Mechanisms of ATII-to-ATI Cell Differentiation during Lung Regeneration


{kind=link}
Abstract
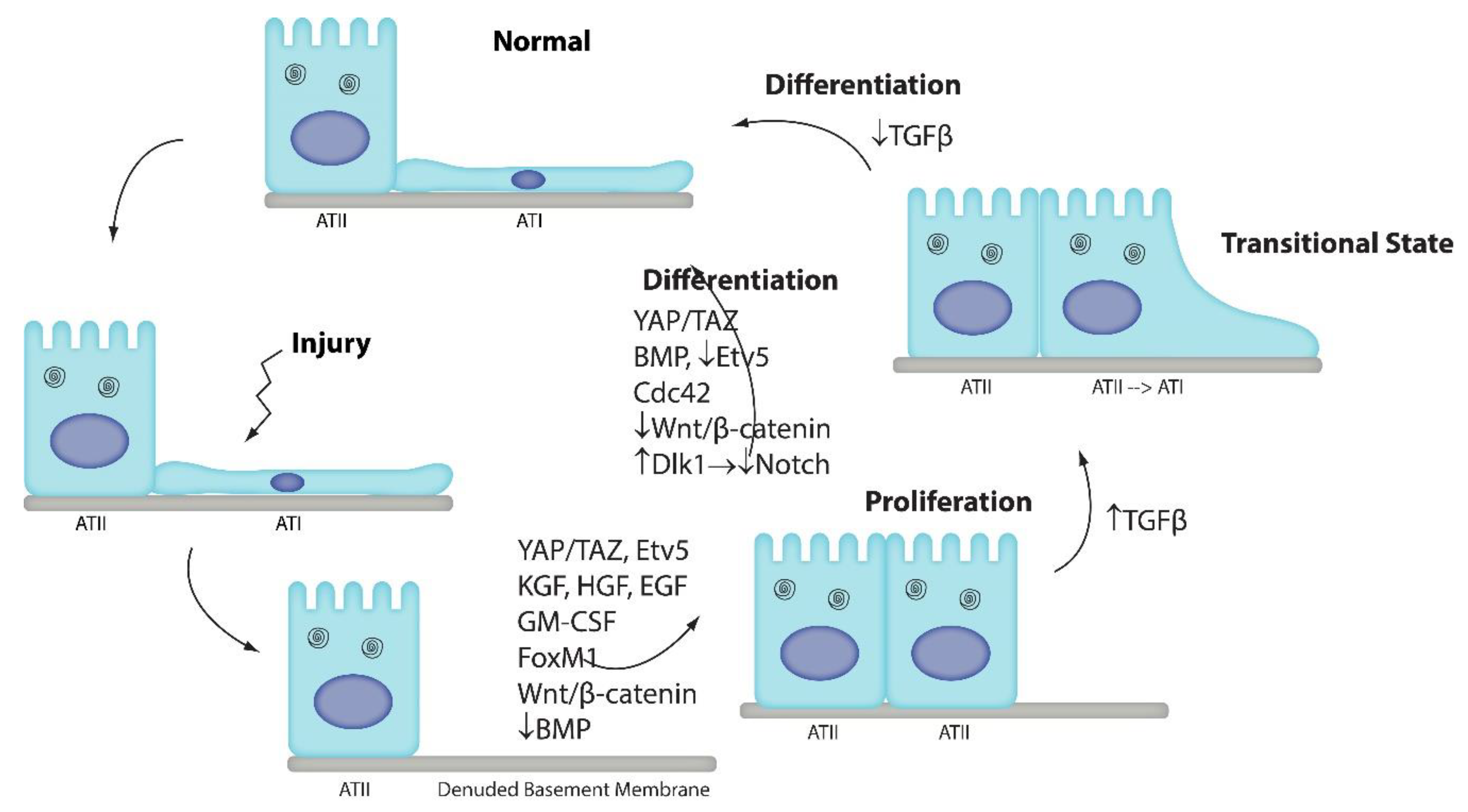
1. Introduction
2. Wnt/β-Catenin Signaling
3. Notch Signaling
4. BMP/SMAD Signaling
5. Yap/Taz Signaling
6. Recruited Macrophages
7. Cdc42
8. Etv5 Signaling
9. TGFβ
10. Limitations
11. Future Directions
12. Conclusions
Funding
Conflicts of Interest
References
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, L.G.; Johnson, M.D.; Vanderbilt, J.; Allen, L.; Gonzalez, R. The great big alveolar TI cell: Evolving concepts and paradigms. Cell Physiol. Biochem. 2010, 25, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.P.; Wrede, C.; Hegermann, J.; Weibel, E.R.; Mühlfeld, C.; Ochs, M. On the Topological Complexity of Human Alveolar Epithelial Type 1 Cells. Am. J Respir Crit. Care Med. 2019, 199, 1153–1156. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Cabral, L.J.; Stephens, R.J.; Freeman, G. Transformation of alveolar type 2 cells to type 1 cells following exposure to NO2. Exp. Mol. Pathol. 1975, 22, 142–150. [Google Scholar] [CrossRef]
- Evans, M.J.; Dekker, N.P.; Cabral-Anderson, L.J.; Freeman, G. Quantitation of damage to the alveolar epithelium by means of type 2 cell proliferation. Am. Rev. Respir Dis. 1978, 118, 787–790. [Google Scholar] [CrossRef]
- Evans, M.J.; Cabral, L.J.; Stephens, R.J.; Freeman, G. Renewal of alveolar epithelium in the rat following exposure to NO2. Am. J. Pathol. 1973, 70, 175–198. [Google Scholar]
- Kathiriya, J.J.; Brumwell, A.N.; Jackson, J.R.; Tang, X.; Chapman, H.A. Distinct Airway Epithelial Stem Cells Hide among Club Cells but Mobilize to Promote Alveolar Regeneration. Cell Stem Cell 2020, 26, 346–358. [Google Scholar] [CrossRef]
- Vaughan, A.E.; Brumwell, A.N.; Xi, Y.; Gotts, J.E.; Brownfield, D.G.; Treutlein, B.; Tan, K.; Tan, V.; Liu, F.C.; Looney, M.R.; et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 2015, 517, 621–625. [Google Scholar] [CrossRef]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef]
- Liu, Y.; Sadikot, R.T.; Adami, G.R.; Kalinichenko, V.V.; Pendyala, S.; Natarajan, V.; Zhao, Y.Y.; Malik, A.B. FoxM1 mediates the progenitor function of type II epithelial cells in repairing alveolar injury induced by Pseudomonas aeruginosa. J. Exp. Med. 2011, 208, 1473–1484. [Google Scholar] [CrossRef]
- Mock, J.R.; Dial, C.F.; D’Alessio, F.R.; Doerschuk, C.M. Foxp3+ Regulatory T Cell Expression Of Keratinocyte Growth Factor During Acute Lung Injury Resolution. Am. J. Respir Crit. Care Med. 2016, 193, A6143. [Google Scholar]
- Rafii, S.; Cao, Z.; Lis, R.; Siempos, I.I.; Chavez, D.; Shido, K.; Rabbany, S.Y.; Ding, B.S. Platelet-derived SDF-1 primes the pulmonary capillary vascular niche to drive lung alveolar regeneration. Nat. Cell Biol. 2015, 17, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Zemans, R.L.; Briones, N.; Campbell, M.; McClendon, J.; Young, S.K.; Suzuki, T.; Yang, I.V.; De Langhe, S.; Reynolds, S.D.; Mason, R.J.; et al. Neutrophil transmigration triggers repair of the lung epithelium via beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 15990–15995. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef]
- Panos, R.J.; Rubin, J.S.; Csaky, K.G.; Aaronson, S.A.; Mason, R.J. Keratinocyte growth factor and hepatocyte growth factor/scatter factor are heparin-binding growth factors for alveolar type II cells in fibroblast-conditioned medium. J. Clin. Invest. 1993, 92, 969–977. [Google Scholar] [CrossRef]
- Mason, R.J.; Leslie, C.C.; McCormick-Shannon, K.; Deterding, R.R.; Nakamura, T.; Rubin, J.S.; Shannon, J.M. Hepatocyte growth factor is a growth factor for rat alveolar type II cells. Am. J. Respir Cell Mol. Biol. 1994, 11, 561–567. [Google Scholar] [CrossRef]
- Fehrenbach, H.; Kasper, M.; Tschernig, T.; Pan, T.; Schuh, D.; Shannon, J.M.; Muller, M.; Mason, R.J. Keratinocyte growth factor-induced hyperplasia of rat alveolar type II cells in vivo is resolved by differentiation into type I cells and by apoptosis. Eur. Respir J. 1999, 14, 534–544. [Google Scholar] [CrossRef][Green Version]
- Ding, B.S.; Nolan, D.J.; Guo, P.; Babazadeh, A.O.; Cao, Z.; Rosenwaks, Z.; Crystal, R.G.; Simons, M.; Sato, T.N.; Worgall, S.; et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell 2011, 147, 539–553. [Google Scholar] [CrossRef]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef]
- Jansing, N.L.; McClendon, J.; Henson, P.M.; Tuder, R.M.; Hyde, D.M.; Zemans, R.L. Unbiased Quantitation of ATII to ATI Cell Transdifferentiation During Repair After Lung Injury in Mice. Am. J. Respir Cell Mol. Biol. 2017, 57, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.; Sottoriva, K.; Pajcini, K.V.; Kitajewski, J.K.; Chen, C.; Zhang, W.; Malik, A.B.; Liu, Y. Dlk1-Mediated Temporal Regulation of Notch Signaling Is Required for Differentiation of Alveolar Type II to Type I Cells during Repair. Cell Rep. 2019, 26, 2942–2954. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.I.; Bujnis, M.; Barkauskas, C.E.; Kobayashi, Y.; Hogan, B.L.M. Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation. Development 2018, 145, dev163014. [Google Scholar] [CrossRef] [PubMed]
- LaCanna, R.; Liccardo, D.; Zhang, P.; Tragesser, L.; Wang, Y.; Cao, T.; Chapman, H.A.; Morrisey, E.E.; Shen, H.; Koch, W.J.; et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J. Clin. Invest. 2019, 130, 2107–2122. [Google Scholar] [CrossRef]
- Sun, T.; Huang, Z.; Zhang, H.; Posner, C.; Jia, G.; Ramalingam, T.R.; Xu, M.; Brightbill, H.; Egen, J.G.; Dey, A.; et al. TAZ is required for lung alveolar epithelial cell differentiation after injury. JCI Insigh 2019, 5. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, H.; Jiang, K.; Wang, Y.; Zhang, W.; Chu, Q.; Li, J.; Huang, H.; Cai, T.; Ji, H.; et al. MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep. 2016, 16, 1810–1819. [Google Scholar] [CrossRef]
- Zhou, B.; Flodby, P.; Luo, J.; Castillo, D.R.; Liu, Y.; Yu, F.X.; McConnell, A.; Varghese, B.; Li, G.; Chimge, N.O.; et al. Claudin-18-mediated YAP activity regulates lung stem and progenitor cell homeostasis and tumorigenesis. J. Clin. Invest. 2018, 128, 970–984. [Google Scholar] [CrossRef]
- Mould, K.J.; Jackson, N.D.; Henson, P.M.; Seibold, M.; Janssen, W.J. Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight. 2019, 4, e126556. [Google Scholar] [CrossRef]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- Moore, B.B.; Paine, R.; Christensen, P.J.; Moore, T.A.; Sitterding, S.; Ngan, R.; Wilke, C.A.; Kuziel, W.A.; Toews, G.B. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J. Immunol. 2001, 167, 4368–4377. [Google Scholar] [CrossRef]
- Herold, S.; Steinmueller, M.; von Wulffen, W.; Cakarova, L.; Pinto, R.; Pleschka, S.; Mack, M.; Kuziel, W.A.; Corazza, N.; Brunner, T.; et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 2008, 205, 3065–3077. [Google Scholar] [CrossRef] [PubMed]
- Lechner, A.J.; Driver, I.H.; Lee, J.; Conroy, C.M.; Nagle, A.; Locksley, R.M.; Rock, J.R. Recruited Monocytes and Type 2 Immunity Promote Lung Regeneration following Pneumonectomy. Cell Stem Cell 2017, 21, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Cakarova, L.; Marsh, L.M.; Wilhelm, J.; Mayer, K.; Grimminger, F.; Seeger, W.; Lohmeyer, J.; Herold, S. Macrophage tumor necrosis factor-alpha induces epithelial expression of granulocyte-macrophage colony-stimulating factor: impact on alveolar epithelial repair. Am. J. Respir Crit. Care Med. 2009, 180, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.-Y.; Sen, D.; Oniskey, T.K.; Katzen, J.; Cohen, N.A.; Vaughan, A.E.; Nieves, W.; Urisman, A.; Beers, M.F.; Krummel, M.F.; et al. Macrophages promote epithelial proliferation following infectious and non-infectious lung injury through a Trefoil factor 2-dependent mechanism. Mucosal Immunology 2019, 12, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, H.; Crawford, H.C.; Hogan, B.L. Role for ETS domain transcription factors Pea3/Erm in mouse lung development. Dev. Biol. 2003, 261, 10–24. [Google Scholar] [CrossRef]
- Zhang, Z.; Newton, K.; Kummerfeld, S.K.; Webster, J.; Kirkpatrick, D.S.; Phu, L.; Eastham-Anderson, J.; Liu, J.; Lee, W.P.; Wu, J.; et al. Transcription factor Etv5 is essential for the maintenance of alveolar type II cells. Proc. Natl. Acad. Sci. USA 2017, 114, 3903–3908. [Google Scholar] [CrossRef]
- Riemondy, K.A.; Jansing, N.L.; Jiang, P.; Redente, E.F.; Gillen, A.E.; Fu, R.; Miller, A.J.; Spence, J.R.; Gerber, A.N.; Hesselberth, J.R.; et al. Single cell RNA sequencing identifies TGFβ as a key regenerative cue following LPS-induced lung injury. JCI Insight. 2019, 4, 123637. [Google Scholar] [CrossRef]
- Strunz, M.; Simon, L.M.; Ansari, M.; Mattner, L.F.; Angelidis, I.; Mayr, C.H.; Kathiriya, J.; Yee, M.; Ogar, P.; Sengupta, A.; et al. Longitudinal single cell transcriptomics reveals Krt8+ alveolar epithelial progenitors in lung regeneration. bioRxiv 2019. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a novel regeneration-associated transitional cell state in pulmonary fibrosis. biorxiv 2020. [Google Scholar] [CrossRef]
- Joshi, N.; Watanabe, S.; Verma, R.; Jablonski, R.P.; Chen, C.I.; Cheresh, P.; Markov, N.S.; Reyfman, P.A.; McQuattie-Pimentel, A.C.; Sichizya, L.; et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur. Respir J. 2020, 55, 1900646. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gil de Rubio, R.; Hrycaj, S.M.; Gurczynski, S.J.; Riemondy, K.A.; Moore, B.B.; Omary, M.B.; Ridge, K.M.; Zemans, R.L. Ineffectual AEC2-to-AEC1 Differentiation in IPF: Persistence of KRT8. Am. J. Respir Crit. Care Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jansing, N.L.; Patel, N.; McClendon, J.; Redente, E.F.; Henson, P.M.; Tuder, R.M.; Hyde, D.M.; Nyengaard, J.R.; Zemans, R.L. Flow Cytometry Underestimates and Planimetry Overestimates Alveolar Epithelial Type 2 Cell Expansion after Lung Injury. Am. J. Respir Crit. Care Med. 2018, 198, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Borok, Z.; Danto, S.I.; Lubman, R.L.; Cao, Y.; Williams, M.C.; Crandall, E.D. Modulation of t1alpha expression with alveolar epithelial cell phenotype in vitro. Am. J. Physiol. 1998, 275, L155–L164. [Google Scholar]
- Marconett, C.N.; Zhou, B.; Rieger, M.E.; Selamat, S.A.; Dubourd, M.; Fang, X.; Lynch, S.K.; Stueve, T.R.; Siegmund, K.D.; Berman, B.P.; et al. Integrated transcriptomic and epigenomic analysis of primary human lung epithelial cell differentiation. PLoS Genet 2013, 9, e1003513. [Google Scholar] [CrossRef]
- Wang, J.; Edeen, K.; Manzer, R.; Chang, Y.; Wang, S.; Chen, X.; Funk, C.J.; Cosgrove, G.P.; Fang, X.; Mason, R.J. Differentiated human alveolar epithelial cells and reversibility of their phenotype in vitro. Am. J. Respir Cell Mol. Biol. 2007, 36, 661–668. [Google Scholar] [CrossRef]
- Correll, K.A.; Edeen, K.E.; Zemans, R.L.; Redente, E.F.; Serban, K.A.; Curran-Everett, D.; Edelman, B.L.; Mikels-Vigdal, A.; Mason, R.J. Transitional human alveolar type II epithelial cells suppress extracellular matrix and growth factor gene expression in lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L283–L294. [Google Scholar] [CrossRef]
- Mock, J.R.; Garibaldi, B.T.; Aggarwal, N.R.; Jenkins, J.; Limjunyawong, N.; Singer, B.D.; Chau, E.; Rabold, R.; Files, D.C.; Sidhaye, V.; et al. Foxp3+ regulatory T cells promote lung epithelial proliferation. Mucosal Immunol. 2014, 7, 1440–1451. [Google Scholar] [CrossRef]
- Dial, C.F.; Tune, M.K.; Doerschuk, C.M.; Mock, J.R. Foxp3+ Regulatory T Cell Expression of Keratinocyte Growth Factor Enhances Lung Epithelial Proliferation. Am. J. Respir Cell Mol. Biol. 2017, 57, 162–173. [Google Scholar] [CrossRef]
- Kosmider, B.; Messier, E.M.; Janssen, W.J.; Nahreini, P.; Wang, J.; Hartshorn, K.L.; Mason, R.J. Nrf2 protects human alveolar epithelial cells against injury induced by influenza A virus. Respir Res. 2012, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, W.J.; Morrisey, E.E. Isolation and culture of human alveolar epithelial progenitor cells. Protoc. Exch. 2018. [Google Scholar] [CrossRef]
- Alsafadi, H.N.; Uhl, F.E.; Pineda, R.H.; Bailey, K.E.; Rojas, M.; Wagner, D.E.; Königshoff, M. Applications and Approaches for 3D Precision-cut Lung Slices: Disease Modeling and Drug Discovery. Am. J. Respir Cell Mol. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Leslie, C.C.; McCormick-Shannon, K.; Shannon, J.M.; Garrick, B.; Damm, D.; Abraham, J.A.; Mason, R.J. Heparin-binding EGF-like growth factor is a mitogen for rat alveolar type II cells. Am. J. Respir Cell Mol. Biol. 1997, 16, 379–387. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aspal, M.; Zemans, R.L. Mechanisms of ATII-to-ATI Cell Differentiation during Lung Regeneration. Int. J. Mol. Sci. 2020, 21, 3188. https://doi.org/10.3390/ijms21093188
Aspal M, Zemans RL. Mechanisms of ATII-to-ATI Cell Differentiation during Lung Regeneration. International Journal of Molecular Sciences. 2020; 21(9):3188. https://doi.org/10.3390/ijms21093188
Chicago/Turabian StyleAspal, Mohit, and Rachel L. Zemans. 2020. "Mechanisms of ATII-to-ATI Cell Differentiation during Lung Regeneration" International Journal of Molecular Sciences 21, no. 9: 3188. https://doi.org/10.3390/ijms21093188
APA StyleAspal, M., & Zemans, R. L. (2020). Mechanisms of ATII-to-ATI Cell Differentiation during Lung Regeneration. International Journal of Molecular Sciences, 21(9), 3188. https://doi.org/10.3390/ijms21093188