Partners in Crime: The Interplay of Proteins and Membranes in Regulated Necrosis

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Plasma Membrane Permeabilization: A Common Feature of Regulated Necrosis
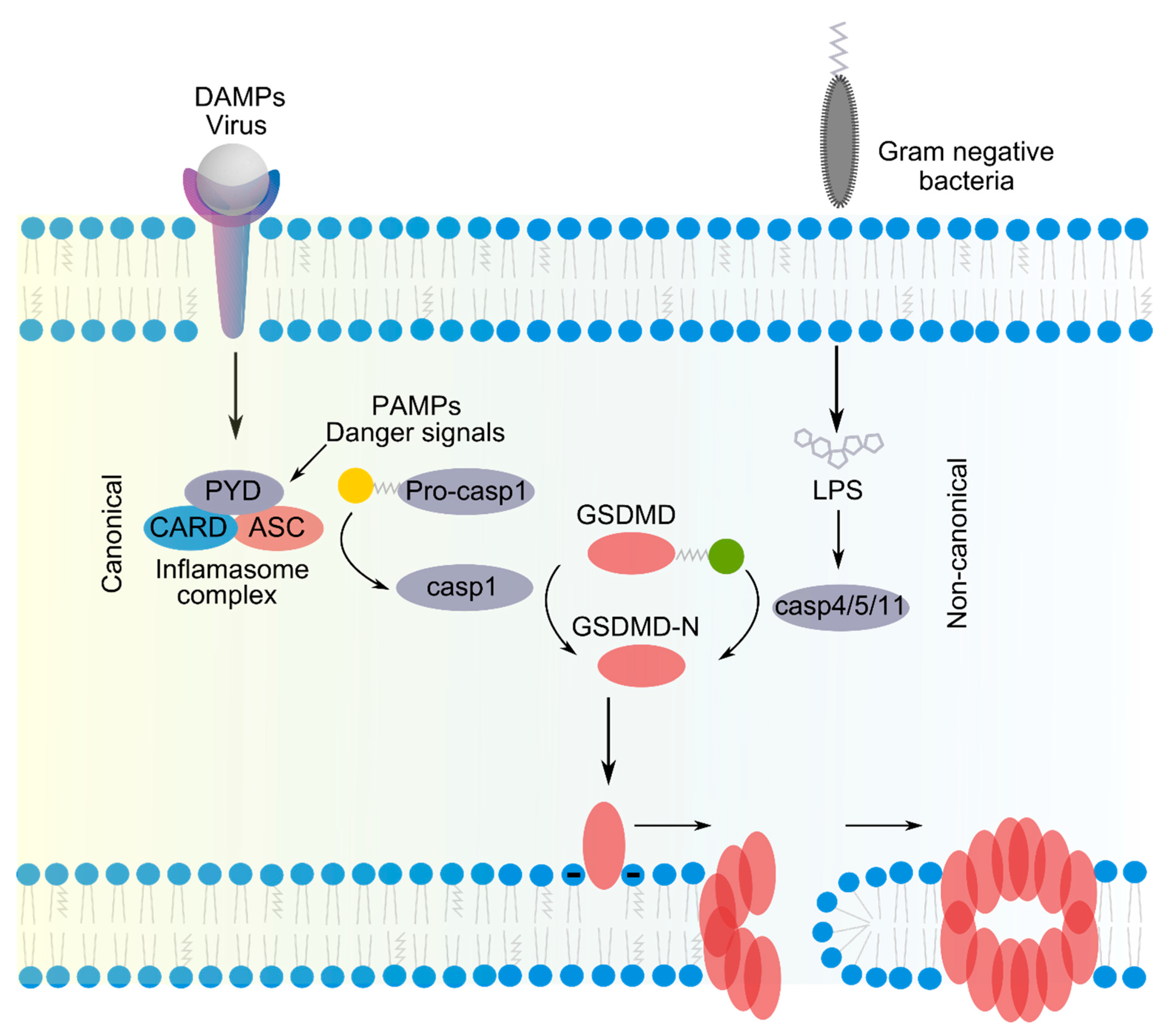
2.1. Pore Formation by GSDMs in Pyroptosis
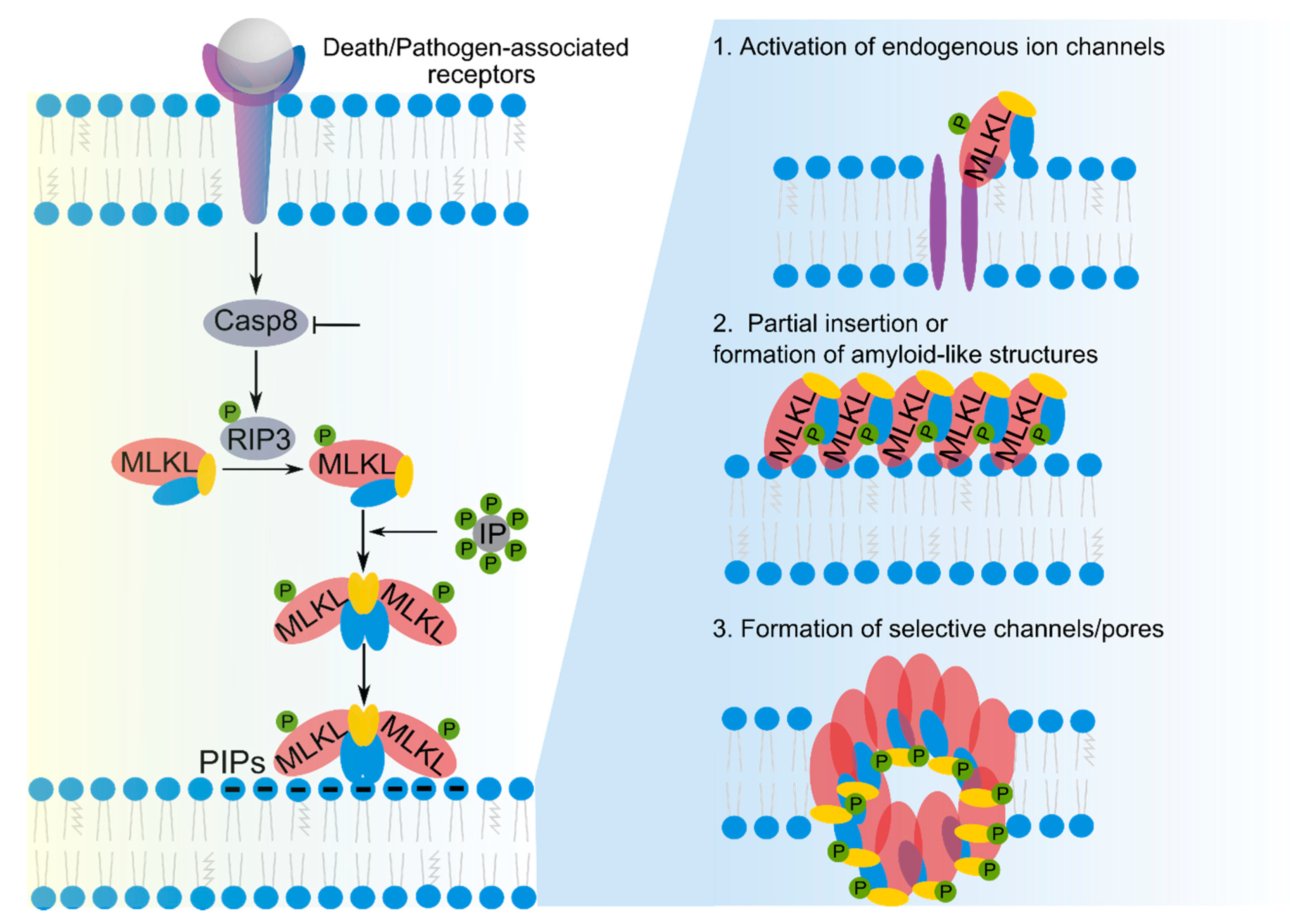
2.2. Membrane Permeabilization by MLKL in Necroptosis
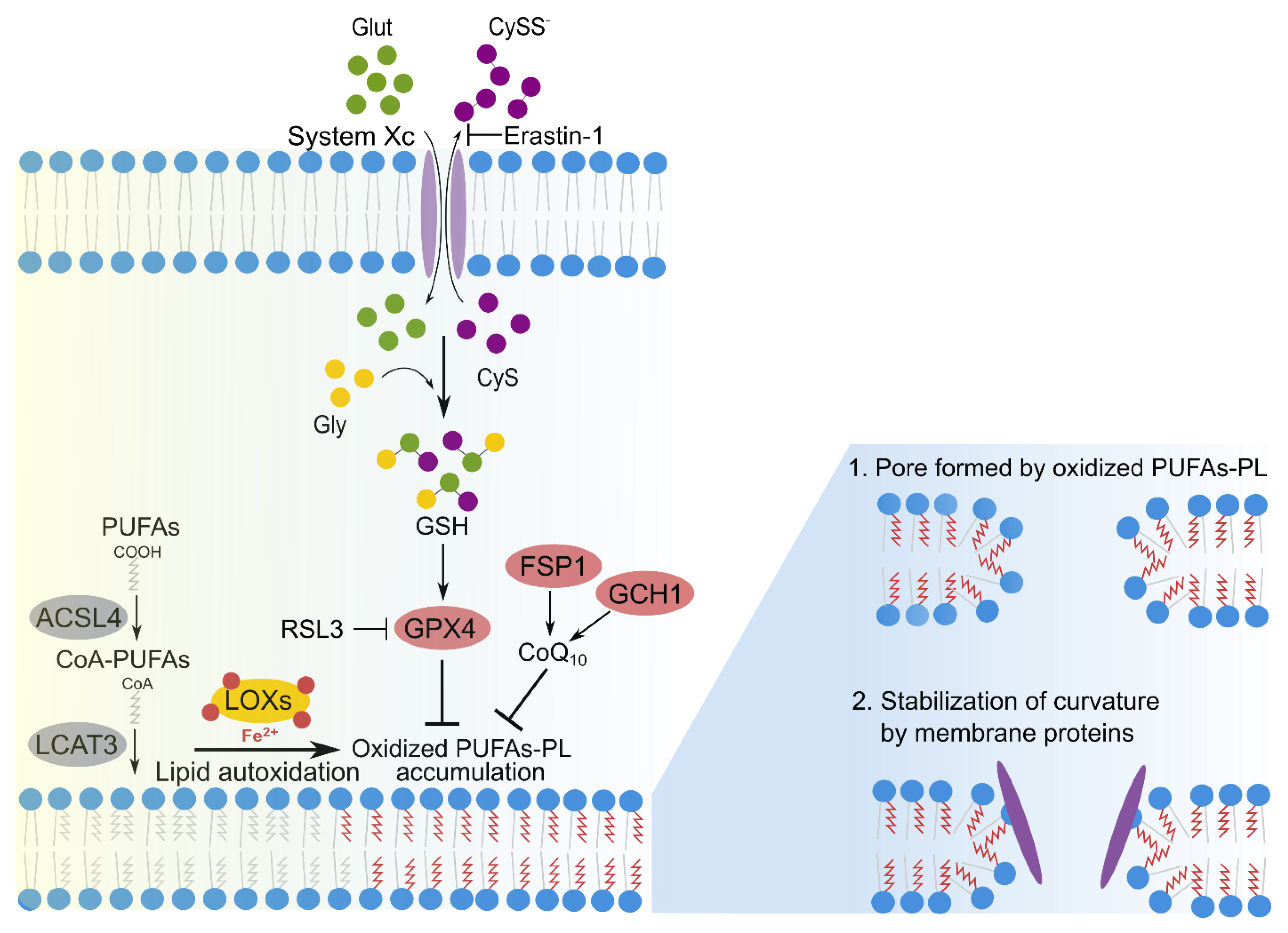
2.3. Lipid Peroxidation in Membranes during Ferroptosis
3. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature committee on cell death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Espiritu, R.A.; Pedrera, L.; Ros, U. Tuning the way to die: Implications of membrane perturbations in necroptosis. In Advances in Biomembranes and Lipid Self-Assembly; Iglič, A., Garcia-Sáez, A., Rappolt, M., Eds.; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Conrad, M.; Angeli, J.P.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and relevance to disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Therapeutic exploitation of necroptosis for cancer therapy. Semin. Cell Dev. Biol. 2014, 35, 51–56. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrin, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2019, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Bleriot, C.; Lecuit, M. The interplay between regulated necrosis and bacterial infection. Cell. Mol. Life Sci. 2016, 73, 2369–2378. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Proneth, B.; Conrad, M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ. 2019, 26, 14–24. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Sborgi, L.; Ruhl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Muller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. MLKL regulates necrotic plasma membrane permeabilization. Cell Res. 2014, 24, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.N.; Guy, C.; Crawford, J.C.; Green, D.R. Biological events and molecular signaling following MLKL activation during necroptosis. Cell Cycle 2017, 16, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.R.; Li, N.; Atilla-Gokcumen, G.E. Very long chain fatty acids are functionally involved in necroptosis. Cell Chem. Biol. 2017, 24, 1445–1454.e8. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Ko, P.J.; Dixon, S.J. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. 2016, 23, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.R.; Morrow, L.M.; Visser, M.B.; Atilla-Gokcumen, G.E. Turning the spotlight on lipids in non-apoptotic cell death. ACS Chem. Biol. 2018, 13, 506–515. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef]
- Gong, Y.N.; Guy, C.; Olauson, H.; Becker, J.U.; Yang, M.; Fitzgerald, P.; Linkermann, A.; Green, D.R. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 2017, 169, 286–300.e16. [Google Scholar] [CrossRef]
- Pedrera, L.; Espiritu, R.; Ros, U.; Garcia-Saez, A.J. Plasma membrane damage and calcium fluxes activate ESCRT-III. Eur. Biophys. J. 2019, 48, S161. [Google Scholar]
- Ruhl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Ros, U.; Garcia-Saez, A.J. More Than a Pore: The interplay of pore-forming proteins and lipid membranes. J. Membr. Biol. 2015, 248, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Aglietti, R.A.; Estevez, A.; Gupta, A.; Ramirez, M.G.; Liu, P.S.; Kayagaki, N.; Ciferri, C.; Dixit, V.M.; Dueber, E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 2016, 113, 7858–7863. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Xia, S.; Liu, X.; Lieberman, J.; Wu, H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature 2018, 557, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, T.; Zhou, Y.; Li, J.; Liang, X.; Zhou, N.; Lv, J.; Xie, J.; Cheng, F.; Fang, Y.; et al. Visualization of perforin/gasdermin/complement-formed pores in real cell membranes using atomic force microscopy. Cell. Mol. Immunol. 2019, 16, 611–620. [Google Scholar] [CrossRef]
- Gilbert, R.J.; Dalla Serra, M.; Froelich, C.J.; Wallace, M.I.; Anderluh, G. Membrane pore formation at protein-lipid interfaces. Trends Biochem. Sci. 2014, 39, 510–516. [Google Scholar] [CrossRef]
- Salvador-Gallego, R.; Mund, M.; Cosentino, K.; Schneider, J.; Unsay, J.; Schraermeyer, U.; Engelhardt, J.; Ries, J.; Garcia-Saez, A.J. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016, 35, 389–401. [Google Scholar] [CrossRef]
- Heilig, R.; Dick, M.S.; Sborgi, L.; Meunier, E.; Hiller, S.; Broz, P. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur. J. Immunol. 2018, 48, 584–592. [Google Scholar] [CrossRef]
- de Torre-Minguela, C.; Barbera-Cremades, M.; Gomez, A.I.; Martin-Sanchez, F.; Pelegrin, P. Macrophage activation and polarization modify P2X7 receptor secretome influencing the inflammatory process. Sci. Rep. 2016, 6, 22586. [Google Scholar] [CrossRef] [PubMed]
- de Vasconcelos, N.M.; Van Opdenbosch, N.; Van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2018, 26, 146–161. [Google Scholar] [CrossRef]
- Banerjee, I.; Behl, B.; Mendonca, M.; Shrivastava, G.; Russo, A.J.; Menoret, A.; Ghosh, A.; Vella, A.T.; Vanaja, S.K.; Sarkar, S.N.; et al. Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity 2018, 49, 413–426.e5. [Google Scholar] [CrossRef]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Petrie, E.J.; Hildebrand, J.M.; Murphy, J.M. Insane in the membrane: A structural perspective of MLKL function in necroptosis. Immunol. Cell Biol. 2017, 95, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Quarato, G.; Guy, C.S.; Grace, C.R.; Llambi, F.; Nourse, A.; Rodriguez, D.A.; Wakefield, R.; Frase, S.; Moldoveanu, T.; Green, D.R. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol. Cell 2016, 61, 589–601. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef]
- Dovey, C.M.; Diep, J.; Clarke, B.P.; Hale, A.T.; McNamara, D.E.; Guo, H.; Brown, N.W., Jr.; Cao, J.Y.; Grace, C.R.; Gough, P.J.; et al. MLKL Requires the Inositol Phosphate Code to Execute Necroptosis. Mol. Cell 2018, 70, 936–948.e7. [Google Scholar] [CrossRef]
- McNamara, D.E.; Dovey, C.M.; Hale, A.T.; Quarato, G.; Grace, C.R.; Guibao, C.D.; Diep, J.; Nourse, A.; Cai, C.R.; Wu, H.; et al. Direct Activation of human MLKL by a select repertoire of inositol phosphate metabolites. Cell Chem. Biol. 2019, 26, 863–877.e7. [Google Scholar] [CrossRef]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef]
- Fan, W.; Guo, J.; Gao, B.; Zhang, W.; Ling, L.; Xu, T.; Pan, C.; Li, L.; Chen, S.; Wang, H.; et al. Flotillin-mediated endocytosis and ALIX-syntenin-1-mediated exocytosis protect the cell membrane from damage caused by necroptosis. Sci. Signal. 2019, 12, eaaw3423. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Pan, C.; Shao, T.; Liu, L.; Li, L.; Guo, D.; Zhang, S.; Yuan, T.; Cao, R.; Jiang, Z.; et al. Mixed lineage kinase domain-like protein MLKL breaks down myelin following nerve injury. Mol. Cell 2018, 72, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Ousingsawat, J.; Cabrita, I.; Wanitchakool, P.; Sirianant, L.; Krautwald, S.; Linkermann, A.; Schreiber, R.; Kunzelmann, K. Ca(2+) signals, cell membrane disintegration, and activation of TMEM16F during necroptosis. Cell Mol. Life Sci. 2017, 74, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, H.; Johnston, A.; Hanna-Addams, S.; Reynoso, E.; Xiang, Y.; Wang, Z. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E7450–E7459. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL forms cation channels. Cell Res. 2016, 26, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef]
- Ros, U.; Pena-Blanco, A.; Hanggi, K.; Kunzendorf, U.; Krautwald, S.; Wong, W.W.; Garcia-Saez, A.J. Necroptosis execution is mediated by plasma membrane nanopores independent of calcium. Cell Rep. 2017, 19, 175–187. [Google Scholar] [CrossRef]
- Yoon, S.; Kovalenko, A.; Bogdanov, K.; Wallach, D. MLKL, the Protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 2017, 47, 51–65.e7. [Google Scholar] [CrossRef]
- Yoon, S.; Bogdanov, K.; Kovalenko, A.; Wallach, D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016, 23, 253–260. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Seiler, A.; Schneider, M.; Forster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Radmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-Mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Muller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kossl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Agmon, E.; Stockwell, B.R. Lipid homeostasis and regulated cell death. Curr. Opin. Chem. Biol. 2017, 39, 83–89. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Boil. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Boil. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem. Boil. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Ye, L.F.; Chaudhary, K.R.; Zandkarimi, F.; Harken, A.D.; Kinslow, C.J.; Upadhyayula, P.S.; Dovas, A.; Higgins, D.M.; Tan, H.; Zhang, Y.; et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem. Biol. 2020, 15, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem. Biol. 2019, 26, 623–633.e9. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.Y.; Deasy, R.; Kost-Alimova, M.; Dancik, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 2019, 10, 1617. [Google Scholar] [CrossRef] [PubMed]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Pedrera, L.; Espiritu, R.A.; Ros, U.; Schmitt, A.; Hailfinger, S.; García-Sáez, A.J. Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics. bioRxiv 2019. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ros, U.; Pedrera, L.; Garcia-Saez, A.J. Partners in Crime: The Interplay of Proteins and Membranes in Regulated Necrosis. Int. J. Mol. Sci. 2020, 21, 2412. https://doi.org/10.3390/ijms21072412
Ros U, Pedrera L, Garcia-Saez AJ. Partners in Crime: The Interplay of Proteins and Membranes in Regulated Necrosis. International Journal of Molecular Sciences. 2020; 21(7):2412. https://doi.org/10.3390/ijms21072412
Chicago/Turabian StyleRos, Uris, Lohans Pedrera, and Ana J. Garcia-Saez. 2020. "Partners in Crime: The Interplay of Proteins and Membranes in Regulated Necrosis" International Journal of Molecular Sciences 21, no. 7: 2412. https://doi.org/10.3390/ijms21072412
APA StyleRos, U., Pedrera, L., & Garcia-Saez, A. J. (2020). Partners in Crime: The Interplay of Proteins and Membranes in Regulated Necrosis. International Journal of Molecular Sciences, 21(7), 2412. https://doi.org/10.3390/ijms21072412