Transitional States in Ligand-Dependent Transformation of the Aryl Hydrocarbon Receptor into Its DNA-Binding Form


Abstract
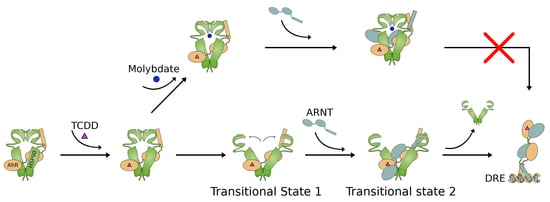
1. Introduction
2. Results
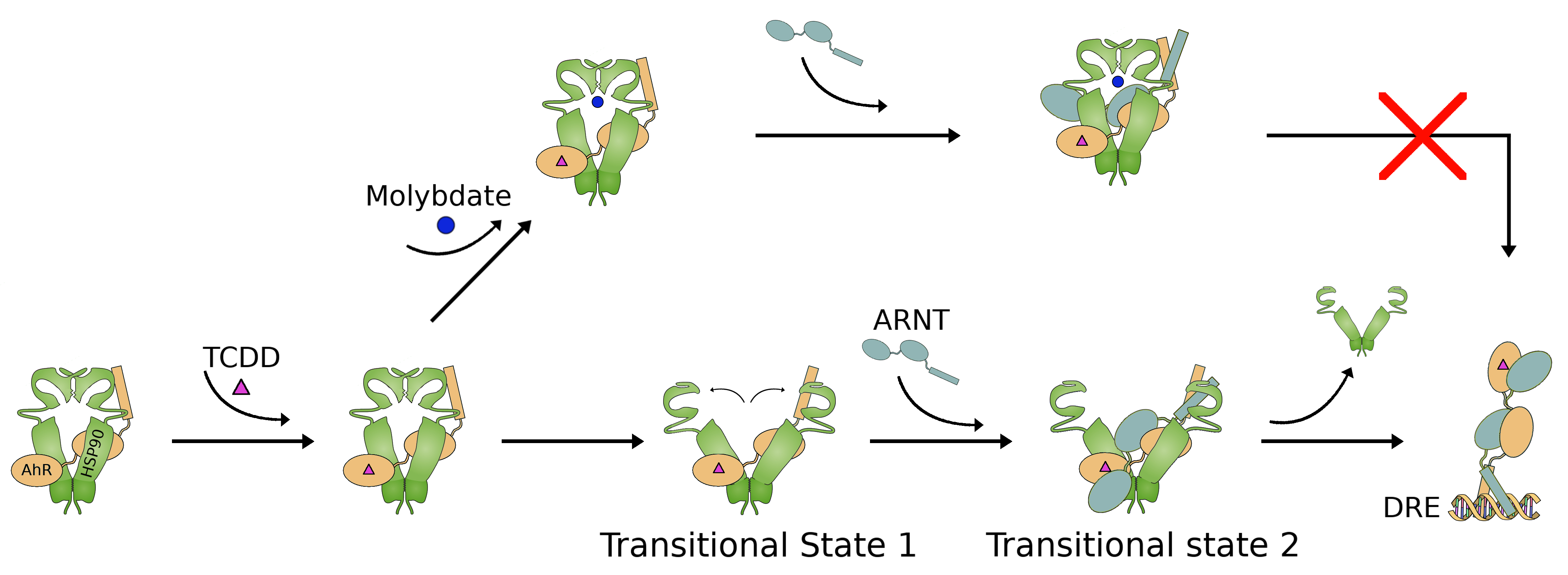
2.1. Study of AhR Transformation through Mutagenesis within the PASB Domain
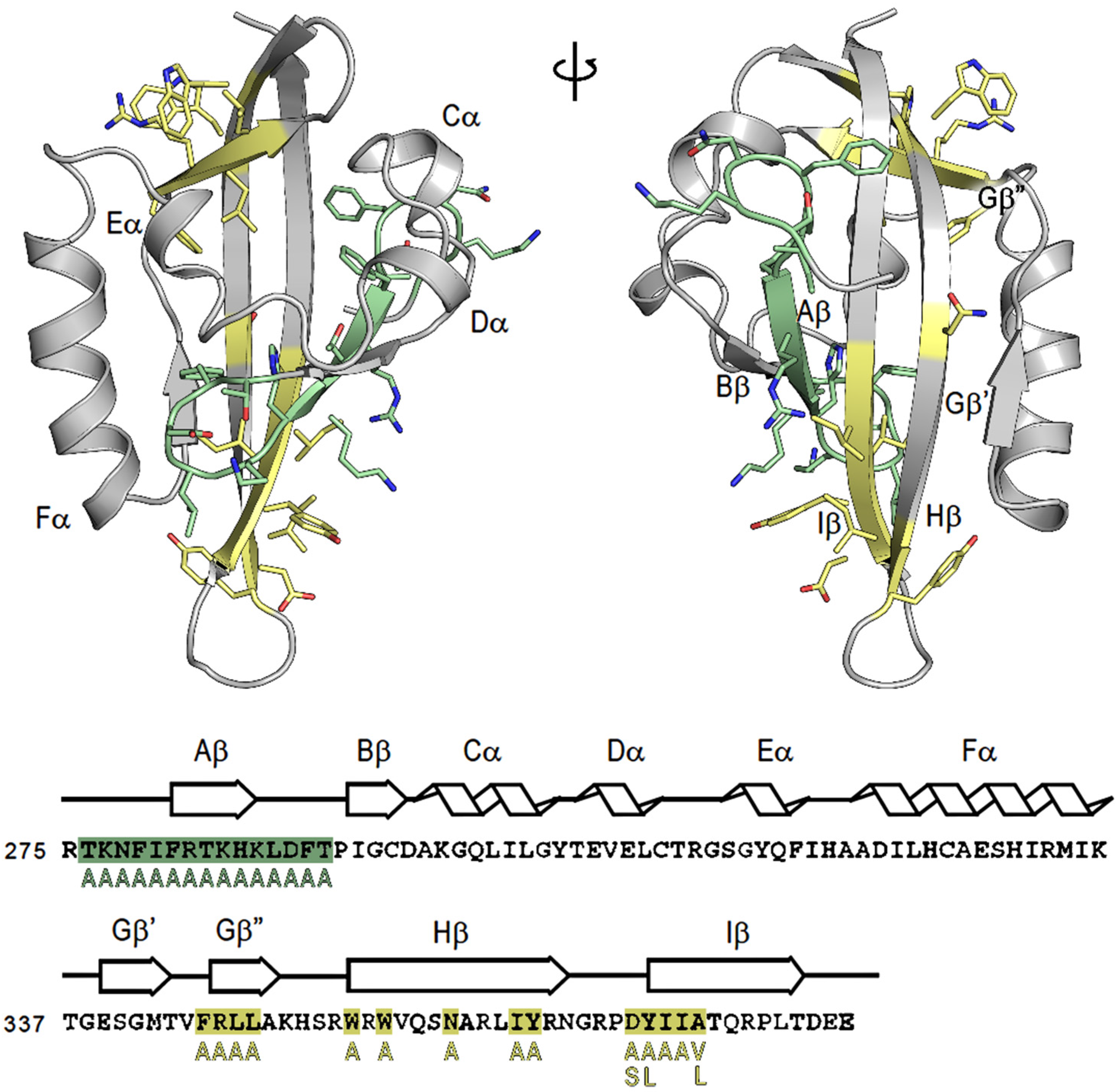
2.2. N-Terminal Mutants: F281A Results in Constitutive Activation of AhR Transformation/DNA Binding
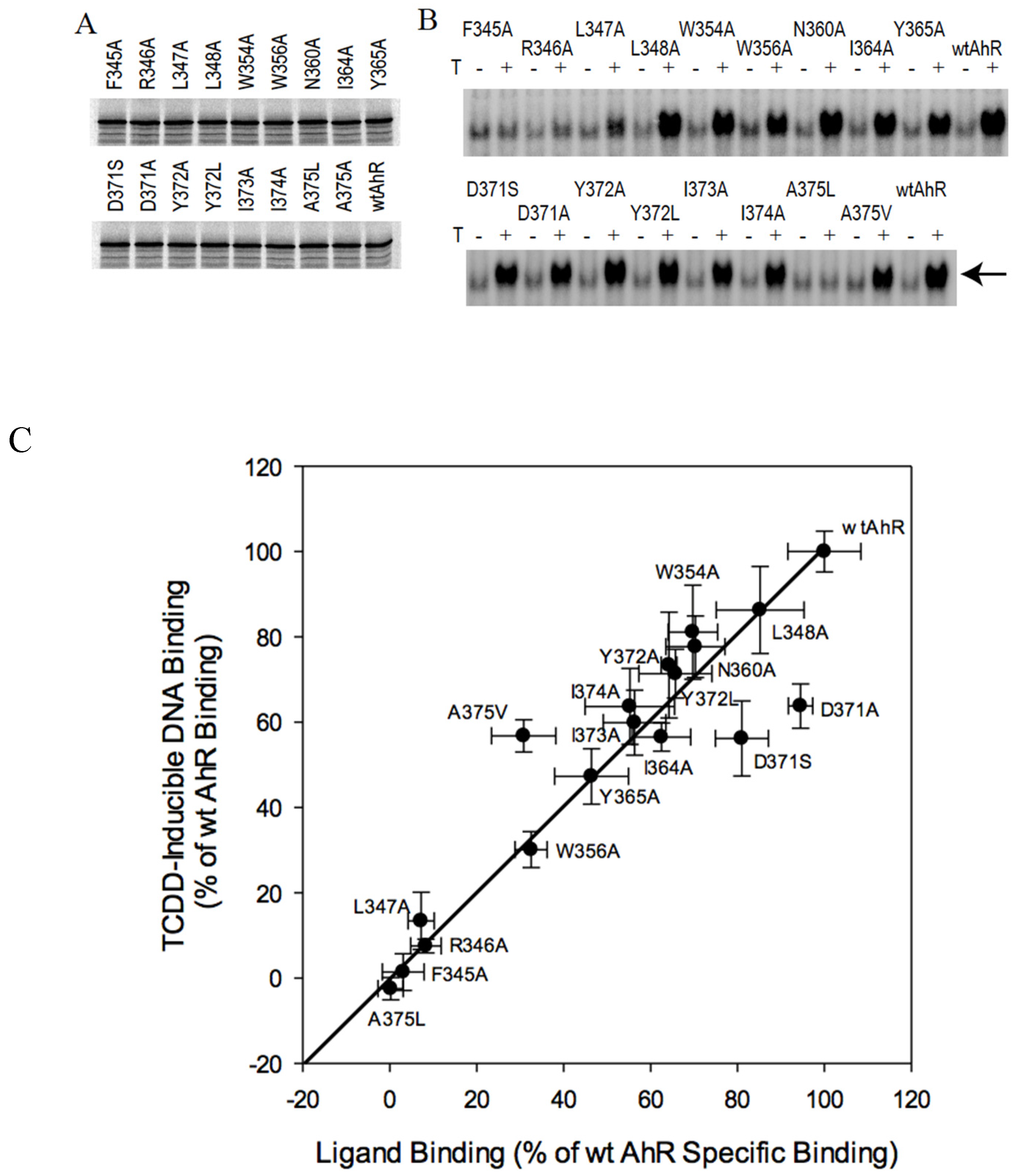
2.3. C-Terminal Mutants: D371A Impairs AhR Transformation
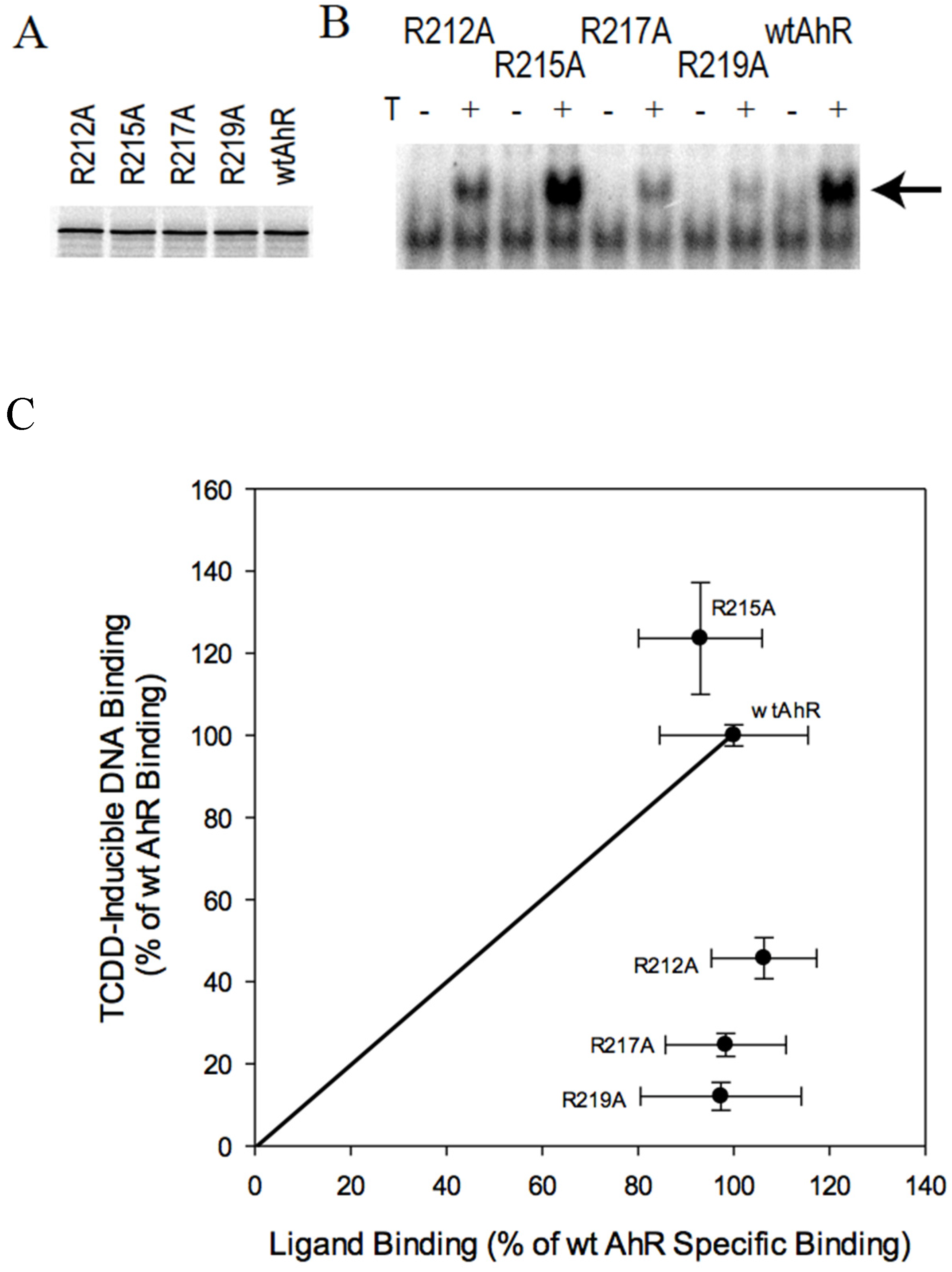
2.4. AhR Transformation Mutations in the PASA Domain
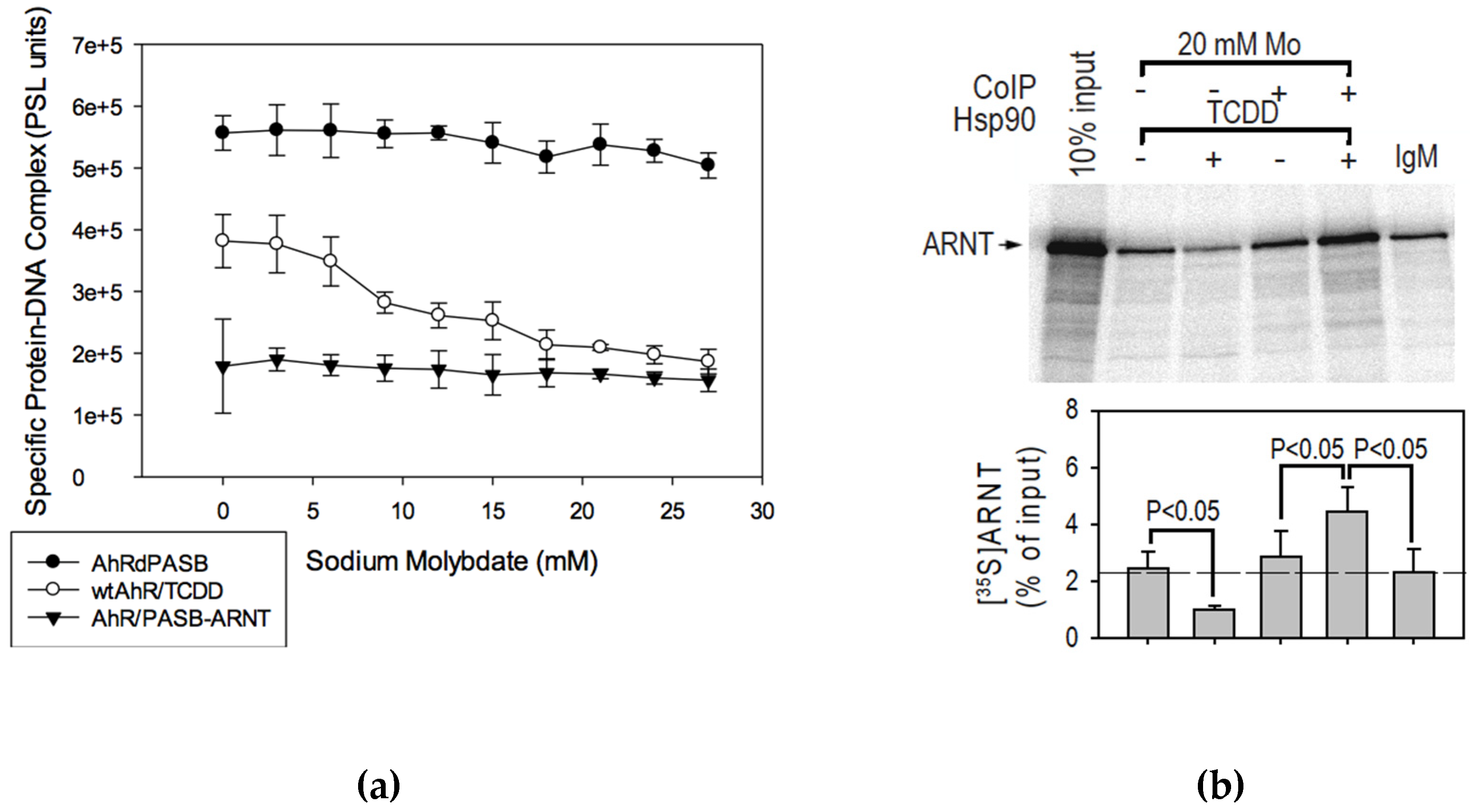
2.5. Effect of Sodium Molybdate on AhR Transformation
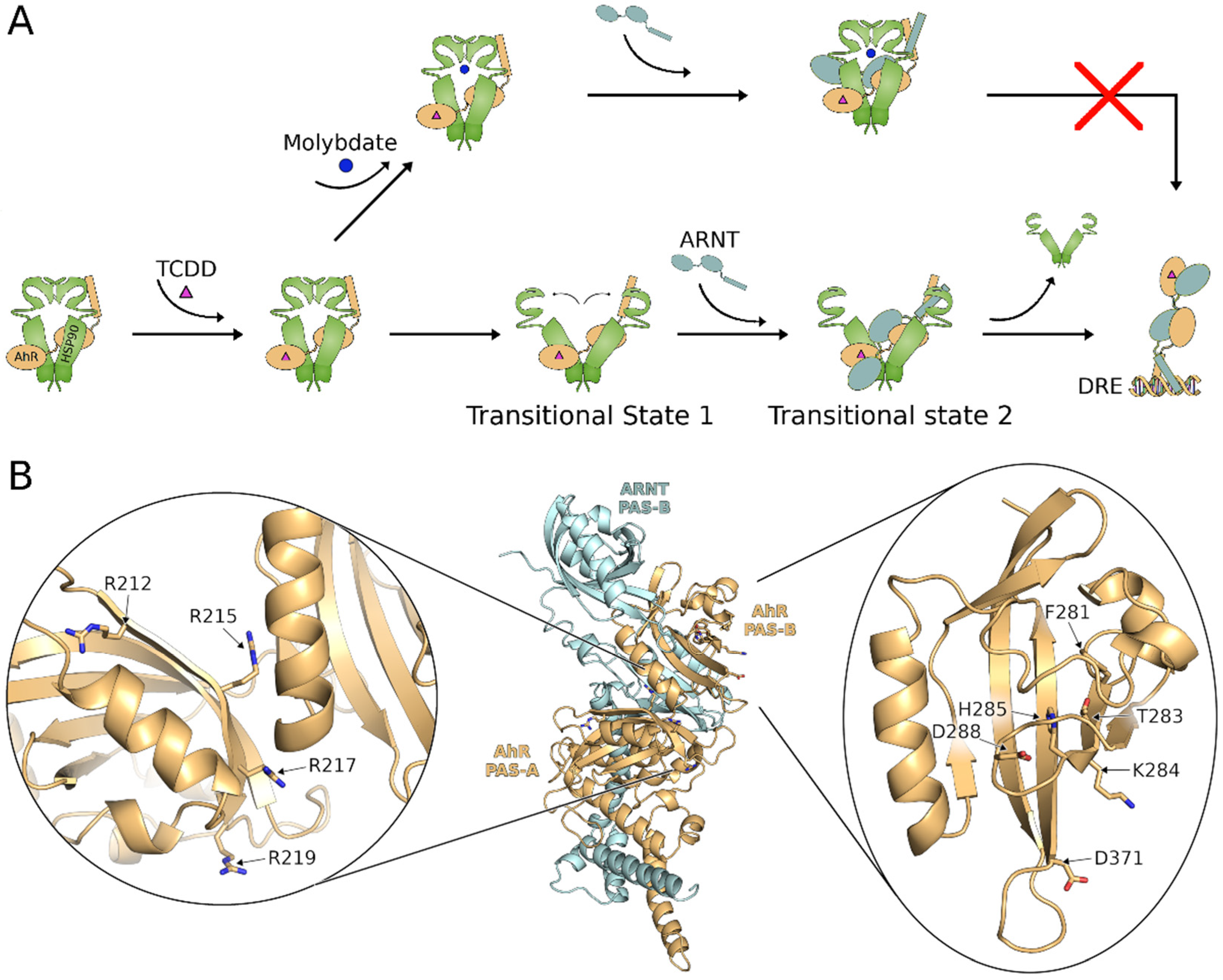
2.6. Molybdate Stabilization of An AhR:hsp90:ARNT Transitional Complex
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Plasmid Constructs
4.3. In vitro Expression
4.4. Hydroxyapatite (HAP) Ligand Binding Assays
4.5. Gel Retardation Assays
4.6. Co-Immunoprecipitation Assay
4.7. Statistical Analysis
4.8. AhR PASB LBD Homology Model
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef]
- Rothammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receprtor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef]
- Roman, Á.C.; Carvajal-Gonzalez, J.M.; Merino, J.M.; Mulero-Navarro, S.; Fernándex-Salguero, P.M. The aryl hydrocarbon receptor in the crossroad of signaling networks with therapeutic value. Pharmacol. Ther. 2018, 185, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.P.; Bradfield, C.A. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 2008, 21, 102–116. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and tryptophan metabolism: Endogenous and dietary routes to ah receptor activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef]
- Bunger, M.K.; Glover, E.; Moran, S.M.; Walisser, J.A.; Lahvis, G.P.; Hsu, E.L.; Bradfield, C.A. Abnormal liver development and resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in mice carrying a mutation in the DNA-binding domain of the aryl hydrocarbon receptor. Toxicol. Sci. 2008, 106, 83–92. [Google Scholar] [CrossRef]
- Fernández-Salguero, P.M.; Hilbert, D.M.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 1996, 140, 173–179. [Google Scholar] [CrossRef]
- Birnbaum, L.S. The mechanism of dioxin toxicity: Relationship to risk assessment. Environ. Health Perspect. 1994, 102, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Singh, S.S.; Perdew, G.H. The Ah receptor is a sensitive target of geldanamycin-induced protein turnover. Arch. Biochem. Biophys. 1997, 348, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.R.; Poland, A. Binding of aryl hydrocarbon receptor (AhR) to AhR-interacting protein. The role of hsp90. J. Biol. Chem. 2000, 275, 36407–36414. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, T.; Eguchi, H.; Tachibana, T.; Yoneda, Y.; Kawajiri, K. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J. Biol. Chem. 1998, 273, 2895–2904. [Google Scholar] [CrossRef]
- Henry, E.C.; Gasiewicz, T.A. Transformation of the aryl hydrocarbon receptor to a DNA-binding form is accompanied by release of the 90 kDa heat-shock protein and increased affinity for 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. J. 1993, 294, 95–101. [Google Scholar] [CrossRef]
- Fukunaga, B.N.; Probst, M.R.; Reisz-Porszasz, S.; Hankinson, O. Identification of functional domains of the aryl hydrocarbon receptor. J. Biol. Chem. 1995, 270, 29270–29278. [Google Scholar] [CrossRef]
- Denison, M.S.; Fisher, J.M.; Whitlock, J.P., Jr. The DNA recognition site for the dioxin-Ah receptor complex. Proc. Natl. Acad. Sci. USA 1988, 263, 17221–17224. [Google Scholar] [CrossRef]
- Probst, M.R.; Reisz-Porszasz, S.; Agbunag, R.V.; Ong, M.S.; Hankinson, O. Role of the aryl hydrocarbon receptor nuclear translocator protein in aryl hydrocarbon (dioxin) receptor action. Mol. Pharmacol. 1993, 44, 511–518. [Google Scholar]
- Heid, S.E.; Pollenz, R.S.; Swanson, H.I. Role of heat shock protein 90 dissociation in mediating agonist-induced activation of the aryl hydrocarbon receptor. Mol. Pharmacol. 2000, 57, 82–92. [Google Scholar]
- Perdew, G.H. Comparison of the nuclear and cytosolic forms of the Ah receptor from Hepa 1c1c7 cells: Charge heterogeneity and ATP binding properties. Arch. Biochem. Biophys. 1991, 291, 284–290. [Google Scholar] [CrossRef]
- Soshilov, A.; Denison, M.S. Role of the Per/Arnt/Sim domains in ligand-dependent transformation of the aryl hydrocarbon receptor. J. Biol. Chem. 2008, 283, 32995–33005. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, N.; Fukuda, K.; Nagata, Y.; Okada, H.; Haga, A.; Hatakeyama, S.; Yoshida, S.; Okamoto, T.; Hosaka, M.; Sekine, K.; et al. The activation mechanism of the aryl hydrocarbon receptor (AhR) by molecular chaperone HSP90. FEBS Open Bio 2014, 4, 796–803. [Google Scholar] [CrossRef]
- Soshilov, A.; Denison, M.S. Ligand displaces heat shock protein 90 from overlapping binding sites within the aryl hydrocarbon receptor ligand binding domain. J. Biol. Chem. 2011, 286, 35275–35282. [Google Scholar] [CrossRef] [PubMed]
- Perdew, G.H.; Bradfield, C.A. Mapping the 90 kDa heat shock protein binding region of the Ah receptor. Biochem. Mol. Biol. Int. 1996, 39, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Moglich, A.; Ayers, R.A.; Moffat, K. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure 2009, 17, 1282–1294. [Google Scholar] [CrossRef]
- Blankenburg, L.; Schroeder, L.; Habenstein, F.; Błasiak, B.; Kottke, T.; Bredenbeck, J. Following local light-induced structure changes and dynamics of the photoreceptor PYP with the thiocyanate IR label. Phys. Chem. Chem. Phys. 2019, 21, 6622–6634. [Google Scholar] [CrossRef]
- Halavaty, A.S.; Moffat, K. N- and C-terminal flanking regions modulate light-induced signal transduction in the LOV2 domain of the blue light sensor phototropin 1 from Avena sativa. Biochemistry 2007, 46, 14001–14009. [Google Scholar] [CrossRef]
- Key, J.; Moffat, K. Crystal structures of deoxy and CO-bound bjFixLH reveal details of ligand recognition and signaling. Biochemistry 2005, 44, 4627–4635. [Google Scholar] [CrossRef]
- Sevvana, M.; Vijayan, V.; Zweckstetter, M.; Reinelt, S.; Madden, D.R.; Herbst-Irmer, R.; Sheldrick, G.M.; Bott, M.; Griesinger, C.; Becker, S. A ligand-induced switch in the periplasmic domain of sensor histidine kinase CitA. J. Mol. Biol. 2008, 377, 512–523. [Google Scholar] [CrossRef]
- Motto, I.; Bordogna, A.; Soshilov, A.; Denison, M.S.; Bonati, L. New aryl hydrocarbon receptor homology model targeted to improve docking reliability. J. Chem. Inf. Model. 2011, 51, 2868–2881. [Google Scholar] [CrossRef]
- Giani Tagliabue, S.; Faber, S.C.; Motta, S.; Denison, M.S.; Bonati, L. Modeling the binding of diverse ligands within the Ah receptor ligand binding domain. Sci. Rep. 2019, 9, 10693. [Google Scholar] [CrossRef] [PubMed]
- Corrada, D.; Soshilov, A.A.; Denison, M.S.; Bonati, L. Deciphering dimerization modes of PAS domains: Computational and experimental analyses of the AhR:ARNT complex reveal new insights into the mechanisms of AhR transformation. PLoS Comput. Biol. 2016, 12, e1004981. [Google Scholar] [CrossRef] [PubMed]
- Corrada, D.; Denison, M.S.; Bonati, L. Structural modeling of the AhR:ARNT complex in the bHLH-PASA-PASB region elucidates the key determinants of dimerization. Mol. Biosyst. 2017, 13, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Phelps, C.L.; Dehoog, J.; Kim, H.J.; Bank, P.A.; Yao, E.F. Species Variation in Ah Receptor Transformation and DNA Binding. In Banbury Report 35: Biological Basis for Risk Assessment of Dioxins and Related Compounds; Gallo, M.A., Scheuplein, R.J., van der Heihjden, K.A., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1991; pp. 337–349. [Google Scholar]
- Harper, P.A.; Giannone, J.V.; Okey, A.B.; Denison, M.S. In vitro transformation of the human Ah receptor and its binding to a dioxin response element. Mol. Pharmacol. 1992, 42, 603–612. [Google Scholar] [PubMed]
- Whitelaw, M.L.; Gottlicher, M.; Gustafsson, J.A.; Poellinger, L. Definition of a novel ligand binding domain of a nuclear bHLH receptor: Co-localization of ligand and hsp90 binding activities within the regulable inactivation domain of the dioxin receptor. EMBO J. 1993, 12, 4169–4179. [Google Scholar] [CrossRef] [PubMed]
- Coumailleau, P.; Poellinger, L.; Gustafsson, J.A.; Whitelaw, M.L. Definition of a minimal domain of the dioxin receptor that is associated with Hsp90 and maintains wild type ligand binding affinity and specificity. J. Biol. Chem. 1995, 270, 25291–25300. [Google Scholar] [CrossRef]
- Whitelaw, M.L.; Gustafsson, J.A.; Poellinger, L. Identification of transactivation and repression functions of the dioxin receptor and its basic helix-loop-helix/PAS partner factor Arnt: Inducible versus constitutive modes of regulation. Mol. Cell. Biol. 1994, 14, 8343–8355. [Google Scholar] [CrossRef]
- Pandini, A.; Soshilov, A.A.; Song, Y.; Zhao, J.; Bonati, L.; Denison, M.S. Detection of the TCDD binding-fingerprint within the Ah receptor ligand binding domain by structurally driven mutagenesis and functional analysis. Biochemistry 2009, 48, 5972–5983. [Google Scholar] [CrossRef]
- Poland, A.; Palen, D.; Glover, E. Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol. Pharmacol. 1994, 46, 915–921. [Google Scholar]
- Sun, W.; Zhang, J.; Hankinson, O. A mutation in the aryl hydrocarbon receptor (AHR) in a cultured mammalian cell line identifies a novel region of AHR that affects DNA binding. J. Biol. Chem. 1997, 272, 31845–31854. [Google Scholar] [CrossRef]
- Denison, M.S.; Vella, L.M.; Okey, A.B. Hepatic Ah receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin. Partial stabilization by molybdate. J. Biol. Chem. 1986, 261, 10189–10195. [Google Scholar] [PubMed]
- Karchner, S.I.; Franks, D.G.; Kennedy, S.W.; Hahn, M.E. The molecular basis for differential dioxin sensitivity in birds: Role of the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 6252–6257. [Google Scholar] [CrossRef] [PubMed]
- Fraccalvieri, D.; Soshilov, A.A.; Karchner, S.I.; Franks, D.G.; Pandini, A.; Bonati, L.; Hahn, M.E.; Denison, M.S. Comparative analysis of homology models of the Ah receptor ligand binding domain: Verification of structure-function predictions by site-directed mutagenesis of a nonfunctional receptor. Biochemistry 2013, 52, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Faber, S.C. And now for something completely different: Diversity in ligand-dependent activation of Ah receptor responses. Curr. Opin. Toxicol. 2017, 2, 124–131. [Google Scholar] [CrossRef]
- Murray, I.A.; Morales, J.L.; Flaveny, C.A.; DiNatale, B.C.; Chiaro, C.; Gowdahalli, K.; Amin, S.; Perdew, G.H. Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity. Mol. Pharmacol. 2010, 77, 247–254. [Google Scholar] [CrossRef]
- Nault, R.; Forgacs, A.L.; Dere, E.; Zacharewski, T.R. Comparisons of differential gene expression elicited by TCDD, PCB126, betaNF, or ICZ in mouse hepatoma Hepa1c1c7 cells and C57BL/6 mouse liver. Toxicol. Lett. 2013, 223, 52–59. [Google Scholar] [CrossRef]
- Dere, E.; Lo, R.; Celius, T.; Matthews, J.; Zacharewski, T.R. Integration of genome-wide computation DRE search, AhR ChiP-chip and gene expression analyses of TCDD-elicited responses in the mouse liver. BMC Genomics 2011, 12, 365. [Google Scholar] [CrossRef]
- Bohonowych, J.E.; Denison, M.S. Persistent binding of ligands to the aryl hydrocarbon receptor. Toxicol. Sci. 2007, 98, 99–109. [Google Scholar] [CrossRef]
- Lees, M.J.; Whitelaw, M.L. Multiple roles of ligand in transforming the dioxin receptor to an active basic helix-loop-helix/PAS transcription factor complex with the nuclear protein Arnt. Mol Cell. Biol. 1999, 19, 5811–5822. [Google Scholar] [CrossRef]
- DeGroot, D.E.; He, G.; Fraccalvieri, D.; Bonati, L.; Pandini, A.; Denison, M.S. AhR Ligands: Promiscuity in Binding and Diversity in Response. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 63–79. [Google Scholar]
- Verba, K.A.; Wang, R.Y.-R.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell. Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Kudo, I.; Hosaka, M.; Haga, A.; Tsuji, N.; Nagata, Y.; Okada, H.; Fukuda, K.; Kakizaki, Y.; Okamoto, T.; Grave, E.; et al. The regulation mechanisms of AhR by molecular chaperone complex. J. Biochem. 2018, 163, 223–232. [Google Scholar] [CrossRef]
- Lindebro, M.C.; Poellinger, L.; Whitelaw, M.L. Protein-protein interaction via PAS domains: Role of the PAS domain in positive and negative regulation of the bHLH/PAS dioxin receptor-Arnt transcription factor complex. EMBO J. 1995, 14, 3528–3539. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Echeverría, P.C.; Picard, D. Overview of Molecular Chaperones in Health and Disease. In Inhibitors of Molecular Chaperones as Therapeutic Agents; Machajewski, T.D., Gao, Z., Eds.; Royal Society of Chemistry: Cambridge, UK, 2013; pp. 1–36. [Google Scholar]
- Motta, S.; Minici, C.; Corrada, D.; Bonati, L.; Pandini, A. Ligand-induced perturbation of the HIF-2 α:ARNT dimer dynamics. PLoS Compl. Biol. 2018, 14, e1006021. [Google Scholar] [CrossRef]
- Dolwick, K.M.; Swanson, H.I.; Bradfield, C.A. In vitro analysis of Ah receptor domains involved in ligand-activated DNA recognition. Proc. Natl. Acad. Sci. USA 1993, 90, 8566–8570. [Google Scholar] [CrossRef]
- Fukunaga, B.N.; Hankinson, O. Identification of a novel domain in the aryl hydrocarbon receptor required for DNA binding. J. Biol. Chem. 1996, 271, 3743–3749. [Google Scholar] [CrossRef]
- Denison, M.S.; Rogers, J.M.; Rushing, S.R.; Jones, C.L.; Tetangco, S.C.; Heath-Pagliuso, S. Analysis of the Aryl Hydrocarbon Receptor (AhR) Signal Transduction Pathway. In Current Protocols in Toxicology; Morgan, K.S., Ed.; John Wiley: New York, NY, USA, 2002. [Google Scholar]
- Soshilov, A.A.; Denison, M.S. DNA Binding (Gel Retardation Assay) Analysis for Identification of Aryl Hydrocarbon (Ah) Receptor Agonists and Antagonists. In Optimization of Drug Discovery: In Vitro Methods, 2nd ed.; Yan, A., Caldwell, G.W., Eds.; Humana Press: New York, NY, USA, 2014; pp. 207–219. [Google Scholar]
- Wu, D.; Potluri, N.; Lu, J.; Kim, Y.; Rastinejad, F. Structural integration in hypoxia-inducible factors. Nature 2015, 524, 303–308. [Google Scholar] [CrossRef]
- PyMOL. The PyMOL Molecular Graphics System, Version 1.6; Schrödinger LLC: New York, NY, USA, 2010. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AhR Mutation | Mode of Activation | Percent Inhibition by Molybdate |
|---|---|---|
| AhRdPASB | Constitutively Active | 4.3 ± 5.8 a |
| AhR/PASB-ARNT | Constitutively Active | 9.1 ± 4.1 |
| AhR F281A | Constitutively Active | 5.0 ± 12.1 |
| AhR R217A | Ligand-Dependent | 67.1 ± 7.3 b |
| AhR D371A | Ligand-Dependent | 61.7 ± 11.5 b |
| wtAhR | Ligand Dependent | 69.6 ± 6.6 b |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soshilov, A.A.; Motta, S.; Bonati, L.; Denison, M.S. Transitional States in Ligand-Dependent Transformation of the Aryl Hydrocarbon Receptor into Its DNA-Binding Form. Int. J. Mol. Sci. 2020, 21, 2474. https://doi.org/10.3390/ijms21072474
Soshilov AA, Motta S, Bonati L, Denison MS. Transitional States in Ligand-Dependent Transformation of the Aryl Hydrocarbon Receptor into Its DNA-Binding Form. International Journal of Molecular Sciences. 2020; 21(7):2474. https://doi.org/10.3390/ijms21072474
Chicago/Turabian StyleSoshilov, Anatoly A., Stefano Motta, Laura Bonati, and Michael S. Denison. 2020. "Transitional States in Ligand-Dependent Transformation of the Aryl Hydrocarbon Receptor into Its DNA-Binding Form" International Journal of Molecular Sciences 21, no. 7: 2474. https://doi.org/10.3390/ijms21072474
APA StyleSoshilov, A. A., Motta, S., Bonati, L., & Denison, M. S. (2020). Transitional States in Ligand-Dependent Transformation of the Aryl Hydrocarbon Receptor into Its DNA-Binding Form. International Journal of Molecular Sciences, 21(7), 2474. https://doi.org/10.3390/ijms21072474