PACAP Modulates the Autophagy Process in an In Vitro Model of Amyotrophic Lateral Sclerosis


 ,
,  ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. PACAP Effect on Human Wild-Type-SOD1 (WT) and G93A Cell Viability after Hypoxic Insult
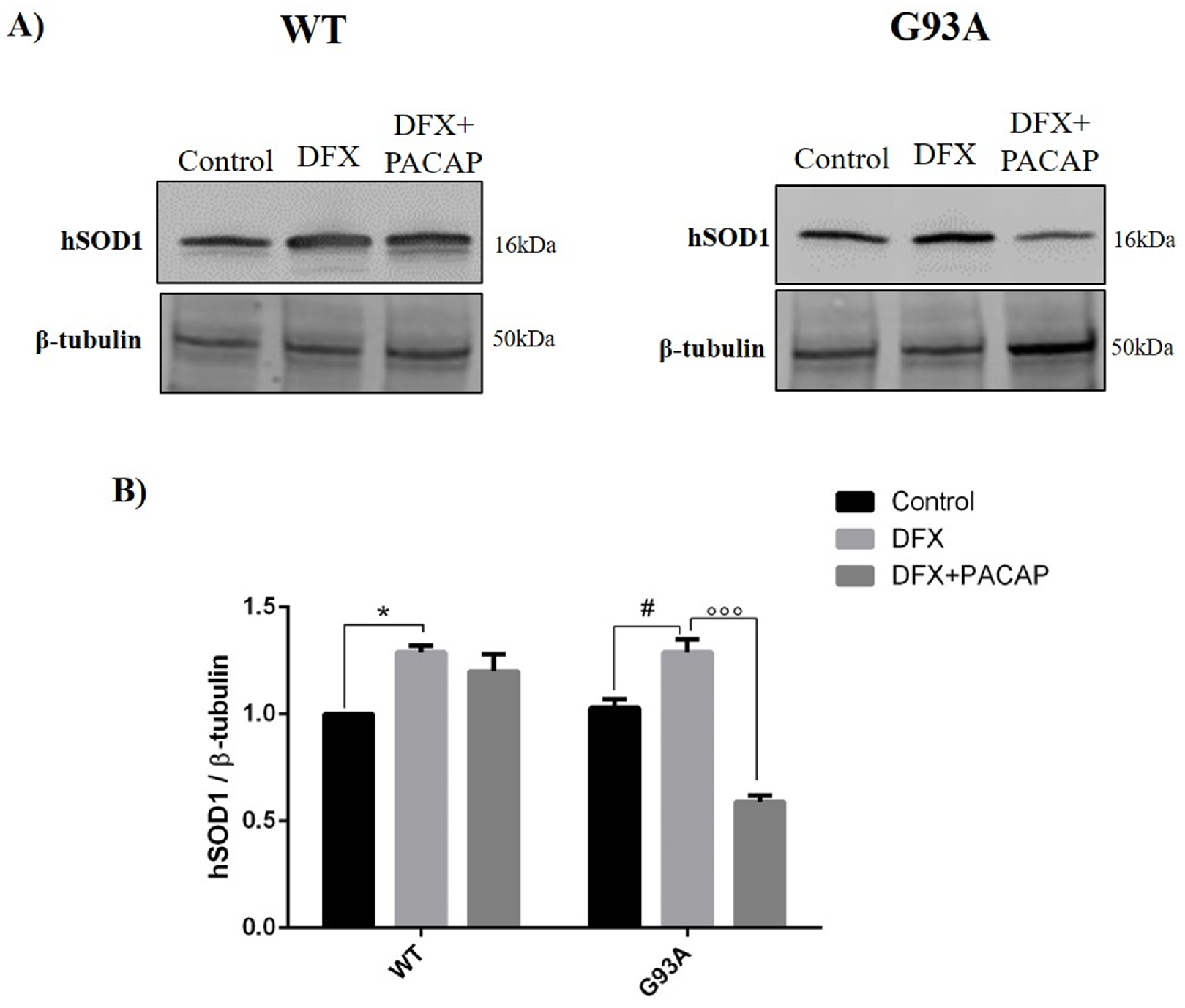
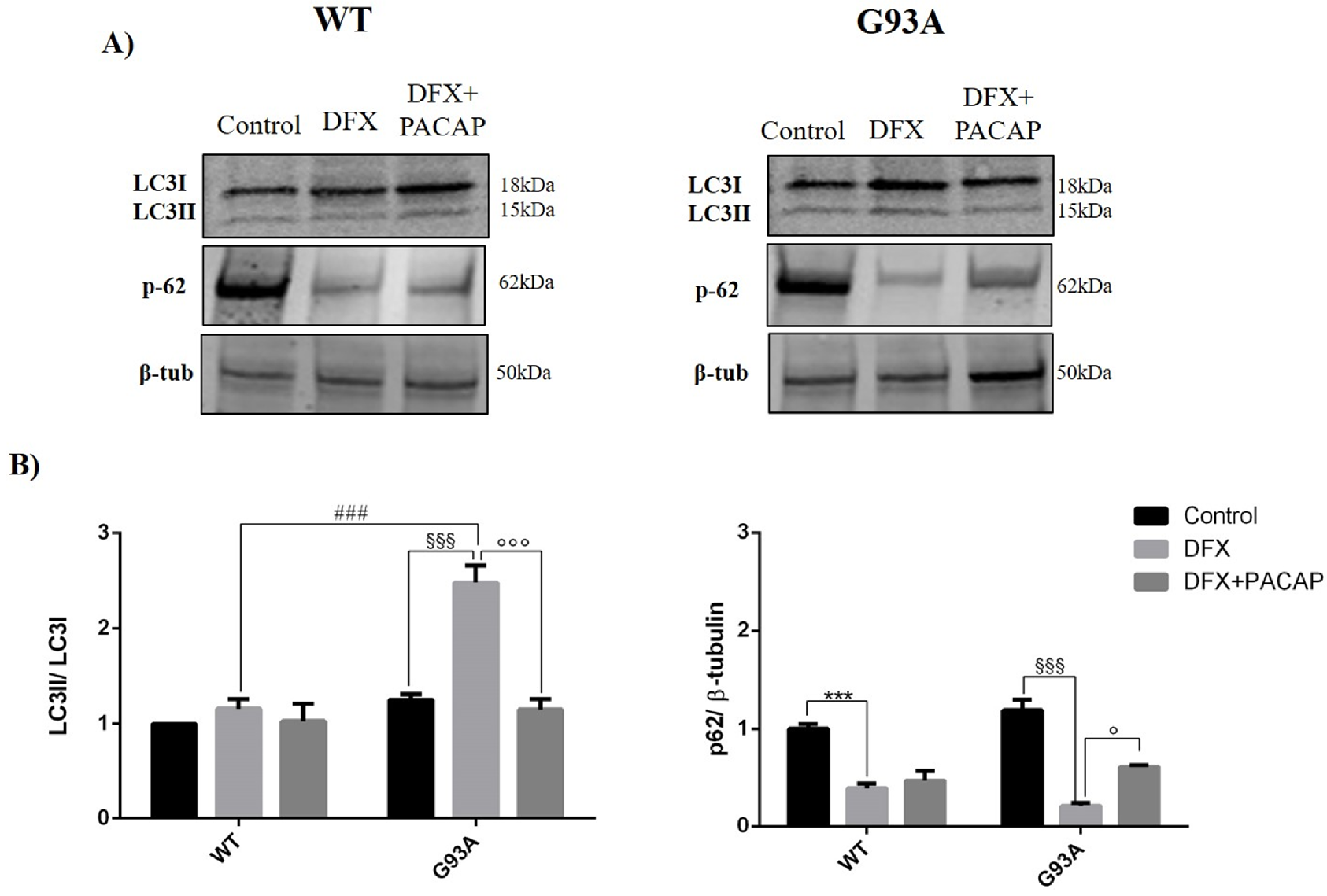
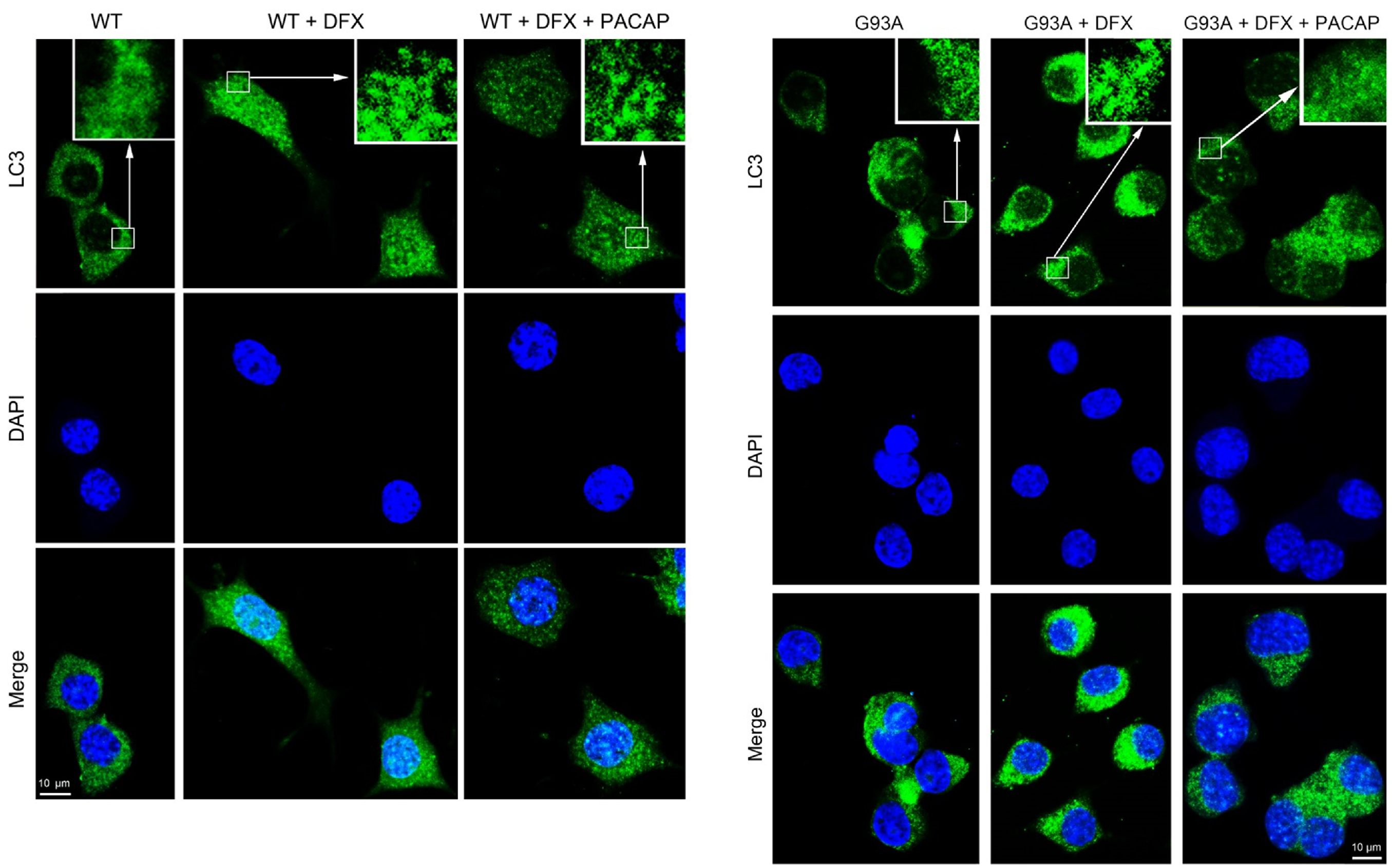
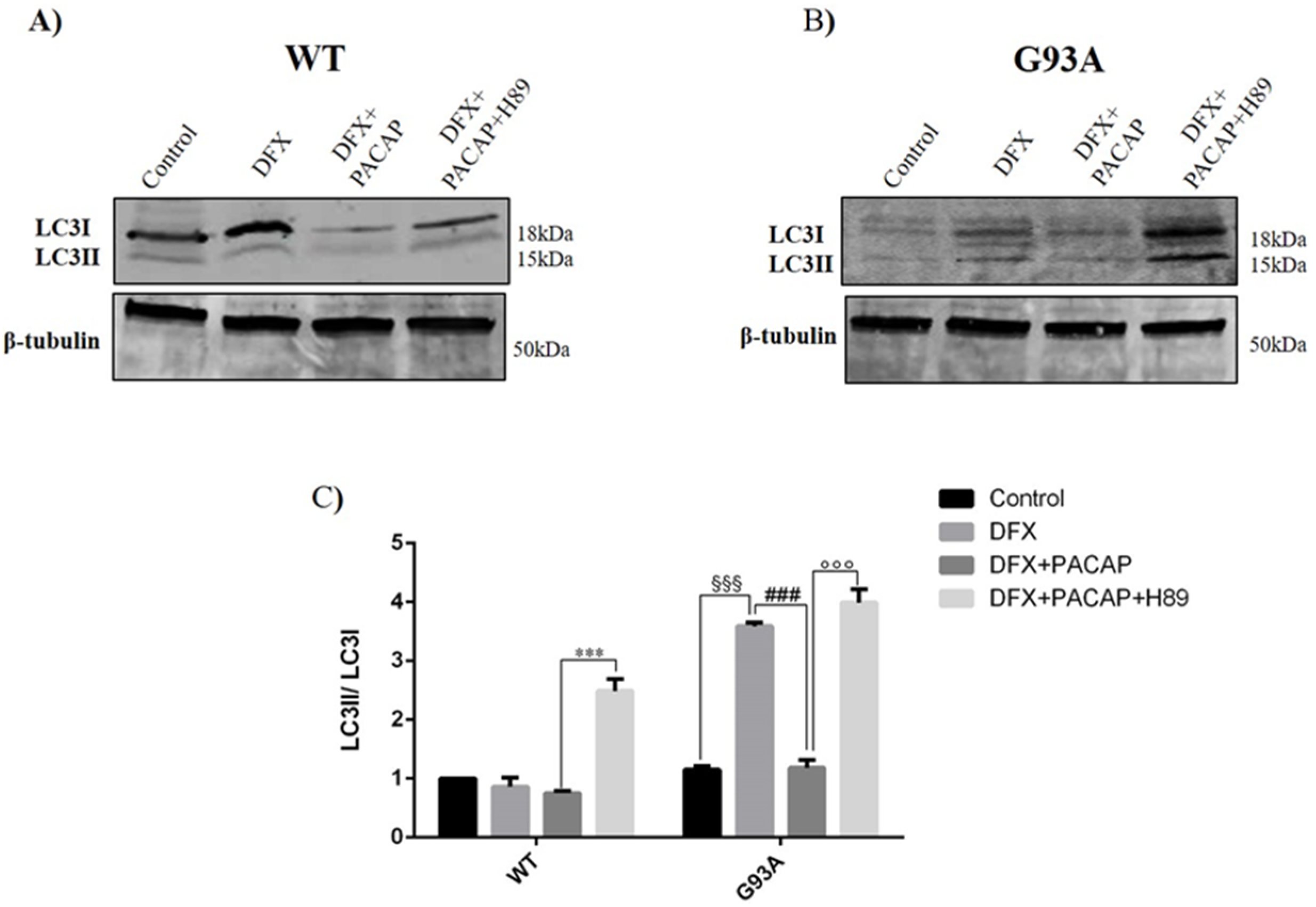
2.2. PACAP Treatment Modulates DFX-Induced Autophagy in WT and G93A Cells
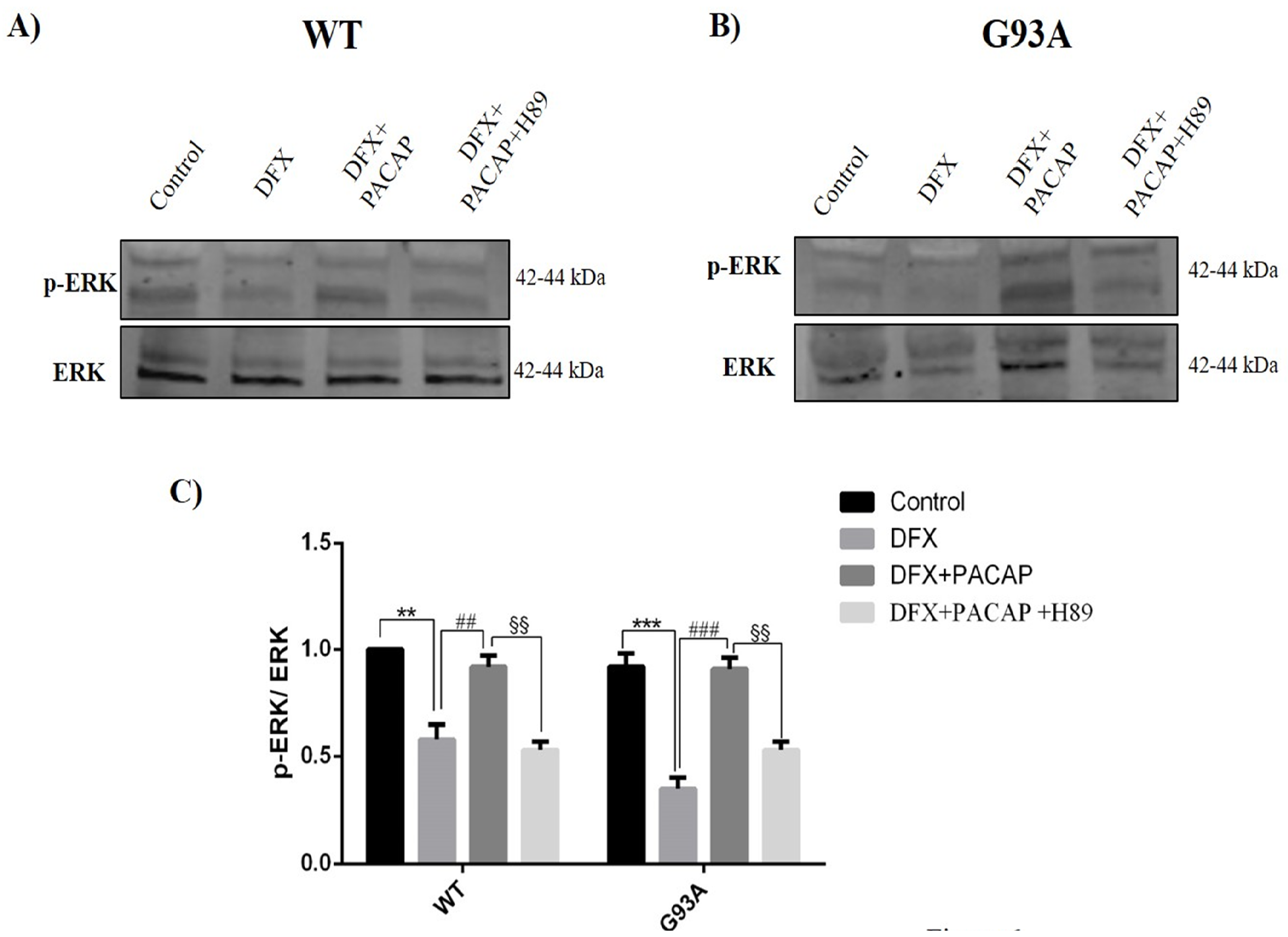
2.3. PACAP Modulates Autophagy in WT and G93A Cells Exposed to Hypoxia through Activation of the MAPK/ERK Pathway
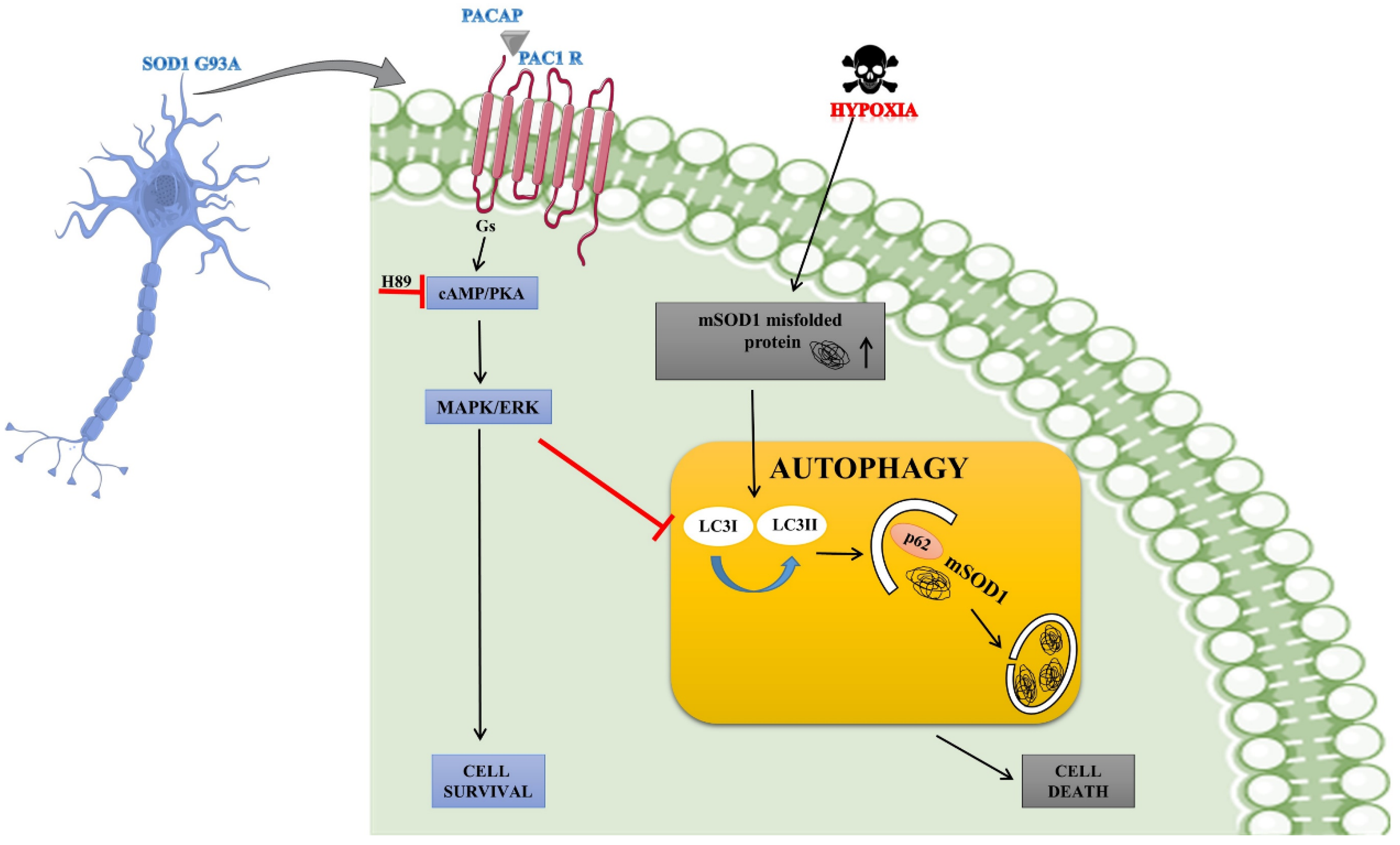
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Treatments
4.3. Cell Viability Assay
4.4. Fluorescence Microscopic Analysis of Cell Death
4.5. Western Blot Analysis
4.6. Immunofluorescence Detection of LC3 Protein
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Wong, P.C.; Rothstein, J.D.; Price, D.L. The genetic and molecular mechanisms of motor neuron disease. Curr. Opin. Neurobiol. 1998, 8, 791–799. [Google Scholar] [CrossRef]
- Nguyen, D.K.H.; Thombre, R.; Wang, J. Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci. Lett. 2018, 697, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D.; Ohama, E.; Reaume, A.G.; Scott, R.W.; Cleveland, D.W. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef]
- Kirby, J.; Halligan, E.; Baptista, M.J.; Allen, S.; Heath, P.R.; Holden, H.; Barber, S.C.; Loynes, C.A.; Wood-Allum, C.A.; Lunec, J.; et al. Mutant SOD1 alters the motor neuronal transcriptome: Implications for familial ALS. Brain 2005, 128, 1686–1706. [Google Scholar] [CrossRef]
- Song, C.-Y.; Guo, J.; Liu, Y.; Tang, B.-S. Autophagy and Its Comprehensive Impact on ALS. Int. J. Neurosci. 2012, 122, 695–703. [Google Scholar] [CrossRef]
- Wei, Y.-M. Autophagic induction of amyotrophic lateral sclerosis-linked Cu/Zn superoxide dismutase 1 G93A mutant in NSC34 cells. Neural Regen. Res. 2014, 9, 16–24. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 15, 885–889. [Google Scholar] [CrossRef]
- Hsueh, K.-W.; Chiou, T.-W.; Chiang, S.-F.; Yamashita, T.; Abe, K.; Borlongan, C.V.; Sanberg, P.R.; Huang, A.; Yu, H.; Lin, S.-Z.; et al. Autophagic down-regulation in motor neurons remarkably prolongs the survival of ALS mice. Neuropharmacology 2016, 108, 152–160. [Google Scholar] [CrossRef]
- Tu, P.H.; Raju, P.; Robinson, K.A.; Gurney, M.E.; Trojanowski, J.Q.; Lee, V.M. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc. Natl. Acad. Sci. USA 1996, 93, 3155–3160. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Rothstein, J.D. Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol. 2015, 69. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Gosset, P.; Kluza, J.; Brunaud-Danel, V.; Lassalle, P.; Marchetti, P.; Defebvre, L.; Destée, A.; Devos, D.P. Deregulation of the hypoxia inducible factor-1α pathway in monocytes from sporadic amyotrophic lateral sclerosis patients. Neuroscience 2011, 172, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yan, J.; Chang, Y.; ShiDu Yan, S.; Shi, H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr. Med. Chem. 2011, 18, 4335–4343. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1813, 1263–1268. [Google Scholar] [CrossRef]
- Skene, P.; Cleveland, D.W. Hypoxia and Lou Gehrig. Nat. Genet. 2001, 28, 107–108. [Google Scholar] [CrossRef]
- Tankersley, C.G.; Haenggeli, C.; Rothstein, J.D. Respiratory impairment in a mouse model of amyotrophic lateral sclerosis. J. Appl. Physiol. 2007, 102, 926–932. [Google Scholar] [CrossRef]
- Cimini, S.; Rizzardini, M.; Biella, G.; Cantoni, L. Hypoxia causes autophagic stress and derangement of metabolic adaptation in a cell model of amyotrophic lateral sclerosis. J. Neurochem. 2014, 129, 413–425. [Google Scholar] [CrossRef]
- Vanacore, N.; Cocco, P.; Fadda, D.; Dosemeci, M. Job strain, hypoxia and risk of amyotrophic lateral sclerosis: Results from a death certificate study. Amyotroph. Lateral Scler. 2010, 11, 430–434. [Google Scholar] [CrossRef]
- Rothstein, J.D. Edaravone: A new drug approved for ALS. Cell 2017, 171, 725. [Google Scholar] [CrossRef]
- Morello, G.; Spampinato, A.G.; Conforti, F.; D’Agata, V.; Cavallaro, S. Selection and Prioritization of Candidate Drug Targets for Amyotrophic Lateral Sclerosis Through a Meta-Analysis Approach. J. Mol. Neurosci. 2017, 61, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Vaudry, H.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.C.; Hashimoto, H.; Galas, L.; et al. Pituitary Adenylate Cyclase-Activating Polypeptide and Its Receptors: 20 Years after the Discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef] [PubMed]
- Reglodi, D.; Lubics, A.; Tamás, A.; Szalontay, L.; Lengvári, I. Pituitary adenylate cyclase activating polypeptide protects dopaminergic neurons and improves behavioral deficits in a rat model of Parkinson’s disease. Behav. Brain Res. 2004, 151, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Reglodi, D.; Kiss, P.; Lubics, A.; Tamas, A. Review on the protective effects of PACAP in models of neurodegenerative diseases in vitro and in vivo. Curr. Pharm. Des. 2011, 17, 962–972. [Google Scholar] [CrossRef]
- D’Amico, A.G.; Maugeri, G.; Reitano, R.; Bucolo, C.; Saccone, S.; Drago, F.; D’Agata, V. PACAP Modulates Expression of Hypoxia-Inducible Factors in Streptozotocin-Induced Diabetic Rat Retina. J. Mol. Neurosci. 2015, 57, 501–509. [Google Scholar] [CrossRef]
- D’Amico, A.G.; Maugeri, G.; Rasà, D.M.; Bucolo, C.; Saccone, S.; Federico, C.; Cavallaro, S.; D’Agata, V. Modulation of IL-1β and VEGF expression in rat diabetic retinopathy after PACAP administration. Peptides 2017, 97, 64–69. [Google Scholar] [CrossRef]
- Maugeri, G.; D’Amico, A.G.; Saccone, S.; Federico, C.; Cavallaro, S.; D’Agata, V. PACAP and VIP Inhibit HIF-1α-Mediated VEGF Expression in a Model of Diabetic Macular Edema. J. Cell. Physiol. 2016, 232, 1209–1215. [Google Scholar] [CrossRef]
- Maugeri, G.; Longo, A.; D’Amico, A.G.; Rasà, D.M.; Reibaldi, M.; Russo, A.; Bonfiglio, V.; Avitabile, T.; D’Agata, V. Trophic effect of PACAP on human corneal endothelium. Peptides 2018, 99, 20–26. [Google Scholar] [CrossRef]
- Rat, D.; Schmitt, U.; Tippmann, F.; Dewachter, I.; Theunis, C.; Wieczerzak, E.; Postina, R.; Van Leuven, F.; Fahrenholz, F.; Kojro, E. Neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) slows down Alzheimer’s disease-like pathology in amyloid precursor protein-transgenic mice. FASEB J. 2011, 25, 3208–3218. [Google Scholar] [CrossRef]
- Reglódi, D.; Tamás, A.; Lengvári, I.; Toth, G.; Szalontay, L.; Lubics, A. Comparative study of the effects of PACAP in young, aging, and castrated males in a rat model of Parkinson’s disease. Ann. N. Y. Acad. Sci. 2006, 1070, 518–524. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Hannibal, J.; Zhao, Q.; Moller, K.; Danielsen, N.; Fahrenkrug, J.; Sundler, F. Pituitary adenylate cyclase activating peptide expression in the rat dorsal root ganglia: Upregulation after peripheral nerve injury. Neuroscience 1996, 74, 1099–1110. [Google Scholar] [CrossRef]
- Suarez, V.; Guntinas-Lichius, O.; Streppel, M.; Ingorokva, S.; Grosheva, M.; Neiss, W.F.; Angelov, D.N.; Klimaschewski, L. The axotomy-induced neuropeptides galanin and pituitary adenylate cyclase-activating peptide promote axonal sprouting of primary afferent and cranial motor neurones. Eur. J. Neurosci. 2006, 24, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, B.; Abad, C.; Chhith, S.; Cheung-Lau, G.; Hajji, O.; Nobuta, H.; Waschek, J.A. Impaired nerve regeneration and enhanced neuroinflammatory response in mice lacking pituitary adenylyl cyclase activating peptide. Neuroscience 2007, 151, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, L.M.; Dahlin, L.B.; Danielsen, N. Changes in expression of PACAP in rat sensory neurons in response to sciatic nerve compression. Eur. J. Neurosci. 2004, 20, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Tomimatsu, N.; Arakawa, Y. Survival-promoting activity of pituitary adenylate cyclase-activating polypeptide in the presence of phosphodiesterase inhibitors on rat motoneurons in culture: cAMP-protein kinase A-mediated survival. J. Neurochem. 2008, 107, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Ringer, C.; Büning, L.-S.; Schäfer, M.K.; Eiden, L.E.; Weihe, E.; Schütz, B. PACAP signaling exerts opposing effects on neuroprotection and neuroinflammation during disease progression in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 54, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, G.; Iemmolo, R.; D’Amico, A.G.; La Cognata, V.; Costanzo, E.; Zappia, M.; D’Agata, V.; Conforti, F.; Aronica, E.; Cavallaro, S. PACAP and PAC1R are differentially expressed in motor cortex of amyotrophic lateral sclerosis patients and support survival of iPSC-derived motor neurons. J. Cell. Physiol. 2017, 233, 3343–3351. [Google Scholar] [CrossRef]
- Maugeri, G.; D’Amico, A.G.; Rasà, D.M.; Federico, C.; Saccone, S.; Morello, G.; La Cognata, V.; Cavallaro, S.; D’Agata, V. Molecular mechanisms involved in the protective effect of pituitary adenylate cyclase-activating polypeptide in an in vitro model of amyotrophic lateral sclerosis. J. Cell. Physiol. 2018, 234, 5203–5521. [Google Scholar] [CrossRef]
- Arciello, M.; Capo, C.R.; Cozzolino, M.; Ferri, A.; Nencini, M.; Carri, M.T.; Rossi, L. Inactivation of cytochromecoxidase by mutant SOD1s in mouse motoneuronal NSC-34 cells is independent from copper availability but is because of nitric oxide. J. Neurochem. 2010, 112, 183–192. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef]
- Barth, S.; Glick, D.; MacLeod, K. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Boil. 2004, 36, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Peters, O.M.; Ghasemi, M.; Brown, R.H. Emerging mechanisms of molecular pathology in ALS. J. Clin. Investig. 2015, 125, 1767–1779. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, T.-Y.; Cho, K.-S.; Na Kim, H.; Koh, J.-Y. Autophagy activation and neuroprotection by progesterone in the G93A-SOD1 transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 59, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, X.; Song, L.; Le, W. Autophagy Dysregulation in Amyotrophic Lateral Sclerosis. Brain Pathol. 2011, 22, 110–116. [Google Scholar] [CrossRef]
- Valbuena, G.; Rizzardini, M.; Cimini, S.; Siskos, A.; Bendotti, C.; Cantoni, L.; Keun, H. Metabolomic Analysis Reveals Increased Aerobic Glycolysis and Amino Acid Deficit in a Cellular Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2015, 53, 2222–2240. [Google Scholar] [CrossRef] [PubMed]
- Shvets, E.; Fass, E.; Scherz-Shouval, R.; Elazar, Z. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J. Cell Sci. 2008, 121, 2685–2695. [Google Scholar] [CrossRef]
- Gal, J.; Strom, A.-L.; Kwinter, D.M.; Kilty, R.; Zhang, J.; Shi, P.; Fu, W.; Wooten, M.W.; Zhu, H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J. Neurochem. 2009, 111, 1062–1073. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, J.; Li, S.; Chen, S.; Le, W. n-butylidenephthalide treatment prolongs life span and attenuates motor neuron loss in SOD1G93A mouse model of amyotrophic lateral sclerosis. CNS Neurosci. Ther. 2017, 23, 375–385. [Google Scholar] [CrossRef]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.; Cashman, N.; Morris-Valero, R.; Bulock, J.; Adams, S.; Sanes, J. An LRE (leucine-arginine-glutamate)-dependent mechanism for adhesion of neurons to S-laminin. J. Neurosci. 1991, 11, 3960–3971. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eggett, C.J.; Crosier, S.; Manning, P.; Cookson, M.R.; Menzies, F.M.; McNeil, C.J.; Shaw, P.J. Development and Characterisation of a Glutamate-Sensitive Motor Neurone Cell Line. J. Neurochem. 2008, 74, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.G.; Maugeri, G.; Rasà, D.M.; La Cognata, V.; Saccone, S.; Federico, C.; Cavallaro, S.; D’Agata, V. NAP counteracts hyperglycemia/hypoxia induced retinal pigment epithelial barrier breakdown through modulation of HIFs and VEGF expression. J. Cell. Physiol. 2017, 233, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.G.; Maugeri, G.; Magro, G.; Salvatorelli, L.; Drago, F.; D’Agata, V. Expression pattern of parkin isoforms in lung adenocarcinomas. Tumor Boil. 2015, 36, 5133–5141. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, G.; D’Amico, A.G.; Rasà, D.M.; Saccone, S.; Federico, C.; Cavallaro, S.; D’Agata, V. PACAP and VIP regulate hypoxia-inducible factors in neuroblastoma cells exposed to hypoxia. Neuropeptides 2018, 69, 84–91. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amico, A.G.; Maugeri, G.; Saccone, S.; Federico, C.; Cavallaro, S.; Reglodi, D.; D’Agata, V. PACAP Modulates the Autophagy Process in an In Vitro Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2943. https://doi.org/10.3390/ijms21082943
D’Amico AG, Maugeri G, Saccone S, Federico C, Cavallaro S, Reglodi D, D’Agata V. PACAP Modulates the Autophagy Process in an In Vitro Model of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2020; 21(8):2943. https://doi.org/10.3390/ijms21082943
Chicago/Turabian StyleD’Amico, Agata Grazia, Grazia Maugeri, Salvatore Saccone, Concetta Federico, Sebastiano Cavallaro, Dora Reglodi, and Velia D’Agata. 2020. "PACAP Modulates the Autophagy Process in an In Vitro Model of Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 21, no. 8: 2943. https://doi.org/10.3390/ijms21082943
APA StyleD’Amico, A. G., Maugeri, G., Saccone, S., Federico, C., Cavallaro, S., Reglodi, D., & D’Agata, V. (2020). PACAP Modulates the Autophagy Process in an In Vitro Model of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 21(8), 2943. https://doi.org/10.3390/ijms21082943