jordan, I.K. Patterns of Transposable Element Expression and Insertion in Cancer. Front. Mol. Biosci 2016, 3, 76. [Google Scholar] [CrossRef]
- Desai, N.; Sajed, D.; Arora, K.S.; Solovyov, A.; Rajurkar, M.; Bledsoe, J.R.; Sil, S.; Amri, R.; Tai, E.; MacKenzie, O.C.; et al. Diverse repetitive element RNA expression defines epigenetic and immunologic features of colon cancer. JCI Insight 2017, 2, e91078. [Google Scholar] [CrossRef]
- Wolff, E.M.; Byun, H.M.; Han, H.F.; Sharma, S.; Nichols, P.W.; Siegmund, K.D.; Yang, A.S.; Jones, P.A.; Liang, G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010, 6, e1000917. [Google Scholar] [CrossRef]
- Anwar, S.L.; Wulaningsih, W.; Lehmann, U. Transposable Elements in Human Cancer: Causes and Consequences of Deregulation. Int. J. Mol. Sci. 2017, 18, 974. [Google Scholar] [CrossRef] [PubMed]
- Oricchio, E.; Sciamanna, I.; Beraldi, R.; Tolstonog, G.V.; Schumann, G.G.; Spadafora, C. Distinct roles for LINE-1 and HERV-K retroelements in cell proliferation, differentiation and tumor progression. Oncogene 2007, 26, 4226–4233. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Sunami, E.; Yamamoto, Y.; Hata, K.; Okada, S.; Murono, K.; Yasuda, K.; Otani, K.; Nishikawa, T.; Tanaka, T.; et al. LINE-1 hypomethylation status of circulating cell-free DNA in plasma as a biomarker for colorectal cancer. Oncotarget 2017, 8, 11906–11916. [Google Scholar] [CrossRef] [PubMed]
- Strissel, P.L.; Ruebner, M.; Thiel, F.; Wachter, D.; Ekici, A.B.; Wolf, F.; Thieme, F.; Ruprecht, K.; Beckmann, M.W.; Strick, R. Reactivation of codogenic endogenous retroviral (ERV) envelope genes in human endometrial carcinoma and prestages: Emergence of new molecular targets. Oncotarget 2012, 3, 1204–1219. [Google Scholar] [CrossRef]
- Babaian, A.; Romanish, M.T.; Gagnier, L.; Kuo, L.Y.; Karimi, M.M.; Steidl, C.; Mager, D.L. Onco-exaptation of an endogenous retroviral LTR drives IRF5 expression in Hodgkin lymphoma. Oncogene 2016, 35, 2542–2546. [Google Scholar] [CrossRef]
- Jang, H.S.; Shah, N.M.; Du, A.Y.; Dailey, Z.Z.; Pehrsson, E.C.; Godoy, P.M.; Zhang, D.; Li, D.; Xing, X.; Kim, S.; et al. Transposable elements drive widespread expression of oncogenes in human cancers. Nat. Genet. 2019, 51, 611–617. [Google Scholar] [CrossRef]
- Cruickshanks, H.A.; Tufarelli, C. Isolation of cancer-specific chimeric transcripts induced by hypomethylation of the LINE-1 antisense promoter. Genomics 2009, 94, 397–406. [Google Scholar] [CrossRef]
- Cruickshanks, H.A.; Vafadar-Isfahani, N.; Dunican, D.S.; Lee, A.; Sproul, D.; Lund, J.N.; Meehan, R.R.; Tufarelli, C. Expression of a large LINE-1-driven antisense RNA is linked to epigenetic silencing of the metastasis suppressor gene TFPI-2 in cancer. Nucleic Acids Res. 2013, 41, 6857–6869. [Google Scholar] [CrossRef]
- Scarfo, I.; Pellegrino, E.; Mereu, E.; Kwee, I.; Agnelli, L.; Bergaggio, E.; Garaffo, G.; Vitale, N.; Caputo, M.; Machiorlatti, R.; et al. Identification of a new subclass of ALK-negative ALCL expressing aberrant levels of ERBB4 transcripts. Blood 2016, 127, 221–232. [Google Scholar] [CrossRef]
- Lock, F.E.; Rebollo, R.; Miceli-Royer, K.; Gagnier, L.; Kuah, S.; Babaian, A.; Sistiaga-Poveda, M.; Lai, C.B.; Nemirovsky, O.; Serrano, I.; et al. Distinct isoform of FABP7 revealed by screening for retroelement-activated genes in diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E3534–E3543. [Google Scholar] [CrossRef]
- Miki, Y.; Nishisho, I.; Horii, A.; Miyoshi, Y.; Utsunomiya, J.; Kinzler, K.W.; Vogelstein, B.; Nakamura, Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992, 52, 643–645. [Google Scholar] [PubMed]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Aschacher, T.; Wolf, B.; Enzmann, F.; Kienzl, P.; Messner, B.; Sampl, S.; Svoboda, M.; Mechtcheriakova, D.; Holzmann, K.; Bergmann, M. LINE-1 induces hTERT and ensures telomere maintenance in tumour cell lines. Oncogene 2016, 35, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhao, Z. Binding of LINE-1 RNA to PSF transcriptionally promotes GAGE6 and regulates cell proliferation and tumor formation in vitro. Exp. Ther. Med. 2017, 14, 1685–1691. [Google Scholar] [CrossRef]
- Colombo, A.R.; Zubair, A.; Thiagarajan, D.; Nuzhdin, S.; Triche, T.J.; Ramsingh, G. Suppression of Transposable Elements in Leukemic Stem Cells. Sci. Rep. 2017, 7, 7029. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, J.Q.; Zheng, S. Expressional activation and functional roles of human endogenous retroviruses in cancers. Rev. Med. Virol. 2019, 29, e2025. [Google Scholar] [CrossRef]
- Li, H.; Jiang, X.; Niu, X. Long Non-Coding RNA Reprogramming (ROR) Promotes Cell Proliferation in Colorectal Cancer via Affecting P53. Med. Sci. Monit. 2017, 23, 919–928. [Google Scholar] [CrossRef]
- Li, L.; Feng, T.; Lian, Y.; Zhang, G.; Garen, A.; Song, X. Role of human noncoding RNAs in the control of tumorigenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12956–12961. [Google Scholar] [CrossRef]
- Wang, G.; Cui, Y.; Zhang, G.; Garen, A.; Song, X. Regulation of proto-oncogene transcription, cell proliferation, and tumorigenesis in mice by PSF protein and a VL30 noncoding RNA. Proc. Natl. Acad. Sci. USA 2009, 106, 16794–16798. [Google Scholar] [CrossRef]
- Xu, L.; Elkahloun, A.G.; Candotti, F.; Grajkowski, A.; Beaucage, S.L.; Petricoin, E.F.; Calvert, V.; Juhl, H.; Mills, F.; Mason, K.; et al. A novel function of RNAs arising from the long terminal repeat of human endogenous retrovirus 9 in cell cycle arrest. J. Virol. 2013, 87, 25–36. [Google Scholar] [CrossRef]
- Benatti, P.; Basile, V.; Merico, D.; Fantoni, L.I.; Tagliafico, E.; Imbriano, C. A balance between NF-Y and p53 governs the pro- and anti-apoptotic transcriptional response. Nucleic Acids Res. 2008, 36, 1415–1428. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Galindo, R.; Kaplan, M.H.; Leissner, P.; Verjat, T.; Ferlenghi, I.; Bagnoli, F.; Giusti, F.; Dosik, M.H.; Hayes, D.F.; Gitlin, S.D.; et al. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J. Virol. 2008, 82, 9329–9336. [Google Scholar] [CrossRef] [PubMed]
- Seifarth, W.; Skladny, H.; Krieg-Schneider, F.; Reichert, A.; Hehlmann, R.; Leib-Mosch, C. Retrovirus-like particles released from the human breast cancer cell line T47-D display type B- and C-related endogenous retroviral sequences. J. Virol. 1995, 69, 6408–6416. [Google Scholar] [CrossRef] [PubMed]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 180. [Google Scholar] [CrossRef] [PubMed]
- Mangeney, M.; Renard, M.; Schlecht-Louf, G.; Bouallaga, I.; Heidmann, O.; Letzelter, C.; Richaud, A.; Ducos, B.; Heidmann, T. Placental syncytins: Genetic disjunction between the fusogenic and immunosuppressive activity of retroviral envelope proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 20534–20539. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 2011, 13, 36–46. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Robert, C.; Watson, M. Errors in RNA-Seq quantification affect genes of relevance to human disease. Genome Biol. 2015, 16, 177. [Google Scholar] [CrossRef]
- Kahles, A.; Behr, J.; Ratsch, G. MMR: A tool for read multi-mapper resolution. Bioinformatics 2016, 32, 770–772. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Lassmann, T.; Murata, M.; Carninci, P. 5’ end-centered expression profiling using cap-analysis gene expression and next-generation sequencing. Nat. Protoc. 2012, 7, 542–561. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, G.J.; Forrest, A.R.; Chalk, A.M.; Schroder, K.; Hayashizaki, Y.; Carninci, P.; Hume, D.A.; Grimmond, S.M. A rescue strategy for multimapping short sequence tags refines surveys of transcriptional activity by CAGE. Genomics 2008, 91, 281–288. [Google Scholar] [CrossRef][Green Version]
- McKerrow, W.; Fenyo, D. L1EM: A tool for accurate locus specific LINE-1 RNA quantification. Bioinformatics 2020, 36, 1167–1173. [Google Scholar] [CrossRef]
- Babaian, A.; Thompson, I.R.; Lever, J.; Gagnier, L.; Karimi, M.M.; Mager, D.L. LIONS: Analysis suite for detecting and quantifying transposable element initiated transcription from RNA-seq. Bioinformatics 2019, 35, 3839–3841. [Google Scholar] [CrossRef]
- Criscione, S.W.; Zhang, Y.; Thompson, W.; Sedivy, J.M.; Neretti, N. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genom. 2014, 15, 583. [Google Scholar] [CrossRef]
- Jeong, H.H.; Yalamanchili, H.K.; Guo, C.; Shulman, J.M.; Liu, Z. An ultra-fast and scalable quantification pipeline for transposable elements from next generation sequencing data. Pac. Symp. Biocomput. 2018, 23, 168–179. [Google Scholar]
- Yang, W.R.; Ardeljan, D.; Pacyna, C.N.; Payer, L.M.; Burns, K.H. SQuIRE reveals locus-specific regulation of interspersed repeat expression. Nucleic Acids Res. 2019, 47, e27. [Google Scholar] [CrossRef]
- Valdebenito-Maturana, B.; Riadi, G. TEcandidates: Prediction of genomic origin of expressed transposable elements using RNA-seq data. Bioinformatics 2018, 34, 3915–3916. [Google Scholar] [CrossRef] [PubMed]
- Bendall, M.L.; de Mulder, M.; Iniguez, L.P.; Lecanda-Sanchez, A.; Perez-Losada, M.; Ostrowski, M.A.; Jones, R.B.; Mulder, L.C.F.; Reyes-Teran, G.; Crandall, K.A.; et al. Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLoS Comput. Biol. 2019, 15, e1006453. [Google Scholar] [CrossRef] [PubMed]
- Lerat, E.; Fablet, M.; Modolo, L.; Lopez-Maestre, H.; Vieira, C. TEtools facilitates big data expression analysis of transposable elements and reveals an antagonism between their activity and that of piRNA genes. Nucleic Acids Res. 2017, 45, e17. [Google Scholar] [CrossRef]
- Jin, Y.; Tam, O.H.; Paniagua, E.; Hammell, M. TEtranscripts: A package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 2015, 31, 3593–3599. [Google Scholar] [CrossRef] [PubMed]
- Navarro, F.C.; Hoops, J.; Bellfy, L.; Cerveira, E.; Zhu, Q.; Zhang, C.; Lee, C.; Gerstein, M.B. TeXP: Deconvolving the effects of pervasive and autonomous transcription of transposable elements. PLoS Comput. Biol. 2019, 15, e1007293. [Google Scholar] [CrossRef]
- Jung, H.; Choi, J.K.; Lee, E.A. Immune signatures correlate with L1 retrotransposition in gastrointestinal cancers. Genome Res. 2018, 28, 1136–1146. [Google Scholar] [CrossRef]
- Cebria-Costa, J.P.; Pascual-Reguant, L.; Gonzalez-Perez, A.; Serra-Bardenys, G.; Querol, J.; Cosin, M.; Verde, G.; Cigliano, R.A.; Sanseverino, W.; Segura-Bayona, S.; et al. LOXL2-mediated H3K4 oxidation reduces chromatin accessibility in triple-negative breast cancer cells. Oncogene 2020, 39, 79–121. [Google Scholar] [CrossRef]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2015. [Google Scholar]
- Deininger, P.; Morales, M.E.; White, T.B.; Baddoo, M.; Hedges, D.J.; Servant, G.; Srivastav, S.; Smither, M.E.; Concha, M.; DeHaro, D.L.; et al. A comprehensive approach to expression of L1 loci. Nucleic Acids Res. 2017, 45, e31. [Google Scholar] [CrossRef]
- Zhou, W.; Emery, S.B.; Flasch, D.A.; Wang, Y.; Kwan, K.Y.; Kidd, J.M.; Moran, J.V.; Mills, R.E. Identification and characterization of occult human-specific LINE-1 insertions using long-read sequencing technology. Nucleic Acids Res. 2020, 48, 1146–1163. [Google Scholar] [CrossRef]
- Istace, B.; Friedrich, A.; d’Agata, L.; Faye, S.; Payen, E.; Beluche, O.; Caradec, C.; Davidas, S.; Cruaud, C.; Liti, G.; et al. de novo assembly and population genomic survey of natural yeast isolates with the Oxford Nanopore MinION sequencer. Gigascience 2017, 6, 1–13. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Resolution | TE Specificity | Detection of Active Transcription | Method Description | Reference |
|---|---|---|---|---|---|
| REdiscoverTE | Subfamily | All | Yes (Intergenic TEs are classified as autonomously transcribed) | Pseudo-alignment on a transcriptome of cDNA and individual genomic loci. | [95] |
| L1EM | Locus-level | LINE1 | Yes | Categorizes L1 loci by the presence of promoter and polyA tail; EM-based quantification. | [146] |
| LIONS | Locus-level | TEs initiating transcripts | No | Identify and quantify TE-initiated transcripts based on read coverage on de-novo reconstructed exons and around TEs. | [147] |
| RepEnrich | Subfamily | All | No | Non-spliced alignment on a pseudo-genome of repeats sequences. | [148] |
| SalmonTE | Subfamily | All | No | Pseudo-alignment on TE consensus sequences. | [149] |
| SQuIRE | Locus-level | All | No | Spliced alignment followed by EM-based locus-level quantification. | [150] |
| TEcandidates | Locus-level | All | No | Alignment of de novo assembled contigs of TE-derived reads to the reference genome. | [151] |
| Telescope | Locus-level | All | No | Reassignment of multi-reads to the most probable source of transcript. | [152] |
| TEtools | Subfamily | All | No | Reference-free alignment on a provided set of TE sequences. | [153] |
| TEtranscripts | Subfamily | All | No | EM-based re-distribution of pre-aligned multi-reads. | [154] |
| TeXP | Subfamily | All | Yes | Removes noise derived from non-autonomous transcription of TEs. | [155] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marasca, F.; Gasparotto, E.; Polimeni, B.; Vadalà, R.; Ranzani, V.; Bodega, B. The Sophisticated Transcriptional Response Governed by Transposable Elements in Human Health and Disease. Int. J. Mol. Sci. 2020, 21, 3201. https://doi.org/10.3390/ijms21093201
Marasca F, Gasparotto E, Polimeni B, Vadalà R, Ranzani V, Bodega B. The Sophisticated Transcriptional Response Governed by Transposable Elements in Human Health and Disease. International Journal of Molecular Sciences. 2020; 21(9):3201. https://doi.org/10.3390/ijms21093201
Chicago/Turabian StyleMarasca, Federica, Erica Gasparotto, Benedetto Polimeni, Rebecca Vadalà, Valeria Ranzani, and Beatrice Bodega. 2020. "The Sophisticated Transcriptional Response Governed by Transposable Elements in Human Health and Disease" International Journal of Molecular Sciences 21, no. 9: 3201. https://doi.org/10.3390/ijms21093201
APA StyleMarasca, F., Gasparotto, E., Polimeni, B., Vadalà, R., Ranzani, V., & Bodega, B. (2020). The Sophisticated Transcriptional Response Governed by Transposable Elements in Human Health and Disease. International Journal of Molecular Sciences, 21(9), 3201. https://doi.org/10.3390/ijms21093201