Immunoproteasome Genes Are Modulated in CD34+ JAK2V617F Mutated Cells from Primary Myelofibrosis Patients

 ,
, 
 ,
,  ,
,  ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results
2.1. Identification of Potential Genes Modulated in JAK2V617F Mutated Compared to JAK2 Wild-Type PMF Patients
2.2. Immunoproteasome (IPs) Genes Expression in PMF Patients
2.3. IFNG Pathways Activation in PB CD34+ Cells of PMF Patients
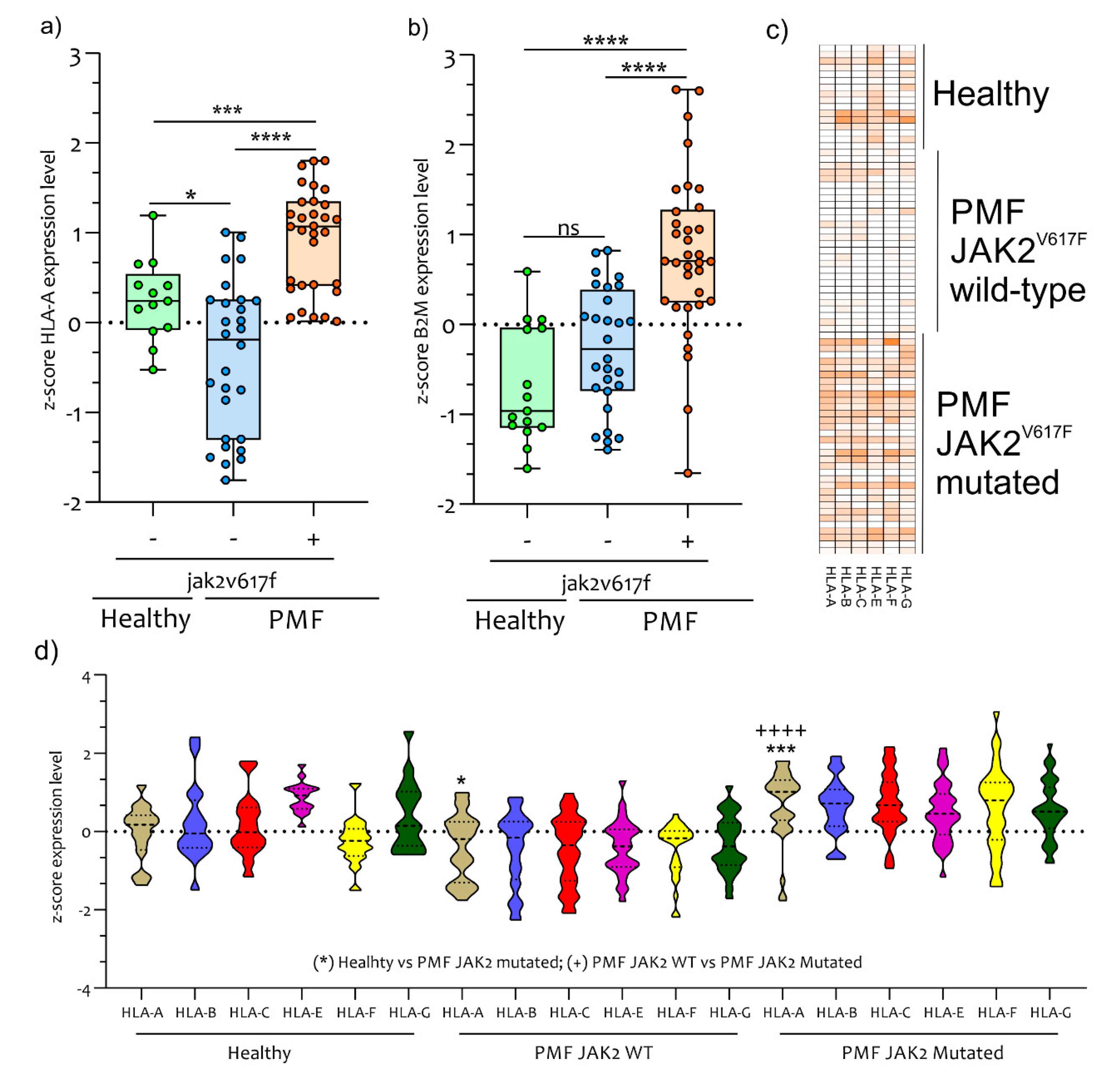
2.4. Antigen Exposition Pathways in PB CD34+ Cells of PMF Patients
2.5. Diagnostic Accuracy of mRNA PSMB8, PSMB9, and PSMB10 for PMF Patients
3. Discussion
4. Materials and Methods
4.1. Data Selection
4.2. Data Processing and Experimental Design
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| c-20S | Proteasomes |
| MM | multiple myeloma |
| IPs | Immunoproteasome |
| TNFα | tumor necrosis factor alpha |
| IFNG | interferon gamma |
| LMP2 or PSMB9 | subunits β1i |
| MECL-1 or PSMB10 | subunits β2i |
| GEO | Gene Expression Omnibus |
| GHATER | Gene Annotation Tool to Help Explain Relationships |
| SDEG | Significantly Different Expressed Genes |
| MeV | MultiExperiment Viewer |
| MHC class I or HLA class I | Major Histocompatibility Complex class I |
| Th | helper T cell |
| TAP1 and TAP2 | Transporter associated with Antigen Processing 1 and 2 |
| PB | Peripheral Blood |
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Arber, D.A. The 2016 WHO classification of acute myeloid leukemia: What the practicing clinician needs to know. Semin. Hematol. 2019, 56, 90–95. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef]
- Palumbo, G.A.; Stella, S.; Pennisi, M.S.; Pirosa, C.; Fermo, E.; Fabris, S.; Cattaneo, D.; Iurlo, A. The Role of New Technologies in Myeloproliferative Neoplasms. Front. Oncol 2019, 9, 321. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients With Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef]
- Barosi, G.; Viarengo, G.; Pecci, A.; Rosti, V.; Piaggio, G.; Marchetti, M.; Frassoni, F. Diagnostic and clinical relevance of the number of circulating CD34(+) cells in myelofibrosis with myeloid metaplasia. Blood 2001, 98, 3249–3255. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Kantarjian, H.M.; Kiladjian, J.J.; Gotlib, J.; Cervantes, F.; Mesa, R.A.; Sarlis, N.J.; Peng, W.; Sandor, V.; Gopalakrishna, P.; et al. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica 2015, 100, 1139–1145. [Google Scholar] [CrossRef]
- Bjorn, M.E.; Hasselbalch, H.C. The impact of ruxolitinib treatment on inflammation-mediated comorbidities in myelofibrosis and related neoplasms. Clin. Case Rep. 2015, 3, 499–503. [Google Scholar] [CrossRef]
- Padron, E.; Dezern, A.; Andrade-Campos, M.; Vaddi, K.; Scherle, P.; Zhang, Q.; Ma, Y.; Balasis, M.E.; Tinsley, S.; Ramadan, H.; et al. A Multi-Institution Phase I Trial of Ruxolitinib in Patients with Chronic Myelomonocytic Leukemia (CMML). Clin. Cancer Res. 2016, 22, 3746–3754. [Google Scholar] [CrossRef]
- Kvasnicka, H.M.; Thiele, J.; Bueso-Ramos, C.E.; Sun, W.; Cortes, J.; Kantarjian, H.M.; Verstovsek, S. Long-term effects of ruxolitinib versus best available therapy on bone marrow fibrosis in patients with myelofibrosis. J. Hematol. Oncol. 2018, 11, 42. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk Res. 2013, 37, 214–220. [Google Scholar] [CrossRef]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef]
- Silver, R.T.; Kiladjian, J.J.; Hasselbalch, H.C. Interferon and the treatment of polycythemia vera, essential thrombocythemia and myelofibrosis. Expert Rev. Hematol. 2013, 6, 49–58. [Google Scholar] [CrossRef]
- Eran, Z.; Zingariello, M.; Bochicchio, M.T.; Bardelli, C.; Migliaccio, A.R. Novel strategies for the treatment of myelofibrosis driven by recent advances in understanding the role of the microenvironment in its etiology. F1000Res 2019, 8. [Google Scholar] [CrossRef]
- Craiu, A.; Gaczynska, M.; Akopian, T.; Gramm, C.F.; Fenteany, G.; Goldberg, A.L.; Rock, K.L. Lactacystin and clasto-lactacystin beta-lactone modify multiple proteasome beta-subunits and inhibit intracellular protein degradation and major histocompatibility complex class I antigen presentation. J. Biol. Chem. 1997, 272, 13437–13445. [Google Scholar] [CrossRef]
- Miller, Z.; Ao, L.; Kim, K.B.; Lee, W. Inhibitors of the immunoproteasome: Current status and future directions. Curr. Pharm. Des. 2013, 19, 4140–4151. [Google Scholar] [CrossRef]
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (Velcadetrade mark) in the Treatment of Multiple Myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef]
- Tibullo, D.; Di Rosa, M.; Giallongo, C.; La Cava, P.; Parrinello, N.L.; Romano, A.; Conticello, C.; Brundo, M.V.; Saccone, S.; Malaguarnera, L.; et al. Bortezomib modulates CHIT1 and YKL40 in monocyte-derived osteoclast and in myeloma cells. Front. Pharmacol. 2015, 6, 226. [Google Scholar] [CrossRef]
- Wang, W.; Lin, X.; Xu, H.; Sun, W.; Bouta, E.M.; Zuscik, M.J.; Chen, D.; Schwarz, E.M.; Xing, L. Attenuated Joint Tissue Damage Associated With Improved Synovial Lymphatic Function Following Treatment With Bortezomib in a Mouse Model of Experimental Posttraumatic Osteoarthritis. Arthritis Rheumatol. 2019, 71, 244–257. [Google Scholar] [CrossRef]
- Mohty, M.; Brissot, E.; Savani, B.N.; Gaugler, B. Effects of bortezomib on the immune system: A focus on immune regulation. Biol Blood Marrow Transplant. 2013, 19, 1416–1420. [Google Scholar] [CrossRef]
- Szychlinska, M.A.; Trovato, F.M.; Di Rosa, M.; Malaguarnera, L.; Puzzo, L.; Leonardi, R.; Castrogiovanni, P.; Musumeci, G. Co-Expression and Co-Localization of Cartilage Glycoproteins CHI3L1 and Lubricin in Osteoarthritic Cartilage: Morphological, Immunohistochemical and Gene Expression Profiles. Int J. Mol. Sci 2016, 17, 359. [Google Scholar] [CrossRef]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Noda, C.; Tanahashi, N.; Shimbara, N.; Hendil, K.B.; Tanaka, K. Tissue distribution of constitutive proteasomes, immunoproteasomes, and PA28 in rats. Biochem. Biophys. Res. Commun. 2000, 277, 348–354. [Google Scholar] [CrossRef]
- Malaguarnera, L.; Imbesi, R.; Di Rosa, M.; Scuto, A.; Castrogiovanni, P.; Messina, A.; Sanfilippo, S. Action of prolactin, IFN-gamma, TNF-alpha and LPS on heme oxygenase-1 expression and VEGF release in human monocytes/macrophages. Int Immunopharmacol 2005, 5, 1458–1469. [Google Scholar] [CrossRef]
- Kammerl, I.E.; Meiners, S. Proteasome function shapes innate and adaptive immune responses. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L328–L336. [Google Scholar] [CrossRef]
- Shin, E.C.; Seifert, U.; Kato, T.; Rice, C.M.; Feinstone, S.M.; Kloetzel, P.M.; Rehermann, B. Virus-induced type I IFN stimulates generation of immunoproteasomes at the site of infection. J. Clin. Invest. 2006, 116, 3006–3014. [Google Scholar] [CrossRef]
- Hallermalm, K.; Seki, K.; Wei, C.; Castelli, C.; Rivoltini, L.; Kiessling, R.; Levitskaya, J. Tumor necrosis factor-alpha induces coordinated changes in major histocompatibility class I presentation pathway, resulting in increased stability of class I complexes at the cell surface. Blood 2001, 98, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Boes, B.; Hengel, H.; Ruppert, T.; Multhaup, G.; Koszinowski, U.H.; Kloetzel, P.M. Interferon gamma stimulation modulates the proteolytic activity and cleavage site preference of 20S mouse proteasomes. J. Exp. Med. 1994, 179, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, H.T.; Conley, T.; Muchamuel, T.; Jiang, J.; Lee, S.; Owen, T.; Barnard, J.; Nevarez, S.; Goldman, B.I.; Kirk, C.J.; et al. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum. 2012, 64, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Dajee, M.; Moll, C.; Groettrup, M.; Kirk, C.J. Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J. Immunol. 2010, 185, 634–641. [Google Scholar] [CrossRef]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef]
- Al-Homsi, A.S.; Lai, Z.; Roy, T.S.; Kouttab, N. Effect of novel proteasome and immunoproteasome inhibitors on dendritic cell maturation, function, and expression of IkappaB and NFkappaB. Transpl. Immunol. 2013, 29, 1–6. [Google Scholar] [CrossRef]
- Mesa, R.A.; Verstovsek, S.; Rivera, C.; Pardanani, A.; Hussein, K.; Lasho, T.; Wu, W.; Tefferi, A. Bortezomib therapy in myelofibrosis: A phase II clinical trial. Leukemia 2008, 22, 1636–1638. [Google Scholar] [CrossRef][Green Version]
- Wagner-Ballon, O.; Pisani, D.F.; Gastinne, T.; Tulliez, M.; Chaligne, R.; Lacout, C.; Aurade, F.; Villeval, J.L.; Gonin, P.; Vainchenker, W.; et al. Proteasome inhibitor bortezomib impairs both myelofibrosis and osteosclerosis induced by high thrombopoietin levels in mice. Blood 2007, 110, 345–353. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic. Acids. Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Musumeci, G.; Castrogiovanni, P.; Barbagallo, I.; Tibullo, D.; Sanfilippo, C.; Nunnari, G.; Pellicano, G.F.; Pavone, P.; Caltabiano, R.; Di Marco, R.; et al. Expression of the OAS Gene Family Is Highly Modulated in Subjects Affected by Juvenile Dermatomyositis, Resembling an Immune Response to a dsRNA Virus Infection. Int. J. Mol. Med. 2018, 19, 2786. [Google Scholar] [CrossRef]
- Sanfilippo, C.; Pinzone, M.R.; Cambria, D.; Longo, A.; Palumbo, M.; Di Marco, R.; Condorelli, F.; Nunnari, G.; Malaguarnera, L.; Di Rosa, M. OAS Gene Family Expression Is Associated with HIV-Related Neurocognitive Disorders. Mol. Neurobiol. 2018, 55, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Nunnari, G.; Lazzara, F.; Longo, A.; Cambria, D.; Distefano, G.; Palumbo, M.; Nicoletti, F.; Malaguarnera, L.; Di Rosa, M. Induction of OAS gene family in HIV monocyte infected patients with high and low viral load. Antiviral. Res. 2016, 131, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, L.; Nunnari, G.; Di Rosa, M. Nuclear import sequence identification in hOAS3 protein. Inflamm. Res. 2016, 65, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, M.; Pinzone, M.R.; Di Rosa, M.; Madeddu, G.; Foca, E.; Martellotta, F.; Schioppa, O.; Ceccarelli, G.; Celesia, B.M.; d’Ettorre, G.; et al. Kidney disease in HIV-infected patients. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2660–2667. [Google Scholar] [PubMed]
- McCarthy, M.K.; Malitz, D.H.; Molloy, C.T.; Procario, M.C.; Greiner, K.E.; Zhang, L.; Wang, P.; Day, S.M.; Powell, S.R.; Weinberg, J.B. Interferon-dependent immunoproteasome activity during mouse adenovirus type 1 infection. Virol. 2016, 498, 57–68. [Google Scholar] [CrossRef]
- Niewerth, D.; Kaspers, G.J.; Assaraf, Y.G.; van Meerloo, J.; Kirk, C.J.; Anderl, J.; Blank, J.L.; van de Ven, P.M.; Zweegman, S.; Jansen, G.; et al. Interferon-gamma-induced upregulation of immunoproteasome subunit assembly overcomes bortezomib resistance in human hematological cell lines. J. Hematol. Oncol. 2014, 7, 7. [Google Scholar] [CrossRef]
- Gaczynska, M.; Rock, K.L.; Goldberg, A.L. Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nat. 1993, 365, 264–267. [Google Scholar] [CrossRef]
- Kalim, K.W.; Basler, M.; Kirk, C.J.; Groettrup, M. Immunoproteasome subunit LMP7 deficiency and inhibition suppresses Th1 and Th17 but enhances regulatory T cell differentiation. J. Immunol. 2012, 189, 4182–4193. [Google Scholar] [CrossRef]
- Basler, M.; Mundt, S.; Bitzer, A.; Schmidt, C.; Groettrup, M. The immunoproteasome: A novel drug target for autoimmune diseases. Clin. Exp. Rheumatol. 2015, 33, S74–S79. [Google Scholar]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef]
- Panteli, K.E.; Hatzimichael, E.C.; Bouranta, P.K.; Katsaraki, A.; Seferiadis, K.; Stebbing, J.; Bourantas, K.L. Serum interleukin (IL)-1, IL-2, sIL-2Ra, IL-6 and thrombopoietin levels in patients with chronic myeloproliferative diseases. Br. J. Haematol. 2005, 130, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Macedo, L.C.; de Cesare Quintero, F.; Pagliari, E.S.S.; Pagnano, K.B.; Rodrigues, C.; de Alencar, J.B.; Sell, A.M.; Visentainer, J.E. Association of TNF polymorphisms with JAK2 (V617F) myeloproliferative neoplasms in Brazilian patients. Blood Cells Mol. Dis. 2016, 57, 54–57. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Zini, R.; Bogani, C.; Salati, S.; Pancrazzi, A.; Bianchi, E.; Mannelli, F.; Ferrari, S.; Le Bousse-Kerdiles, M.C.; Bosi, A.; et al. Molecular profiling of CD34+ cells in idiopathic myelofibrosis identifies a set of disease-associated genes and reveals the clinical significance of Wilms’ tumor gene 1 (WT1). Stem. Cells 2007, 25, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Calura, E.; Pizzini, S.; Bisognin, A.; Coppe, A.; Sales, G.; Gaffo, E.; Fanelli, T.; Mannarelli, C.; Zini, R.; Norfo, R.; et al. A data-driven network model of primary myelofibrosis: Transcriptional and post-transcriptional alterations in CD34+ cells. Blood Cancer J. 2016, 6, e439. [Google Scholar] [CrossRef] [PubMed]
- Steunou, V.; Le Bousse-Kerdiles, M.C.; Colin-Micouin, A.; Clay, D.; Chevillard, S.; Martyre, M.C.; French, I.R.N.o.M.M.M. Altered transcription of the stem cell leukemia gene in myelofibrosis with myeloid metaplasia. Leukemia 2003, 17, 1998–2006. [Google Scholar] [CrossRef][Green Version]
- Pennucci, V.; Zini, R.; Norfo, R.; Guglielmelli, P.; Bianchi, E.; Salati, S.; Sacchi, G.; Prudente, Z.; Tenedini, E.; Ruberti, S.; et al. Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative, I., Abnormal expression patterns of WT1-as, MEG3 and ANRIL long non-coding RNAs in CD34+ cells from patients with primary myelofibrosis and their clinical correlations. Leuk. Lymphoma 2015, 56, 492–496. [Google Scholar]
- Dao, T.; Korontsvit, T.; Zakhaleva, V.; Jarvis, C.; Mondello, P.; Oh, C.; Scheinberg, D.A. An immunogenic WT1-derived peptide that induces T cell response in the context of HLA-A*02:01 and HLA-A*24:02 molecules. Oncoimmunol. 2017, 6, e1252895. [Google Scholar] [CrossRef]
- Stetka, J.; Vyhlidalova, P.; Lanikova, L.; Koralkova, P.; Gursky, J.; Hlusi, A.; Flodr, P.; Hubackova, S.; Bartek, J.; Hodny, Z.; et al. Addiction to DUSP1 protects JAK2V617F-driven polycythemia vera progenitors against inflammatory stress and DNA damage, allowing chronic proliferation. Oncogene 2019, 38, 5627–5642. [Google Scholar] [CrossRef]
- Tefferi, A. Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 93, 1551–1560. [Google Scholar] [CrossRef]
- Keohane, C.; Kordasti, S.; Seidl, T.; Perez Abellan, P.; Thomas, N.S.; Harrison, C.N.; McLornan, D.P.; Mufti, G.J. JAK inhibition induces silencing of T Helper cytokine secretion and a profound reduction in T regulatory cells. Br. J. Haematol. 2015, 171, 60–73. [Google Scholar] [CrossRef]
- Elli, E.M.; Barate, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019, 9, 1186. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G. An immune dysregulation in MPN. Curr. Hematol. Malig. Rep. 2014, 9, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, T.; Osawa, Y.; Izumi, T.; Nagao, S.; Takano, K.; Okada, Y.; Tachi, N.; Teramoto, M.; Kawamura, T.; Horiuchi, T.; et al. Myeloproliferative leukemia protein activation directly induces fibrocyte differentiation to cause myelofibrosis. Leukemia 2017, 31, 2709–2716. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Li, L.; Yang, S.H.; Gao, C.Y.; Liao, L.H.; Xie, Y.Q.; Yin, X.Y.; Yang, Y.Q.; Fei, Y.Y.; Lian, Z.X. CD8(+) T cells and IFN-gamma induce autoimmune myelofibrosis in mice. J. Autoimmun. 2018, 89, 101–111. [Google Scholar] [CrossRef]
- Skov, V.; Riley, C.H.; Thomassen, M.; Kjaer, L.; Stauffer Larsen, T.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. The impact of interferon-alpha2 on HLA genes in patients with polycythemia vera and related neoplasms. Leuk. Lymphoma 2017, 58, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Tibullo, D.; Longo, A.; Vicario, N.; Romano, A.; Barbato, A.; Di Rosa, M.; Barbagallo, I.; Anfuso, C.D.; Lupo, G.; Gulino, R.; et al. Ixazomib Improves Bone Remodeling and Counteracts sonic Hedgehog signaling Inhibition Mediated by Myeloma Cells. Cancers (Basel) 2020, 12, 323. [Google Scholar] [CrossRef]
- Zahr, A.A.; Salama, M.E.; Carreau, N.; Tremblay, D.; Verstovsek, S.; Mesa, R.; Hoffman, R.; Mascarenhas, J. Bone marrow fibrosis in myelofibrosis: Pathogenesis, prognosis and targeted strategies. Haematologica 2016, 101, 660–671. [Google Scholar] [CrossRef]
- Wehenkel, M.; Ban, J.O.; Ho, Y.K.; Carmony, K.C.; Hong, J.T.; Kim, K.B. A selective inhibitor of the immunoproteasome subunit LMP2 induces apoptosis in PC-3 cells and suppresses tumour growth in nude mice. Br. J. Cancer 2012, 107, 53–62. [Google Scholar] [CrossRef]
- Keller, I.E.; Vosyka, O.; Takenaka, S.; Kloss, A.; Dahlmann, B.; Willems, L.I.; Verdoes, M.; Overkleeft, H.S.; Marcos, E.; Adnot, S.; et al. Regulation of immunoproteasome function in the lung. Sci. Rep. 2015, 5, 10230. [Google Scholar] [CrossRef]
- Cabrera, C.M.; Jimenez, P.; Cabrera, T.; Esparza, C.; Ruiz-Cabello, F.; Garrido, F. Total loss of MHC class I in colorectal tumors can be explained by two molecular pathways: Beta2-microglobulin inactivation in MSI-positive tumors and LMP7/TAP2 downregulation in MSI-negative tumors. Tissue Antigens 2003, 61, 211–219. [Google Scholar] [CrossRef]
- Kaur, G.; Batra, S. Emerging role of immunoproteasomes in pathophysiology. Immunol. Cell Biol. 2016, 94, 812–820. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.K.; Weinberg, J.B. The immunoproteasome and viral infection: A complex regulator of inflammation. Front. Microbiol. 2015, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [PubMed]
- Norfo, R.; Zini, R.; Pennucci, V.; Bianchi, E.; Salati, S.; Guglielmelli, P.; Bogani, C.; Fanelli, T.; Mannarelli, C.; Rosti, V.; et al. Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative, I., miRNA-mRNA integrative analysis in primary myelofibrosis CD34+ cells: Role of miR-155/JARID2 axis in abnormal megakaryopoiesis. Blood 2014, 124, e21–e32. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Xiao, J.; Cao, H.; Chen, J. False discovery rate control incorporating phylogenetic tree increases detection power in microbiome-wide multiple testing. Bioinformatics 2017, 33, 2873–2881. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. 2004, 3, Article3. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef]
- Zuberi, K.; Franz, M.; Rodriguez, H.; Montojo, J.; Lopes, C.T.; Bader, G.D.; Morris, Q. GeneMANIA prediction server 2013 update. Nucleic Acids Res. 2013, 41, W115–W122. [Google Scholar] [CrossRef]
- Chang, J.T.; Nevins, J.R. GATHER: A systems approach to interpreting genomic signatures. Bioinformatics 2006, 22, 2926–2933. [Google Scholar] [CrossRef]
- Cheadle, C.; Vawter, M.P.; Freed, W.J.; Becker, K.G. Analysis of microarray data using Z score transformation. J. Mol. Diagn.: JMD 2003, 5, 73–81. [Google Scholar] [CrossRef]
- Sanfilippo, C.; Castrogiovanni, P.; Imbesi, R.; Tibullo, D.; Li Volti, G.; Barbagallo, I.; Vicario, N.; Musumeci, G.; Di Rosa, M. Middle-aged healthy women and Alzheimer’s disease patients present an overlapping of brain cell transcriptional profile. Neurosci. 2019, 406, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N° | Dataset | GPL | Healthy | PMF | CD34+ | JAK2V617F +JAK2V617F − | |
|---|---|---|---|---|---|---|---|
| 1 | GSE53482 | GPL13667 | 16 | 42 | PB | 23 | 19 |
| 2 | GSE41812 | GPL13667 | 0 | 20 | PB | 11 | 9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Rosa, M.; Giallongo, C.; Romano, A.; Tibullo, D.; Li Volti, G.; Musumeci, G.; Barbagallo, I.; Imbesi, R.; Castrogiovanni, P.; Palumbo, G.A. Immunoproteasome Genes Are Modulated in CD34+ JAK2V617F Mutated Cells from Primary Myelofibrosis Patients. Int. J. Mol. Sci. 2020, 21, 2926. https://doi.org/10.3390/ijms21082926
Di Rosa M, Giallongo C, Romano A, Tibullo D, Li Volti G, Musumeci G, Barbagallo I, Imbesi R, Castrogiovanni P, Palumbo GA. Immunoproteasome Genes Are Modulated in CD34+ JAK2V617F Mutated Cells from Primary Myelofibrosis Patients. International Journal of Molecular Sciences. 2020; 21(8):2926. https://doi.org/10.3390/ijms21082926
Chicago/Turabian StyleDi Rosa, Michelino, Cesarina Giallongo, Alessandra Romano, Daniele Tibullo, Giovanni Li Volti, Giuseppe Musumeci, Ignazio Barbagallo, Rosa Imbesi, Paola Castrogiovanni, and Giuseppe A. Palumbo. 2020. "Immunoproteasome Genes Are Modulated in CD34+ JAK2V617F Mutated Cells from Primary Myelofibrosis Patients" International Journal of Molecular Sciences 21, no. 8: 2926. https://doi.org/10.3390/ijms21082926
APA StyleDi Rosa, M., Giallongo, C., Romano, A., Tibullo, D., Li Volti, G., Musumeci, G., Barbagallo, I., Imbesi, R., Castrogiovanni, P., & Palumbo, G. A. (2020). Immunoproteasome Genes Are Modulated in CD34+ JAK2V617F Mutated Cells from Primary Myelofibrosis Patients. International Journal of Molecular Sciences, 21(8), 2926. https://doi.org/10.3390/ijms21082926