Cancer Cachexia and Related Metabolic Dysfunction

 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
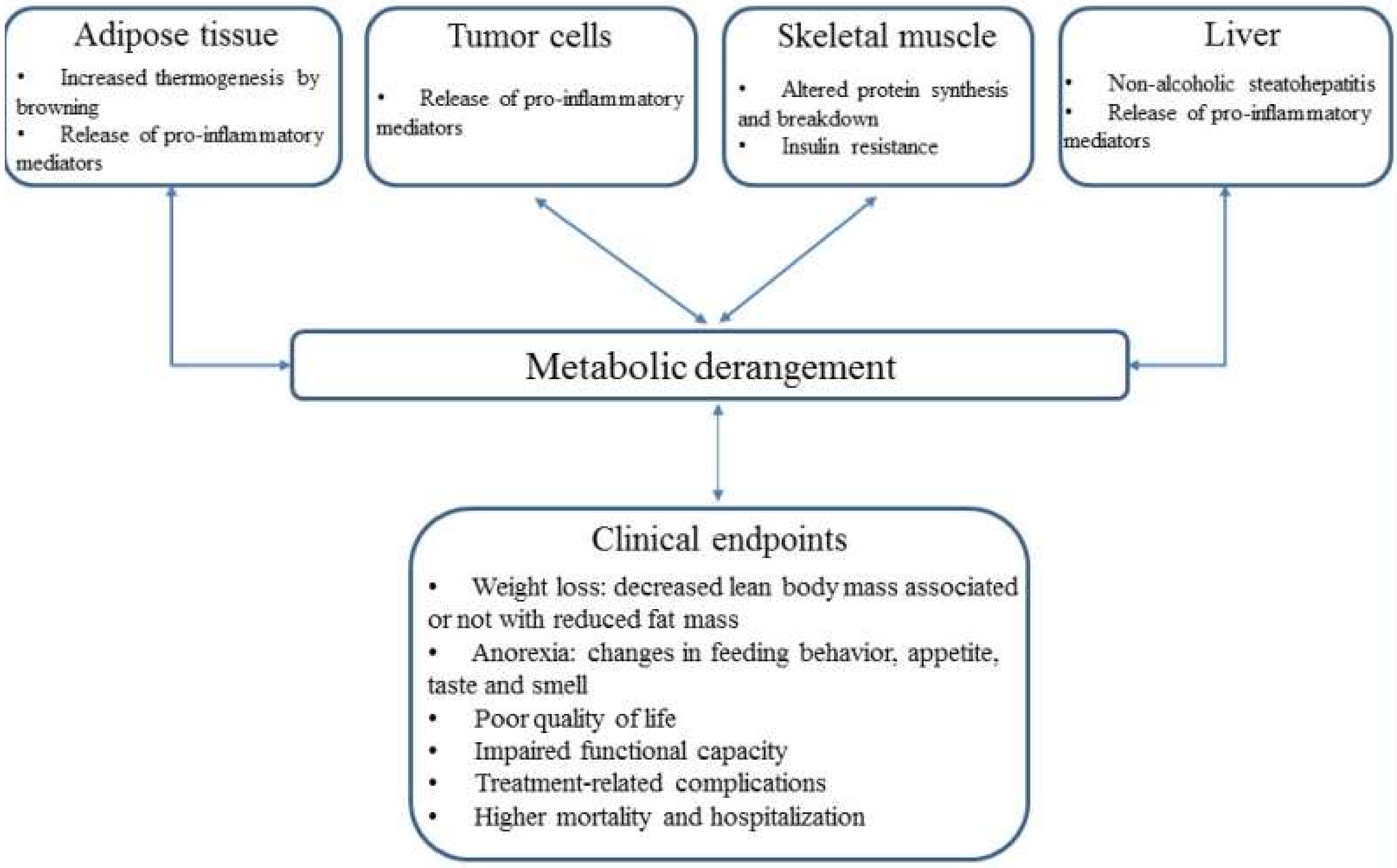
2. Altered Energy Balance
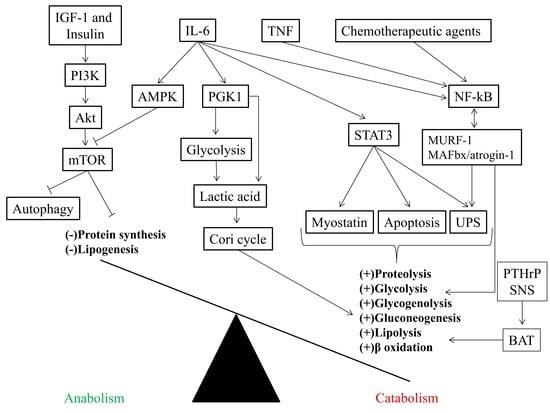
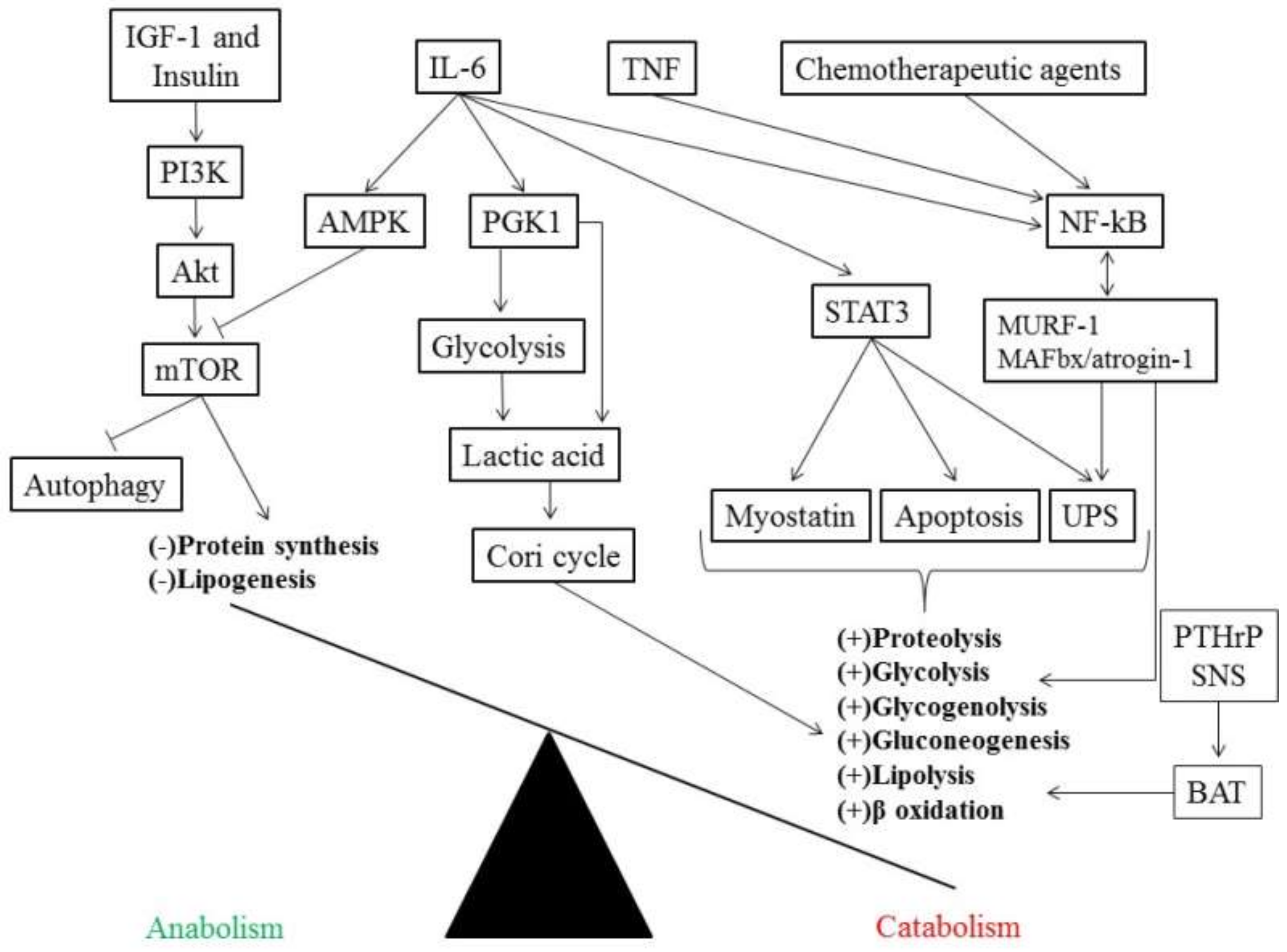
3. Inflammatory Markers and Muscle Mass
4. Insulin Resistance in Cancer Cachexia
5. Brown Adipose Tissue and Metabolism
6. Role of Liver Cells in Metabolism
7. Treatment Perspective
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADP | Adenosine diphosphate |
| Akt | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| ATP | Adenosine triphosphate |
| BAT | Brown adipose tissue |
| BMI | Body mass index |
| CPT I | Carnitine palmitoyltransferase I |
| CPT II | Carnitine palmitoyltransferase II |
| EGF | Epidermal growth factor |
| GH | Growth hormone |
| HSL | Hormone-sensitive lipase |
| IFN-γ | Interferon-gamma |
| IGFBP | Insulin growth factor binding protein |
| IGF-1 | Insulin-like growth factor 1 |
| IL-1β | Interleukin-1 beta |
| IL-2 | Interleukin-2 |
| IL-4 | Interleukin-4 |
| IL-6 | Interleukin-6 |
| IL-10 | Interleukin-10 |
| IR | Insulin resistance |
| JAKs | Janus kinases |
| LBM | Lean body mass |
| MABp1 | Monoclonal antibody |
| MAFbx | Muscle atrophy F-box |
| mTOR | mammalian target of rapamycin |
| mTORC1 | mTOR complex 1 |
| mTORC2 | mTOR complex 2 |
| MuRF-1 | Muscle RING Finger-1 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NSCLC | Non-small cell lung cancer |
| PGK 1 | Phosphoglycerate kinase 1 |
| PI3K | Phosphoinositide 3-kinase |
| PPAR-γ | Peroxisome proliferator-activated receptor-gamma |
| PPAR-α | Peroxisome proliferator-activated receptor-alpha |
| PTHrP | Tumor-derived parathyroid-hormone-related protein |
| SARMs | Selective androgen receptor modulators |
| STAT3 | Activating the signal transducer and activator of transcription 3 |
| TNF | Tumor necrosis factor |
| UCP1 | Uncoupling protein 1 |
| UPS | Ubiquitin-proteasome system |
| WAT | White adipose tissue |
References
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Anker, M.S.; Holcomb, R.; Muscaritoli, M.; von Haehling, S.; Haverkamp, W.; Jatoi, A.; Morley, J.E.; Strasser, F.; Landmesser, U.; Coats, A.J.S.; et al. Orphan disease status of cancer cachexia in the USA and in the European Union: A systematic review. J. Cachexia Sarcopenia Muscle 2019, 10, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Arthur, S.T.; Noone, J.M.; Van Doren, B.A.; Roy, D.; Blanchette, C.M. One-year prevalence, comorbidities and cost of cachexia-related inpatient admissions in the USA. Drugs Context 2014, 3, 212265. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Anker, S.D. Prevalence, incidence and clinical impact of cachexia: Facts and numbers-update 2014. J. Cachexia Sarcopenia Muscle 2014, 5, 261–263. [Google Scholar] [CrossRef]
- Farkas, J.; von Haehling, S.; Kalantar-Zadeh, K.; Morley, J.E.; Anker, S.D.; Lainscak, M. Cachexia as a major public health problem: Frequent, costly, and deadly. J. Cachexia Sarcopenia Muscle 2013, 4, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Caan, B.J.; Weltzien, E.; Cespedes Feliciano, E.M.; Kroenke, C.H.; Meyerhardt, J.A.; Baracos, V.E.; Kwan, M.L.; Castillo, A.L.; Prado, C.M. Associations of pre-existing co-morbidities with skeletal muscle mass and radiodensity in patients with non-metastatic colorectal cancer. J. Cachexia Sarcopenia Muscle 2018, 9, 654–663. [Google Scholar] [CrossRef]
- Blum, D.; Stene, G.B.; Solheim, O.; Fayers, P.; Hjermstad, M.J.; Baracos, V.E.; Fearon, K.; Strasser, F.; Kaasa, S.; Lieve, V.D.B.; et al. Validation of the Consensus-Definition for Cancer Cachexia and evaluation of a classification model—A study based on data from an international multicentre project (EPCRC-CSA). Ann. Oncol. 2014, 25, 1635–1642. [Google Scholar] [CrossRef]
- Kays, J.K.; Shahda, S.; Stanley, M.; Bell, T.M.; O’Neill, B.H.; Kohli, M.D.; Couch, M.E.; Koniaris, L.G.; Zimmers, T.A. Three cachexia phenotypes and the impact of fat-only loss on survival in FOLFIRINOX therapy for pancreatic cancer. J. Cachexia Sarcopenia Muscle 2018, 9, 673–684. [Google Scholar] [CrossRef]
- Brown, J.C.; Cespedes Feliciano, E.M.; Caan, B.J. The evolution of body composition in oncology-epidemiology, clinical trials, and the future of patient care: Facts and numbers. J. Cachexia Sarcopenia Muscle 2018, 9, 1200–1208. [Google Scholar] [CrossRef]
- Argilés, J.M.; Stemmler, B.; López-Soriano, F.J.; Busquets, S. Inter-tissue communication in cancer cachexia. Nat. Rev. Endocrinol. 2018, 15, 9–20. [Google Scholar] [CrossRef]
- Bezuidenhout, N.; Shoshan, M. A Shifty Target: Tumor-Initiating Cells and Their Metabolism. Int. J. Mol. Sci. 2019, 20, 5370. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Witte, D.; Fiedler, C.; Gädeken, T.; Kaufmann, R.; Lehnert, H.; Gieseler, F.; Rauch, B.H. The Role of PAR2 in TGF-β1-Induced ERK Activation and Cell Motility. Int. J. Mol. Sci. 2017, 18, 2776. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Bosaeus, I.; Daneryd, P.; Svanberg, E.; Lundholm, K. Dietary intake and resting energy expenditure in relation to weight loss in unselected cancer patients. Int. J. Cancer 2001, 93, 380–383. [Google Scholar] [CrossRef]
- Staal-van den Brekel, A.J.; Schols, A.M.; ten Velde, G.P.; Buurman, W.A.; Wouters, E.F. Analysis of the energy balance in lung cancer patients. Cancer Res. 1994, 54, 6430–6433. [Google Scholar]
- Nordhausen, K.; Solass, W.; Demtroeder, C.; Tempfer, C.B.; Reymond, M. Cachexia-anorexia syndrome in patients with peritoneal metastasis: An observational study. Pleura Peritoneum 2016, 1, 57–63. [Google Scholar] [CrossRef][Green Version]
- Wang, F.; Liu, H.; Hu, L.; Liu, Y.; Duan, Y.; Cui, R.; Tian, W. The Warburg effect in human pancreatic cancer cells triggers cachexia in athymic mice carrying the cancer cells. BMC Cancer 2018, 18, 360. [Google Scholar] [CrossRef]
- John, A.P. Dysfunctional mitochondria, not oxygen insufficiency, cause cancer cells to produce inordinate amounts of lactic acid: The impact of this on the treatment of cancer. Med. Hypotheses 2001, 57, 429–431. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Archid, R.; Solass, W.; Tempfer, C.; Königsrainer, A.; Adolph, M.; Reymond, M.A.; Wilson, R.B. Cachexia Anorexia Syndrome and Associated Metabolic Dysfunction in Peritoneal Metastasis. Int. J. Mol. Sci. 2019, 20, 5444. [Google Scholar] [CrossRef] [PubMed]
- Sudo, Y.; Otsuka, H.; Miyakawa, R.; Goto, A.; Kashiwase, Y.; Terawaki, K.; Miyano, K.; Hirao, Y.; Taki, K.; Tagawa, R.; et al. Differential Metabolic Responses to Adipose Atrophy Associated with Cancer Cachexia and Caloric Restriction in Rats and the Effect of Rikkunshito in Cancer Cachexia. Int. J. Mol. Sci. 2018, 19, 3852. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.P.; Cooper, S.T.; Parry, B.R.; Tuckey, J.A. Increased expression of the mRNA for hormone-sensitive lipase in adipose tissue of cancer patients. Biochim. Biophys. Acta 1993, 1180, 236–242. [Google Scholar] [CrossRef]
- Dahlman, I.; Mejhert, N.; Linder, K.; Agustsson, T.; Mutch, D.M.; Kulyte, A.; Isaksson, B.; Permert, J.; Petrovic, N.; Nedergaard, J.; et al. Adipose tissue pathways involved in weight loss of cancer cachexia. Br. J. Cancer 2010, 102, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.S.; Manda, G. Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. Int. J. Mol. Sci. 2017, 18, 2755. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef]
- Friesen, D.E.; Baracos, V.E.; Tuszynski, J.A. Modeling the energetic cost of cancer as a result of altered energy metabolism: Implications for cachexia. Theor. Biol. Med. Model. 2015, 12, 17. [Google Scholar] [CrossRef]
- Fredrix, E.W.; Soeters, P.B.; Wouters, E.F.; Deerenberg, I.M.; von Meyenfeldt, M.F.; Saris, W.H. Effect of different tumor types on resting energy expenditure. Cancer Res. 1991, 51, 6138–6141. [Google Scholar]
- Cao, D.X.; Wu, G.H.; Zhang, B.; Quan, Y.J.; Wei, J.; Jin, H.; Jiang, Y.; Yang, Z.A. Resting energy expenditure and body composition in patients with newly detected cancer. Clin. Nutr. 2010, 29, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Roza, A.M.; Shizgal, H.M. The Harris Benedict equation reevaluated: Resting energy requirements and the body cell mass. Am. J. Clin. Nutr. 1984, 40, 168–182. [Google Scholar] [CrossRef]
- Dev, R.; Hui, D.; Chisholm, G.; Delgado-Guay, M.; Dalal, S.; Del Fabbro, E.; Bruera, E. Hypermetabolism and symptom burden in advanced cancer patients evaluated in a cachexia clinic. J. Cachexia Sarcopenia Muscle 2015, 6, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Jouinot, A.; Ulmann, G.; Vazeille, C.; Durand, J.P.; Boudou-Rouquette, P.; Arrondeau, J.; Tlemsani, C.; Fournel, L.; Alifano, M.; Wislez, M.; et al. Hypermetabolism is an independent prognostic factor of survival in metastatic non-small cell lung cancer patients. Clin. Nutr. 2019. [Google Scholar] [CrossRef] [PubMed]
- Silver, H.J.; Dietrich, M.S.; Murphy, B.A. Changes in body mass, energy balance, physical function, and inflammatory state in patients with locally advanced head and neck cancer treated with concurrent chemoradiation after low-dose induction chemotherapy. Head Neck 2007, 29, 893–900. [Google Scholar] [CrossRef]
- Molfino, A.; Papa, A.; Gasperini-Zacco, M.L.; Muscaritoli, M.; Amoroso, A.; Cascino, A.; Catalano, C.; Albanese, C.V.; Laviano, A. Left ventricular mass correlates with lean body mass in patients with disease-associated wasting. J. Cachexia Sarcopenia Muscle 2014, 5, 251–252. [Google Scholar] [CrossRef]
- Loncar, G.; Springer, J.; Anker, M.; Doehner, W.; Lainscak, M. Cardiac cachexia: Hic et nunc. J. Cachexia Sarcopenia Muscle 2016, 7, 246–260. [Google Scholar] [CrossRef]
- Fields, D.P.; Roberts, B.M.; Simon, A.K.; Judge, A.R.; Fuller, D.D.; Mitchell, G.S. Cancer cachexia impairs neural respiratory drive in hypoxia but not hypercapnia. J. Cachexia Sarcopenia Muscle 2019, 10, 63–72. [Google Scholar] [CrossRef]
- Ausoni, S.; Calamelli, S.; Saccà, S.; Azzarello, G. How progressive cancer endangers the heart: An intriguing and underestimated problem. Cancer Metastasis Rev. 2020. [Google Scholar] [CrossRef]
- Wigmore, S.J.; Plester, C.E.; Ross, J.A.; Fearon, K.C. Contribution of anorexia and hypermetabolism to weight loss in anicteric patients with pancreatic cancer. Br. J. Surg. 1997, 84, 196–197. [Google Scholar] [CrossRef]
- Laviano, A.; Meguid, M.M.; Inui, A.; Muscaritoli, M.; Rossi-Fanelli, F. Therapy insight: Cancer anorexia-cachexia syndrome—When all you can eat is yourself. Nat. Clin. Pract. Oncol. 2005, 2, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, A.J.; Scarlett, J.M.; Marks, D.L. Hypothalamic mechanisms in cachexia. Physiol. Behav. 2010, 100, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Molfino, A.; Iannace, A.; Colaiacomo, M.C.; Farcomeni, A.; Emiliani, A.; Gualdi, G.; Laviano, A.; Rossi Fanelli, F. Cancer anorexia: Hypothalamic activity and its association with inflammation and appetite-regulating peptides in lung cancer. J. Cachexia Sarcopenia Muscle 2017, 8, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.H.; Birdsell, L.A.; Martin, L.; Baracos, V.E.; Fearon, K.C. Sarcopenia in an overweight or obese patient is an adverse prognostic factor in pancreatic cancer. Clin. Cancer Res. 2009, 15, 6973–6979. [Google Scholar] [CrossRef] [PubMed]
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676. [Google Scholar] [CrossRef] [PubMed]
- Nasr, R.; Salim Hammoud, M.; Nassar, F.; Mukherji, D.; Shamseddine, A.; Temraz, S. Inflammatory Markers and MicroRNAs: The Backstage Actors Influencing Prognosis in Colorectal Cancer Patients. Int. J. Mol. Sci. 2018, 19, 1867. [Google Scholar] [CrossRef]
- Cocchiola, R.; Rubini, E.; Altieri, F.; Chichiarelli, S.; Paglia, G.; Romaniello, D.; Carissimi, S.; Giorgi, A.; Giamogante, F.; Macone, A.; et al. STAT3 Post-Translational Modifications Drive Cellular Signaling Pathways in Prostate Cancer Cells. Int. J. Mol. Sci. 2019, 20, 1815. [Google Scholar] [CrossRef]
- Fung, M.M.; Rohwer, F.; McGuire, K.L. IL-2 activation of a PI3K-dependent STAT3 serine phosphorylation pathway in primary human T cells. Cell Signal. 2003, 15, 625–636. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef]
- Silva, K.A.; Dong, J.; Dong, Y.; Schor, N.; Tweardy, D.J.; Zhang, L.; Mitch, W.E. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. J. Biol. Chem. 2015, 290, 11177–11187. [Google Scholar] [CrossRef]
- Puppa, M.J.; Gao, S.; Narsale, A.A.; Carson, J.A. Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB J. 2014, 28, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Gilabert, M.; Calvo, E.; Airoldi, A.; Hamidi, T.; Moutardier, V.; Turrini, O.; Iovanna, J. Pancreatic cancer-induced cachexia is Jak2-dependent in mice. J. Cell Physiol. 2014, 229, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pan, J.; Dong, Y.; Tweardy, D.J.; Garibotto, G.; Mitch, W.E. Stat3 activation links a C/EBPδ to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 2013, 18, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Puppa, M.J.; White, J.P.; Velázquez, K.T.; Baltgalvis, K.A.; Sato, S.; Baynes, J.W.; Carson, J.A. The effect of exercise on IL-6-induced cachexia in the Apc (Min/+) mouse. J. Cachexia Sarcopenia Muscle 2012, 3, 117–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, G.; Chu, H.; Wang, X.; Xiong, L.; Cai, G.; Liu, R.; Gao, H.; Tao, B.; Li, W.; et al. Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis. Mol. Cell 2018, 71, 201–215.e7. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef]
- Ahmad, S.S.; Glatzle, J.; Bajaeifer, K.; Bühler, S.; Lehmann, T.; Königsrainer, I.; Vollmer, J.P.; Sipos, B.; Northoff, H.; Königsrainer, A.; et al. Phosphoglycerate kinase 1 as a promoter of metastasis in colon cancer. Int. J. Oncol. 2013, 43, 586–590. [Google Scholar] [CrossRef]
- Knudsen, J.G.; Gudiksen, A.; Bertholdt, L.; Overby, P.; Villesen, I.; Schwartz, C.L.; Pilegaard, H. Skeletal muscle IL-6 regulates muscle substrate utilization and adipose tissue metabolism during recovery from an acute bout of exercise. PLoS ONE 2017, 12, e0189301. [Google Scholar] [CrossRef]
- White, J.P.; Puppa, M.J.; Sato, S.; Gao, S.; Price, R.L.; Baynes, J.W.; Kostek, M.C.; Matesic, L.E.; Carson, J.A. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2012, 2, 14. [Google Scholar] [CrossRef]
- Bohlen, J.; McLaughlin, S.L.; Hazard-Jenkins, H.; Infante, A.M.; Montgomery, C.; Davis, M.; Pistilli, E.E. Dysregulation of metabolic-associated pathways in muscle of breast cancer patients: Preclinical evaluation of interleukin-15 targeting fatigue. J. Cachexia Sarcopenia Muscle 2018, 9, 701–714. [Google Scholar] [CrossRef]
- Lambert, C.P.; Wright, N.R.; Finck, B.N.; Villareal, D.T. Exercise but not diet-induced weight loss decreases skeletal muscle inflammatory gene expression in frail obese elderly persons. J. Appl. Physiol. 2008, 105, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Drummond, M.J.; Timmerman, K.L.; Markofski, M.M.; Walker, D.K.; Dickinson, J.M.; Jamaluddin, M.; Brasier, A.R.; Rasmussen, B.B.; Volpi, E. Short-term bed rest increases TLR4 and IL-6 expression in skeletal muscle of older adults. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R216–R223. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, K.C.; Makena, M.R.; Bhowmick, K.; Pandey, M.K. Advancement of NF-κB Signaling Pathway: A Novel Target in Pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, 3890. [Google Scholar] [CrossRef]
- Cai, D.; Frantz, J.D.; Tawa, N.E.; Melendez, P.A.; Oh, B.C.; Lidov, H.G.; Hasselgren, P.O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef]
- Damrauer, J.S.; Stadler, M.E.; Acharyya, S.; Baldwin, A.S.; Couch, M.E.; Guttridge, D.C. Chemotherapy-induced muscle wasting: Association with NF-κB and cancer cachexia. Eur. J. Transl. Myol. 2018, 28, 7590. [Google Scholar] [CrossRef]
- Daly, L.E.; Ní Bhuachalla, É.; Power, D.G.; Cushen, S.J.; James, K.; Ryan, A.M. Loss of skeletal muscle during systemic chemotherapy is prognostic of poor survival in patients with foregut cancer. J. Cachexia Sarcopenia Muscle 2018, 9, 315–325. [Google Scholar] [CrossRef]
- Tintignac, L.A.; Lagirand, J.; Batonnet, S.; Sirri, V.; Leibovitch, M.P.; Leibovitch, S.A. Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J. Biol. Chem. 2005, 280, 2847–2856. [Google Scholar] [CrossRef]
- Rom, O.; Reznick, A.Z. The role of E3 ubiquitin-ligases MuRF-1 and MAFbx in loss of skeletal muscle mass. Free Radic. Biol. Med. 2016, 98, 218–230. [Google Scholar] [CrossRef]
- Yuan, L.; Han, J.; Meng, Q.; Xi, Q.; Zhuang, Q.; Jiang, Y.; Han, Y.; Zhang, B.; Fang, J.; Wu, G. Muscle-specific E3 ubiquitin ligases are involved in muscle atrophy of cancer cachexia: An in vitro and in vivo study. Oncol. Rep. 2015, 33, 2261–2268. [Google Scholar] [CrossRef]
- Brown, J.L.; Lee, D.E.; Rosa-Caldwell, M.E.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Huseman, K.; Sataranatarajan, K.; Van Remmen, H.; Washington, T.A.; et al. Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2018, 9, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Khal, J.; Hine, A.V.; Fearon, K.C.; Dejong, C.H.; Tisdale, M.J. Increased expression of proteasome subunits in skeletal muscle of cancer patients with weight loss. Int. J. Biochem. Cell Biol. 2005, 37, 2196–2206. [Google Scholar] [CrossRef] [PubMed]
- Duval, A.P.; Jeanneret, C.; Santoro, T.; Dormond, O. mTOR and Tumor Cachexia. Int. J. Mol. Sci. 2018, 19, 2225. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S. mTOR as a Key Regulator in Maintaining Skeletal Muscle Mass. Front. Physiol. 2017, 8, 788. [Google Scholar] [CrossRef]
- White, J.P.; Puppa, M.J.; Gao, S.; Sato, S.; Welle, S.L.; Carson, J.A. Muscle mTORC1 suppression by IL-6 during cancer cachexia: A role for AMPK. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1042–E1052. [Google Scholar] [CrossRef]
- Aversa, Z.; Pin, F.; Lucia, S.; Penna, F.; Verzaro, R.; Fazi, M.; Colasante, G.; Tirone, A.; Rossi Fanelli, F.; Ramaccini, C.; et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 2016, 6, 30340. [Google Scholar] [CrossRef]
- Tardif, N.; Klaude, M.; Lundell, L.; Thorell, A.; Rooyackers, O. Autophagic-lysosomal pathway is the main proteolytic system modified in the skeletal muscle of esophageal cancer patients. Am. J. Clin. Nutr. 2013, 98, 1485–1492. [Google Scholar] [CrossRef]
- Op den Kamp, C.M.; Langen, R.C.; Snepvangers, F.J.; de Theije, C.C.; Schellekens, J.M.; Laugs, F.; Dingemans, A.M.; Schols, A.M. Nuclear transcription factor κ B activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am. J. Clin. Nutr. 2013, 98, 738–748. [Google Scholar] [CrossRef]
- Dev, R.; Bruera, E.; Dalal, S. Insulin resistance and body composition in cancer patients. Ann. Oncol. 2018, 29, ii18–ii26. [Google Scholar] [CrossRef]
- Copeland, G.P.; Leinster, S.J.; Davis, J.C.; Hipkin, L.J. Insulin resistance in patients with colorectal cancer. Br. J. Surg. 1987, 74, 1031–1035. [Google Scholar] [CrossRef]
- Winter, A.; MacAdams, J.; Chevalier, S. Normal protein anabolic response to hyperaminoacidemia in insulin-resistant patients with lung cancer cachexia. Clin. Nutr. 2012, 31, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.L.; Tian, M.; Wendel, A.A.; Belury, M.A. Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int. J. Cancer 2010, 126, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Noguchi, Y.; Doi, C.; Makino, T.; Nomura, K. Insulin resistance in patients with cancer: Relationships with tumor site, tumor stage, body-weight loss, acute-phase response, and energy expenditure. Nutrition 2001, 17, 590–593. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Noguchi, Y.; Matsumoto, A. Effects of tumor removal and body weight loss on insulin resistance in patients with cancer. Surgery 1994, 116, 62–66. [Google Scholar] [PubMed]
- Kwon, Y.; Song, W.; Droujinine, I.A.; Hu, Y.; Asara, J.M.; Perrimon, N. Systemic organ wasting induced by localized expression of the secreted insulin/IGF antagonist ImpL2. Dev. Cell 2015, 33, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Clarevega, A.; Bilder, D. Malignant Drosophila tumors interrupt insulin signaling to induce cachexia-like wasting. Dev. Cell 2015, 33, 47–55. [Google Scholar] [CrossRef]
- Parida, S.; Siddharth, S.; Sharma, D. Adiponectin, Obesity, and Cancer: Clash of the Bigwigs in Health and Disease. Int. J. Mol. Sci. 2019, 20, 2519. [Google Scholar] [CrossRef]
- Orrù, S.; Nigro, E.; Mandola, A.; Alfieri, A.; Buono, P.; Daniele, A.; Mancini, A.; Imperlini, E. A Functional Interplay between IGF-1 and Adiponectin. Int. J. Mol. Sci. 2017, 18, 2145. [Google Scholar] [CrossRef]
- Trobec, K.; von Haehling, S.; Anker, S.D.; Lainscak, M. Growth hormone, insulin-like growth factor 1, and insulin signaling-a pharmacological target in body wasting and cachexia. J. Cachexia Sarcopenia Muscle 2011, 2, 191–200. [Google Scholar] [CrossRef]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Camargo, R.G.; Riccardi, D.M.; Ribeiro, H.Q.; Carnevali, L.C.; de Matos-Neto, E.M.; Enjiu, L.; Neves, R.X.; Lima, J.D.; Figuerêdo, R.G.; de Alcântara, P.S.; et al. NF-κBp65 and Expression of Its Pro-Inflammatory Target Genes Are Upregulated in the Subcutaneous Adipose Tissue of Cachectic Cancer Patients. Nutrients 2015, 7, 4465–4479. [Google Scholar] [CrossRef] [PubMed]
- Maroni, G.; Panetta, D.; Luongo, R.; Krishnan, I.; La Rosa, F.; Campani, D.; Salvadori, P.; Iozzo, P.; Blasi, F.; Penkov, D.; et al. The Role of Prep1 in the Regulation of Mesenchymal Stromal Cells. Int. J. Mol. Sci. 2019, 20, 3639. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; White, J.P.; Kleiner, S.; Kazak, L.; Cohen, P.; Baracos, V.E.; Spiegelman, B.M. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 2014, 513, 100–104. [Google Scholar] [CrossRef]
- Han, J.; Meng, Q.; Shen, L.; Wu, G. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis. 2018, 17, 14. [Google Scholar] [CrossRef]
- Tsoli, M.; Moore, M.; Burg, D.; Painter, A.; Taylor, R.; Lockie, S.H.; Turner, N.; Warren, A.; Cooney, G.; Oldfield, B.; et al. Activation of thermogenesis in brown adipose tissue and dysregulated lipid metabolism associated with cancer cachexia in mice. Cancer Res. 2012, 72, 4372–4382. [Google Scholar] [CrossRef]
- Cvan Trobec, K.; Kerec Kos, M.; Trontelj, J.; Grabnar, I.; Tschirner, A.; Palus, S.; Anker, S.D.; Springer, J.; Lainscak, M. Influence of cancer cachexia on drug liver metabolism and renal elimination in rats. J. Cachexia Sarcopenia Muscle 2015, 6, 45–52. [Google Scholar] [CrossRef]
- Gonçalves, D.C.; Lira, F.S.; Yamashita, A.S.; Carnevali Junior, L.C.; Eder, R.; Laviano, A.; Seelaender, M.C.L. Liver lipid metabolism disruption in cancer cachexia is aggravated by cla supplementation -induced inflammation. Clin. Nutr. 2019, 38, 2219–2230. [Google Scholar] [CrossRef]
- Martignoni, M.E.; Kunze, P.; Hildebrandt, W.; Künzli, B.; Berberat, P.; Giese, T.; Klöters, O.; Hammer, J.; Büchler, M.W.; Giese, N.A.; et al. Role of mononuclear cells and inflammatory cytokines in pancreatic cancer-related cachexia. Clin. Cancer Res. 2005, 11, 5802–5808. [Google Scholar] [CrossRef]
- Martignoni, M.E.; Dimitriu, C.; Bachmann, J.; Krakowski-Rosen, H.; Ketterer, K.; Kinscherf, R.; Friess, H. Liver macrophages contribute to pancreatic cancer-related cachexia. Oncol. Rep. 2009, 21, 363–369. [Google Scholar] [CrossRef]
- Prokopchuk, O.; Steinacker, J.M.; Nitsche, U.; Otto, S.; Bachmann, J.; Schubert, E.C.; Friess, H.; Martignoni, M.E. IL-4 mRNA Is Downregulated in the Liver of Pancreatic Cancer Patients Suffering from Cachexia. Nutr. Cancer 2017, 69, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Dumas, J.F.; Goupille, C.; Julienne, C.M.; Pinault, M.; Chevalier, S.; Bougnoux, P.; Servais, S.; Couet, C. Efficiency of oxidative phosphorylation in liver mitochondria is decreased in a rat model of peritoneal carcinosis. J. Hepatol. 2011, 54, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Peyta, L.; Jarnouen, K.; Pinault, M.; Coulouarn, C.; Guimaraes, C.; Goupille, C.; de Barros, J.P.; Chevalier, S.; Dumas, J.F.; Maillot, F.; et al. Regulation of hepatic cardiolipin metabolism by TNFα: Implication in cancer cachexia. Biochim. Biophys. Acta 2015, 1851, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Teli, M.R.; James, O.F.; Burt, A.D.; Bennett, M.K.; Day, C.P. The natural history of nonalcoholic fatty liver: A follow-up study. Hepatology 1995, 22, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Takaki, A.; Tsuzaki, R.; Yasunaka, T.; Koike, K.; Shimomura, Y.; Seki, H.; Matsushita, H.; Miyake, Y.; Ikeda, F.; et al. L-carnitine prevents progression of non-alcoholic steatohepatitis in a mouse model with upregulation of mitochondrial pathway. PLoS ONE 2014, 9, e100627. [Google Scholar] [CrossRef]
- Liu, S.; Wu, H.J.; Zhang, Z.Q.; Chen, Q.; Liu, B.; Wu, J.P.; Zhu, L. L-carnitine ameliorates cancer cachexia in mice by regulating the expression and activity of carnitine palmityl transferase. Cancer Biol. Ther. 2011, 12, 125–130. [Google Scholar] [CrossRef]
- Jiang, F.; Zhang, Z.; Zhang, Y.; Wu, J.; Yu, L.; Liu, S. L-carnitine ameliorates the liver inflammatory response by regulating carnitine palmitoyltransferase I-dependent PPARγ signaling. Mol. Med. Rep. 2016, 13, 1320–1328. [Google Scholar] [CrossRef]
- Kraft, M.; Kraft, K.; Gärtner, S.; Mayerle, J.; Simon, P.; Weber, E.; Schütte, K.; Stieler, J.; Koula-Jenik, H.; Holzhauer, P.; et al. L-Carnitine-supplementation in advanced pancreatic cancer (CARPAN)—A randomized multicentre trial. Nutr. J. 2012, 11, 52. [Google Scholar] [CrossRef]
- Goncalves, M.D.; Hwang, S.K.; Pauli, C.; Murphy, C.J.; Cheng, Z.; Hopkins, B.D.; Wu, D.; Loughran, R.M.; Emerling, B.M.; Zhang, G.; et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E743–E752. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, B.; Liu, H.; Yang, K.; Thapa, S.; Zhang, H.; Li, L.; Yu, S. Development and validation of a clinically applicable score to classify cachexia stages in advanced cancer patients. J. Cachexia Sarcopenia Muscle 2018, 9, 306–314. [Google Scholar] [CrossRef]
- Bourgeois, B.; Fan, B.; Johannsen, N.; Gonzalez, M.C.; Ng, B.K.; Sommer, M.J.; Shepherd, J.A.; Heymsfield, S.B. Improved strength prediction combining clinically available measures of skeletal muscle mass and quality. J. Cachexia Sarcopenia Muscle 2019, 10, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Ebner, N.; Springer, J.; Kalantar-Zadeh, K.; Lainscak, M.; Doehner, W.; Anker, S.D.; von Haehling, S. Mechanism and novel therapeutic approaches to wasting in chronic disease. Maturitas 2013, 75, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Ebner, N.; Anker, S.D.; von Haehling, S. Recent developments in the field of cachexia, sarcopenia, and muscle wasting: Highlights from the 11th Cachexia Conference. J. Cachexia Sarcopenia Muscle 2019, 10, 218–225. [Google Scholar] [CrossRef]
- Plas, R.L.C.; Poland, M.; Faber, J.; Argilès, J.; van Dijk, M.; Laviano, A.; Meijerink, J.; Witkamp, R.F.; van Helvoort, A.; van Norren, K. A Diet Rich in Fish Oil and Leucine Ameliorates Hypercalcemia in Tumour-Induced Cachectic Mice. Int. J. Mol. Sci. 2019, 20, 4978. [Google Scholar] [CrossRef] [PubMed]
- Trobec, K.; Palus, S.; Tschirner, A.; von Haehling, S.; Doehner, W.; Lainscak, M.; Anker, S.D.; Springer, J. Rosiglitazone reduces body wasting and improves survival in a rat model of cancer cachexia. Nutrition 2014, 30, 1069–1075. [Google Scholar] [CrossRef]
- Nissinen, T.A.; Hentilä, J.; Penna, F.; Lampinen, A.; Lautaoja, J.H.; Fachada, V.; Holopainen, T.; Ritvos, O.; Kivelä, R.; Hulmi, J.J. Treating cachexia using soluble ACVR2B improves survival, alters mTOR localization, and attenuates liver and spleen responses. J. Cachexia Sarcopenia Muscle 2018, 9, 514–529. [Google Scholar] [CrossRef]
- Molinari, F.; Pin, F.; Gorini, S.; Chiandotto, S.; Pontecorvo, L.; Penna, F.; Rizzuto, E.; Pisu, S.; Musarò, A.; Costelli, P.; et al. The mitochondrial metabolic reprogramming agent trimetazidine as an ‘exercise mimetic’ in cachectic C26-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 954–973. [Google Scholar] [CrossRef]
- Penna, F.; Bonetto, A.; Aversa, Z.; Minero, V.G.; Rossi Fanelli, F.; Costelli, P.; Muscaritoli, M. Effect of the specific proteasome inhibitor bortezomib on cancer-related muscle wasting. J. Cachexia Sarcopenia Muscle 2016, 7, 345–354. [Google Scholar] [CrossRef]
- Penna, F.; Camperi, A.; Muscaritoli, M.; Filigheddu, N.; Costelli, P. The role of vitamin D in cancer cachexia. Curr. Opin. Support. Palliat. Care 2017, 11, 287–292. [Google Scholar] [CrossRef]
- Saioth, M.; Ishida, J.; Ebner, N.; Anker, S.D.; Von Haehling, S. Myostatin inhibitors as pharmacological treatment for muscle wasting and muscular dystrophy. JCSM Clin. Rep. 2017, 2, 1–10. [Google Scholar] [CrossRef]
- Ruiz-García, V.; López-Briz, E.; Carbonell-Sanchis, R.; Bort-Martí, S.; Gonzálvez-Perales, J.L. Megestrol acetate for cachexia-anorexia syndrome. A systematic review. J. Cachexia Sarcopenia Muscle 2018, 9, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Wright, T.J.; Dillon, E.L.; Durham, W.J.; Chamberlain, A.; Randolph, K.M.; Danesi, C.; Horstman, A.M.; Gilkison, C.R.; Willis, M.; Richardson, G.; et al. A randomized trial of adjunct testosterone for cancer-related muscle loss in men and women. J. Cachexia Sarcopenia Muscle 2018, 9, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Geva, R.; Richards, D.; Madhusudan, S.; Lin, B.K.; Wang, H.T.; Walgren, R.A.; Stemmer, S.M. LY2495655, an antimyostatin antibody, in pancreatic cancer: A randomized, phase 2 trial. J. Cachexia Sarcopenia Muscle 2018, 9, 871–879. [Google Scholar] [CrossRef]
- Hong, D.S.; Hui, D.; Bruera, E.; Janku, F.; Naing, A.; Falchook, G.S.; Piha-Paul, S.; Wheler, J.J.; Fu, S.; Tsimberidou, A.M.; et al. MABp1, a first-in-class true human antibody targeting interleukin-1α in refractory cancers: An open-label, phase 1 dose-escalation and expansion study. Lancet Oncol. 2014, 15, 656–666. [Google Scholar] [CrossRef]
- Advani, S.M.; Advani, P.G.; VonVille, H.M.; Jafri, S.H. Pharmacological management of cachexia in adult cancer patients: A systematic review of clinical trials. BMC Cancer 2018, 18, 1174. [Google Scholar] [CrossRef] [PubMed]
- Yennurajalingam, S.; Willey, J.S.; Palmer, J.L.; Allo, J.; Del Fabbro, E.; Cohen, E.N.; Tin, S.; Reuben, J.M.; Bruera, E. The role of thalidomide and placebo for the treatment of cancer-related anorexia-cachexia symptoms: Results of a double-blind placebo-controlled randomized study. J. Palliat. Med. 2012, 15, 1059–1064. [Google Scholar] [CrossRef]
- Akamizu, T.; Kangawa, K. Ghrelin for cachexia. J. Cachexia Sarcopenia Muscle 2010, 1, 169–176. [Google Scholar] [CrossRef]
- Colldén, G.; Tschöp, M.H.; Müller, T.D. Therapeutic Potential of Targeting the Ghrelin Pathway. Int. J. Mol. Sci. 2017, 18, 798. [Google Scholar] [CrossRef]
- Temel, J.S.; Abernethy, A.P.; Currow, D.C.; Friend, J.; Duus, E.M.; Yan, Y.; Fearon, K.C. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): Results from two randomised, double-blind, phase 3 trials. Lancet Oncol. 2016, 17, 519–531. [Google Scholar] [CrossRef]
- Currow, D.; Temel, J.S.; Abernethy, A.; Milanowski, J.; Friend, J.; Fearon, K.C. ROMANA 3: A phase 3 safety extension study of anamorelin in advanced non-small-cell lung cancer (NSCLC) patients with cachexia. Ann. Oncol. 2017, 28, 1949–1956. [Google Scholar] [CrossRef]
- Dobs, A.S.; Boccia, R.V.; Croot, C.C.; Gabrail, N.Y.; Dalton, J.T.; Hancock, M.L.; Johnston, M.A.; Steiner, M.S. Effects of enobosarm on muscle wasting and physical function in patients with cancer: A double-blind, randomised controlled phase 2 trial. Lancet Oncol. 2013, 14, 335–345. [Google Scholar] [CrossRef]
- Lainscak, M.; Laviano, A. ACT-ONE - ACTION at last on cancer cachexia by adapting a novel action beta-blocker. J. Cachexia Sarcopenia Muscle 2016, 7, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Lainscak, M.; Springer, J.; Anker, S.D. Cardiac cachexia: A systematic overview. Pharmacol. Ther. 2009, 121, 227–252. [Google Scholar] [CrossRef] [PubMed]
- Stewart Coats, A.J.; Ho, G.F.; Prabhash, K.; von Haehling, S.; Tilson, J.; Brown, R.; Beadle, J.; Anker, S.D.; for and on behalf of the ACT-ONE Study Group. Espindolol for the treatment and prevention of cachexia in patients with stage III/IV non-small cell lung cancer or colorectal cancer: A randomized, double-blind, placebo-controlled, international multicentre phase II study (the ACT-ONE trial). J. Cachexia Sarcopenia Muscle 2016, 7, 355–365. [Google Scholar] [CrossRef]
- Solheim, T.S.; Laird, B.J.A.; Balstad, T.R.; Bye, A.; Stene, G.; Baracos, V.; Strasser, F.; Griffiths, G.; Maddocks, M.; Fallon, M.; et al. Cancer cachexia: Rationale for the MENAC (Multimodal-Exercise, Nutrition and Anti-inflammatory medication for Cachexia) trial. BMJ Support. Palliat. Care 2018, 8, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Muscaritoli, M.; Molfino, A.; Lucia, S.; Rossi Fanelli, F. Cachexia: A preventable comorbidity of cancer. A T.A.R.G.E.T. approach. Crit. Rev. Oncol. Hematol. 2015, 94, 251–259. [Google Scholar] [CrossRef]
- Hojman, P. Exercise protects from cancer through regulation of immune function and inflammation. Biochem. Soc. Trans. 2017, 45, 905–911. [Google Scholar] [CrossRef]
- Hojman, P.; Gehl, J.; Christensen, J.F.; Pedersen, B.K. Molecular Mechanisms Linking Exercise to Cancer Prevention and Treatment. Cell Metab. 2018, 27, 10–21. [Google Scholar] [CrossRef]
- Nilsen, T.S.; Thorsen, L.; Fosså, S.D.; Wiig, M.; Kirkegaard, C.; Skovlund, E.; Benestad, H.B.; Raastad, T. Effects of strength training on muscle cellular outcomes in prostate cancer patients on androgen deprivation therapy. Scand. J. Med. Sci. Sports 2016, 26, 1026–1035. [Google Scholar] [CrossRef]
- Colleluori, G.; Aguirre, L.; Phadnis, U.; Fowler, K.; Armamento-Villareal, R.; Sun, Z.; Brunetti, L.; Hyoung Park, J.; Kaipparettu, B.A.; Putluri, N.; et al. Aerobic Plus Resistance Exercise in Obese Older Adults Improves Muscle Protein Synthesis and Preserves Myocellular Quality Despite Weight Loss. Cell Metab. 2019, 30, 261–273.e6. [Google Scholar] [CrossRef]
- Prado, C.M.; Purcell, S.A.; Laviano, A. Nutrition interventions to treat low muscle mass in cancer. J. Cachexia Sarcopenia Muscle 2020. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseca, G.W.P.d.; Farkas, J.; Dora, E.; von Haehling, S.; Lainscak, M. Cancer Cachexia and Related Metabolic Dysfunction. Int. J. Mol. Sci. 2020, 21, 2321. https://doi.org/10.3390/ijms21072321
Fonseca GWPd, Farkas J, Dora E, von Haehling S, Lainscak M. Cancer Cachexia and Related Metabolic Dysfunction. International Journal of Molecular Sciences. 2020; 21(7):2321. https://doi.org/10.3390/ijms21072321
Chicago/Turabian StyleFonseca, Guilherme Wesley Peixoto da, Jerneja Farkas, Eva Dora, Stephan von Haehling, and Mitja Lainscak. 2020. "Cancer Cachexia and Related Metabolic Dysfunction" International Journal of Molecular Sciences 21, no. 7: 2321. https://doi.org/10.3390/ijms21072321
APA StyleFonseca, G. W. P. d., Farkas, J., Dora, E., von Haehling, S., & Lainscak, M. (2020). Cancer Cachexia and Related Metabolic Dysfunction. International Journal of Molecular Sciences, 21(7), 2321. https://doi.org/10.3390/ijms21072321