Anti-Inflammatory and Anti-Migratory Activities of Isoquinoline-1-Carboxamide Derivatives in LPS-Treated BV2 Microglial Cells via Inhibition of MAPKs/NF-κB Pathway

 ,
,
Abstract
1. Introduction
2. Results
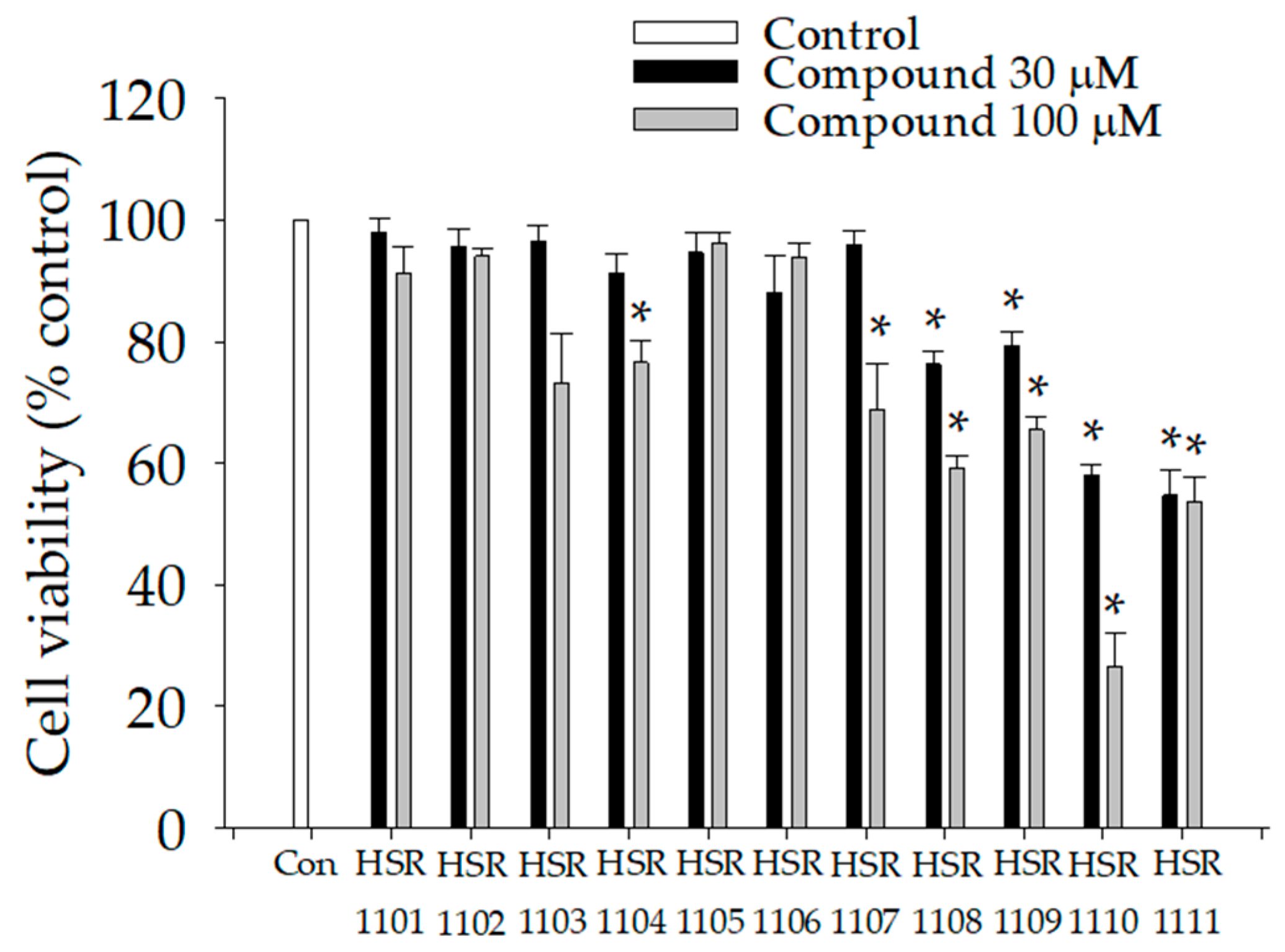
2.1. Effects of Isoquinoline-1-carboxamides on Viabilities of BV2 Cells
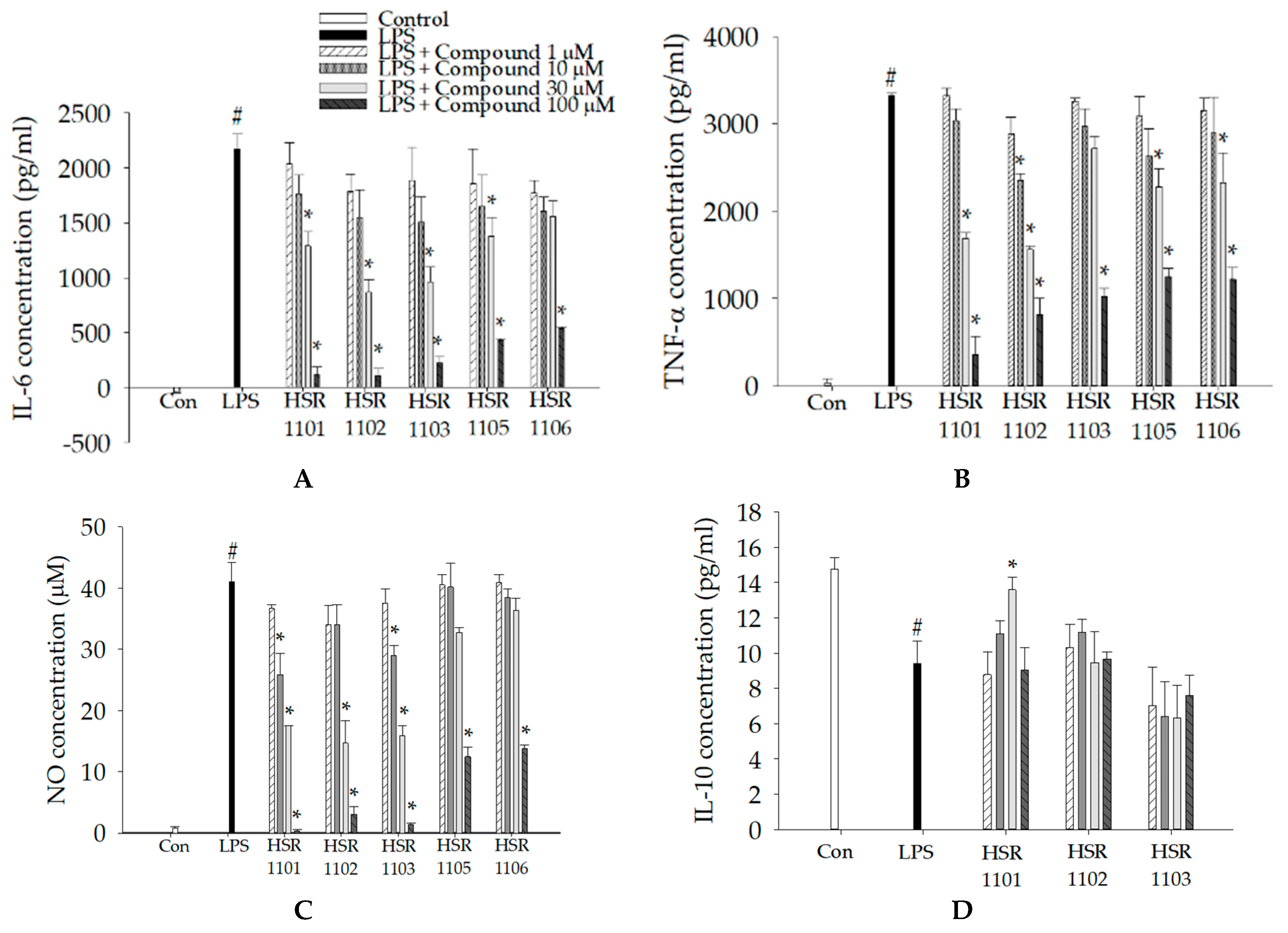
2.2. Effects of Isoquinoline-1-carboxamides on the Production of Pro- and Anti-Inflammatory Mediators in LPS-Treated BV2 Cells
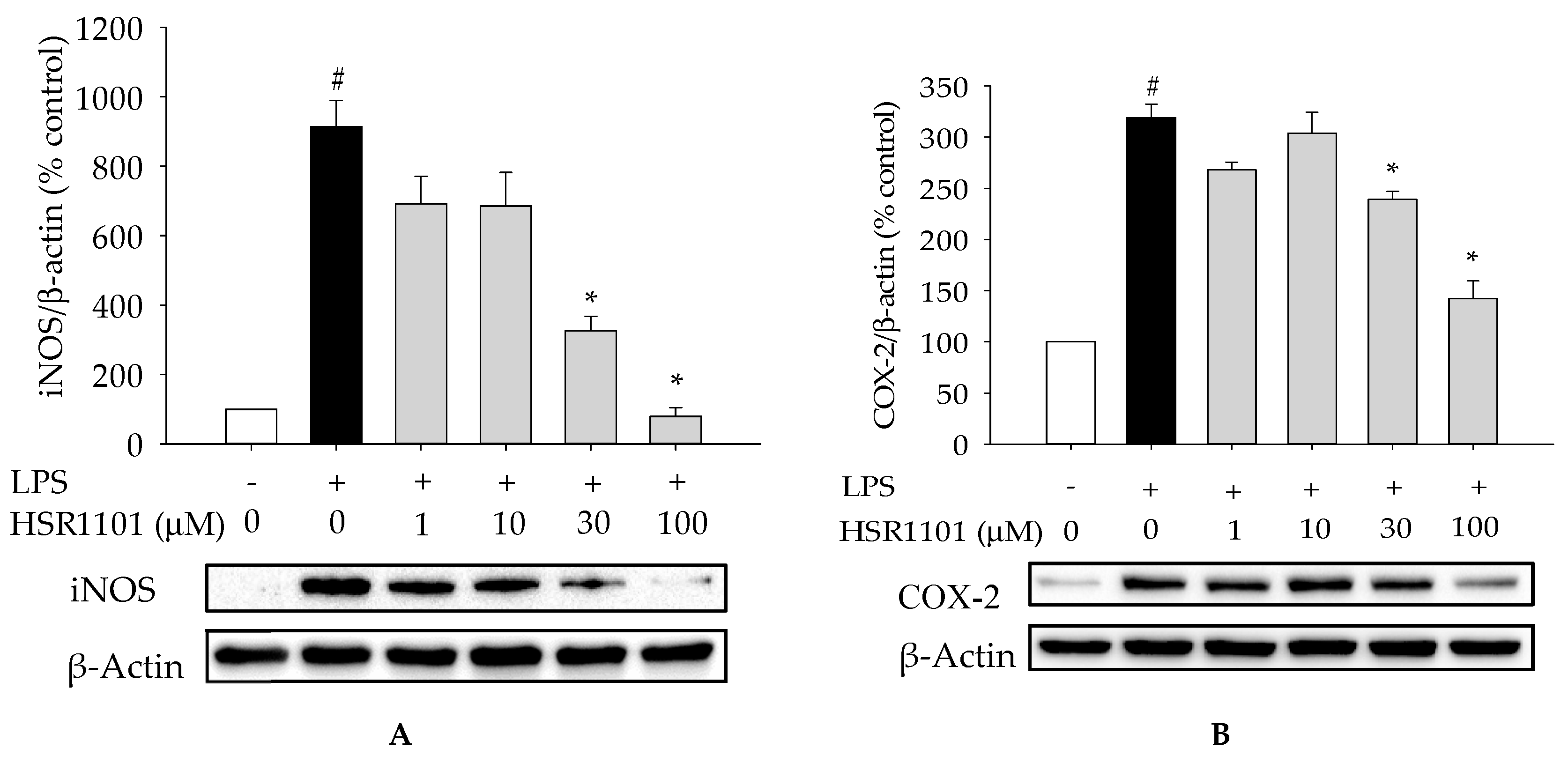
2.3. Effect of HSR1101 on LPS-Stimulated iNOS and COX-2 Expression in BV2 Cells
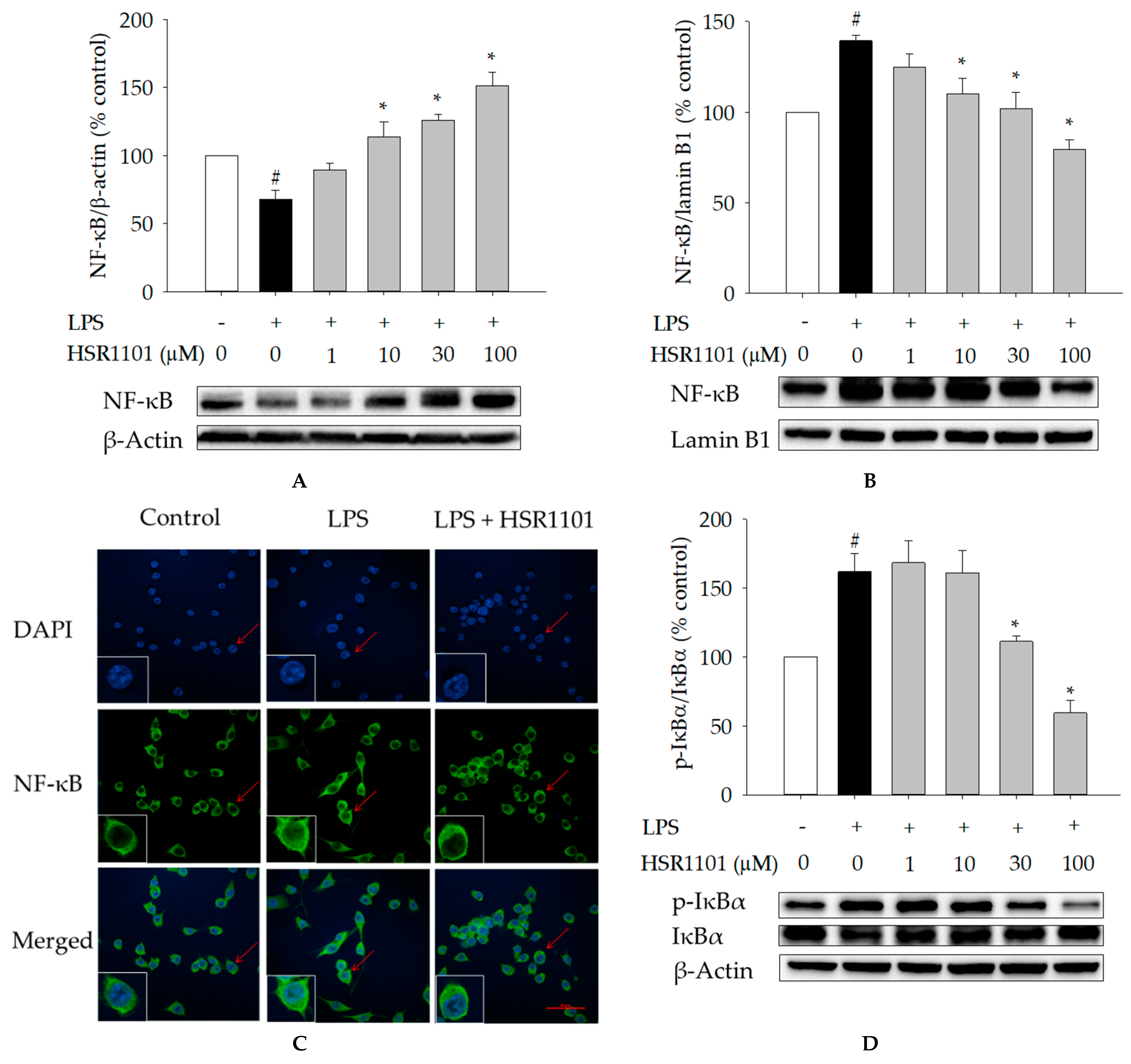
2.4. Effects of HSR1101 on LPS-Induced NF-κB Translocation and IκBα Phosphorylation in BV2 Cells
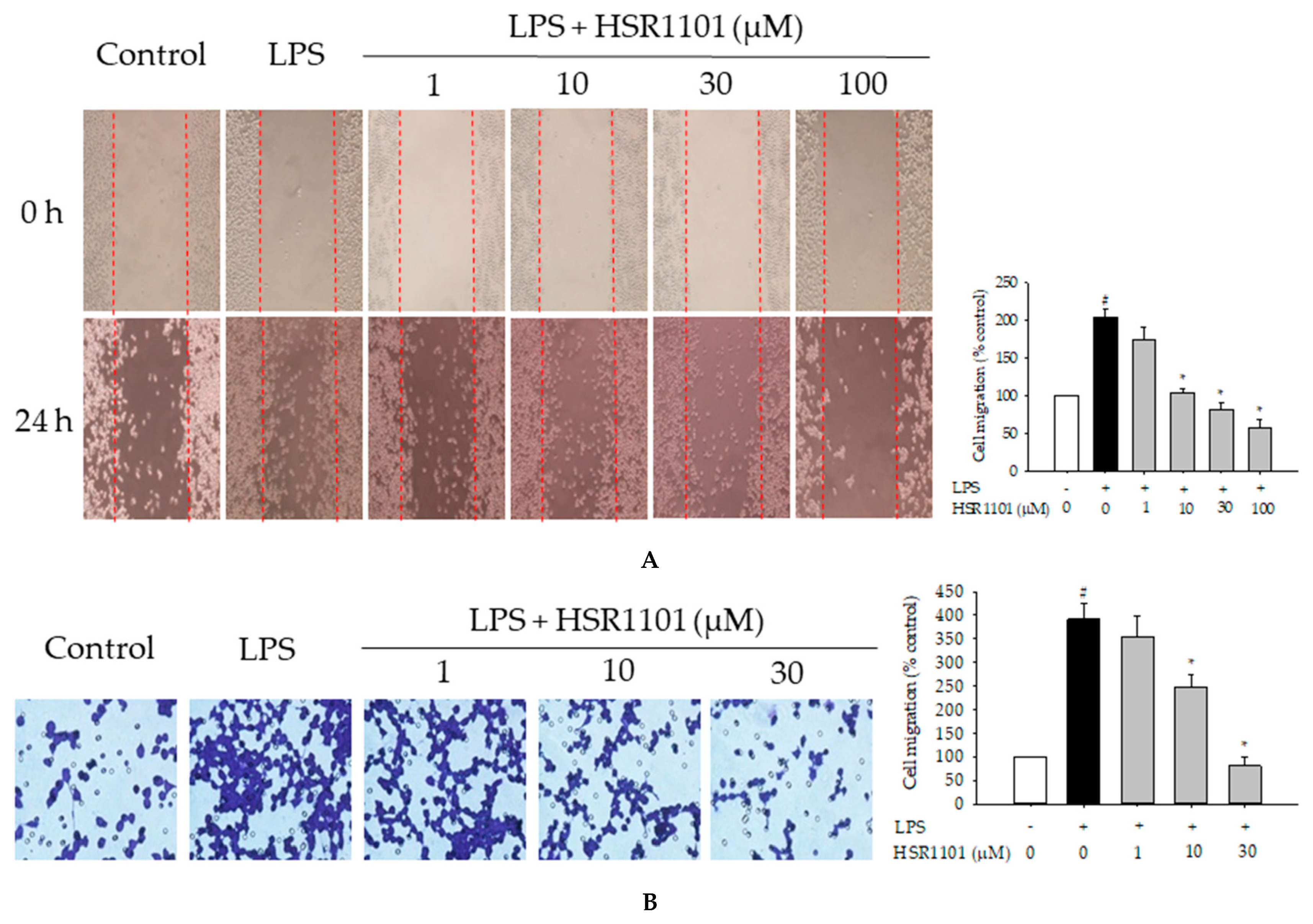
2.5. Effect of HSR1101 on LPS-Induced Cell Migration in BV2 Cells
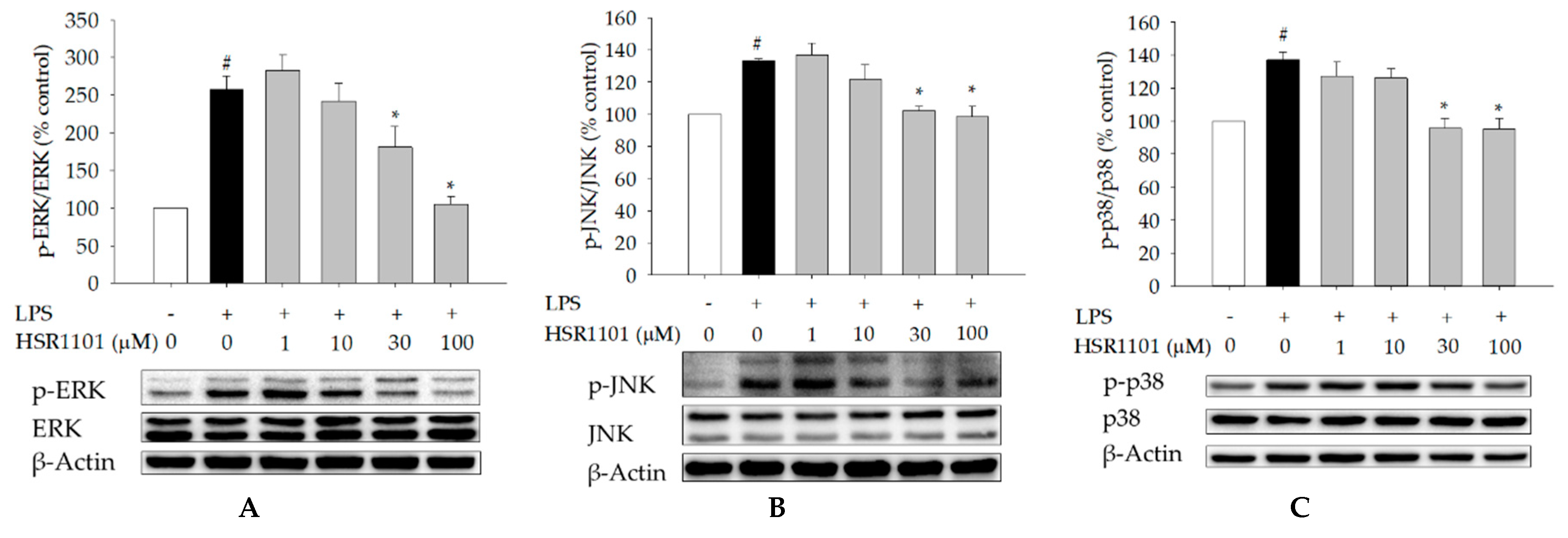
2.6. Effect of HSR1101 on MAPK Phosphorylation in LPS-Treated BV2 Cells
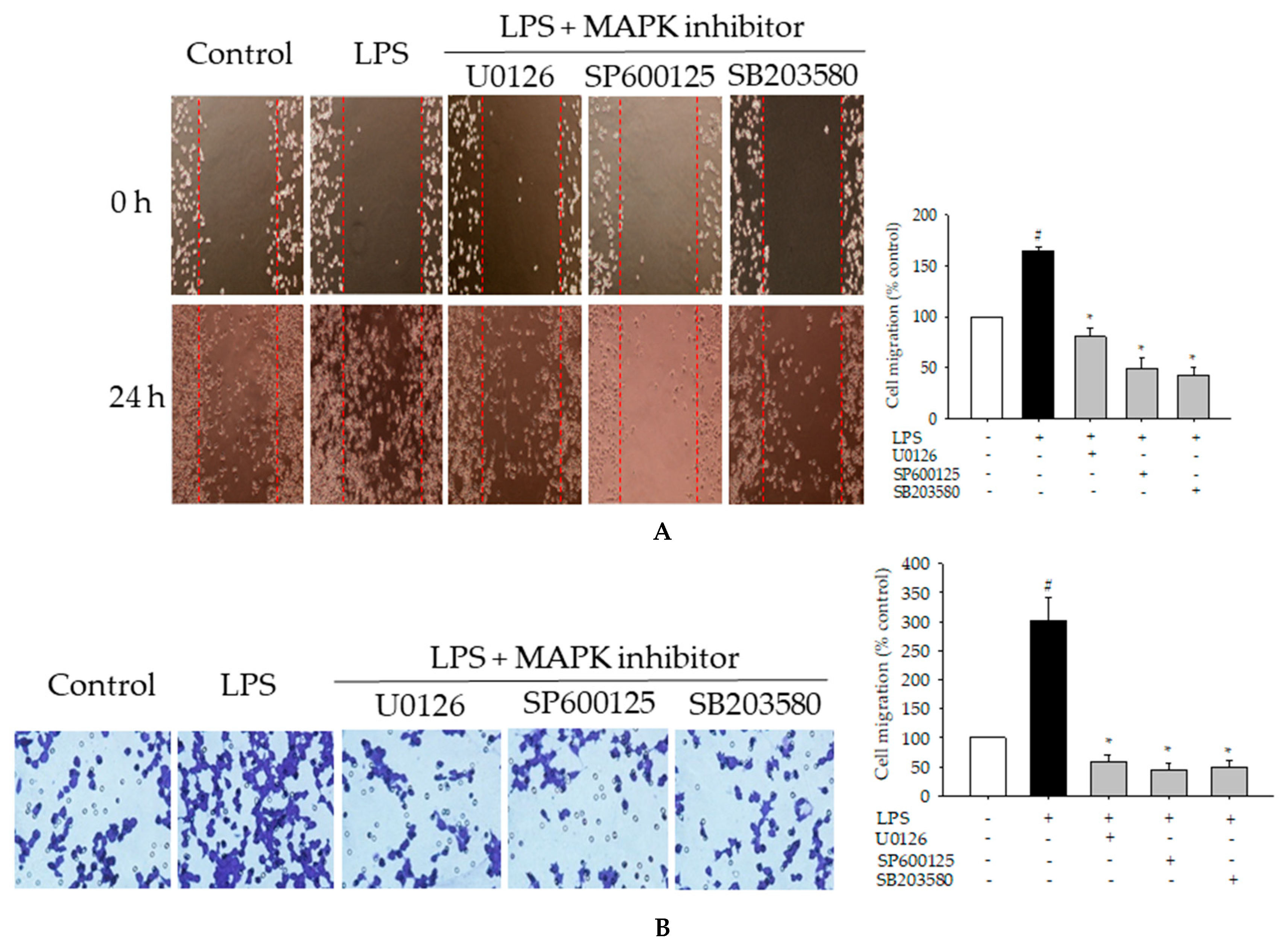
2.7. Effects of MAPK Inhibitors on LPS-Induced Pro-Inflammatory Mediators and Cell Migration in BV2 Cells
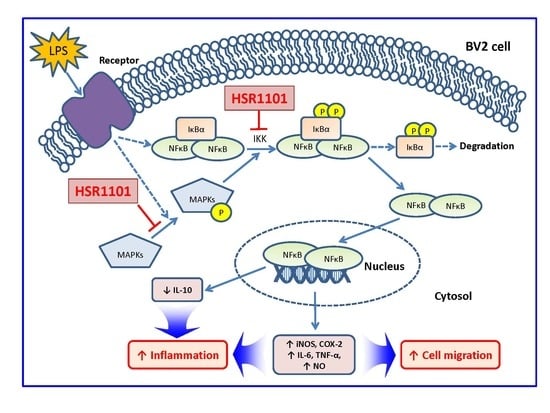
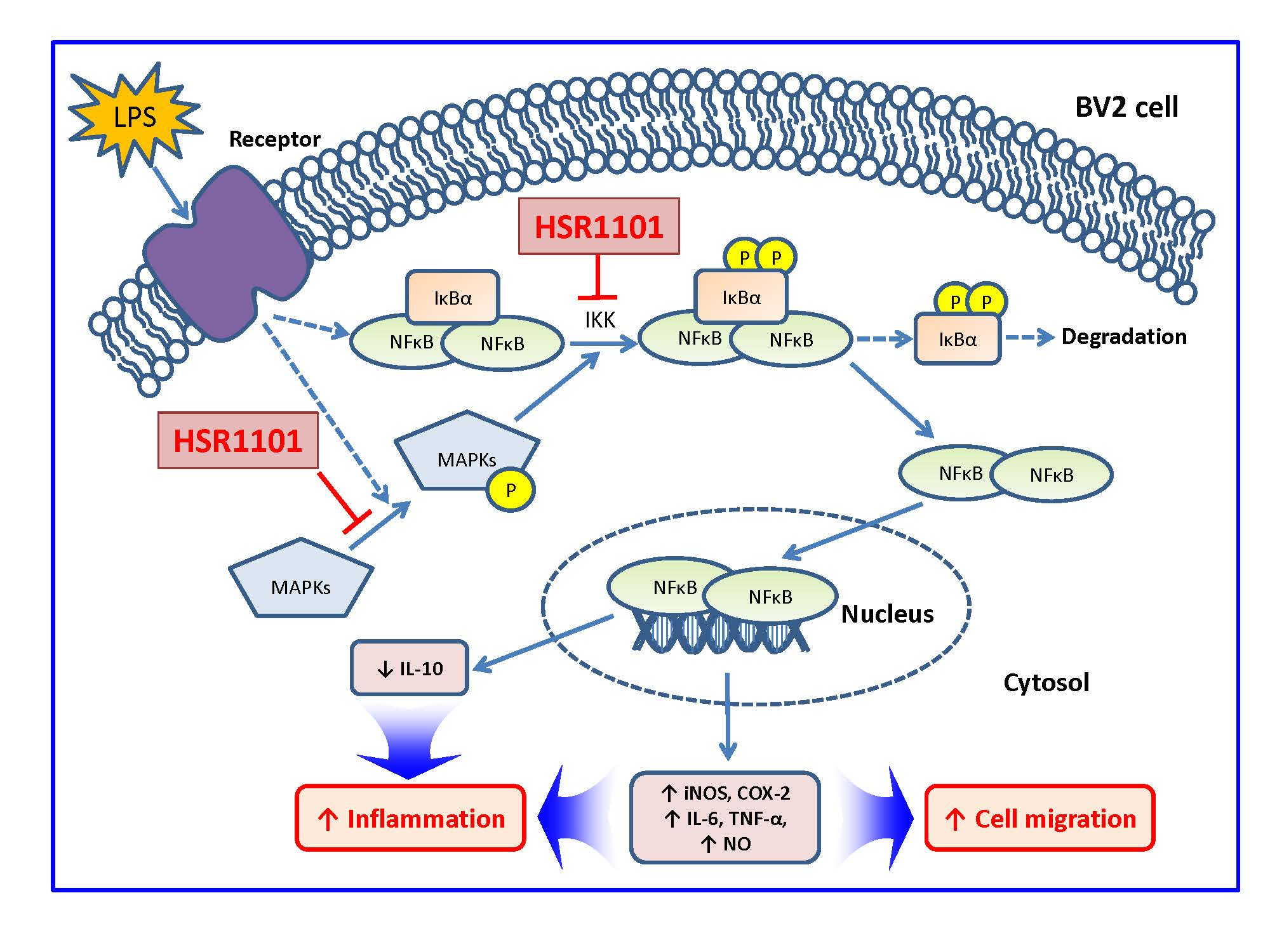
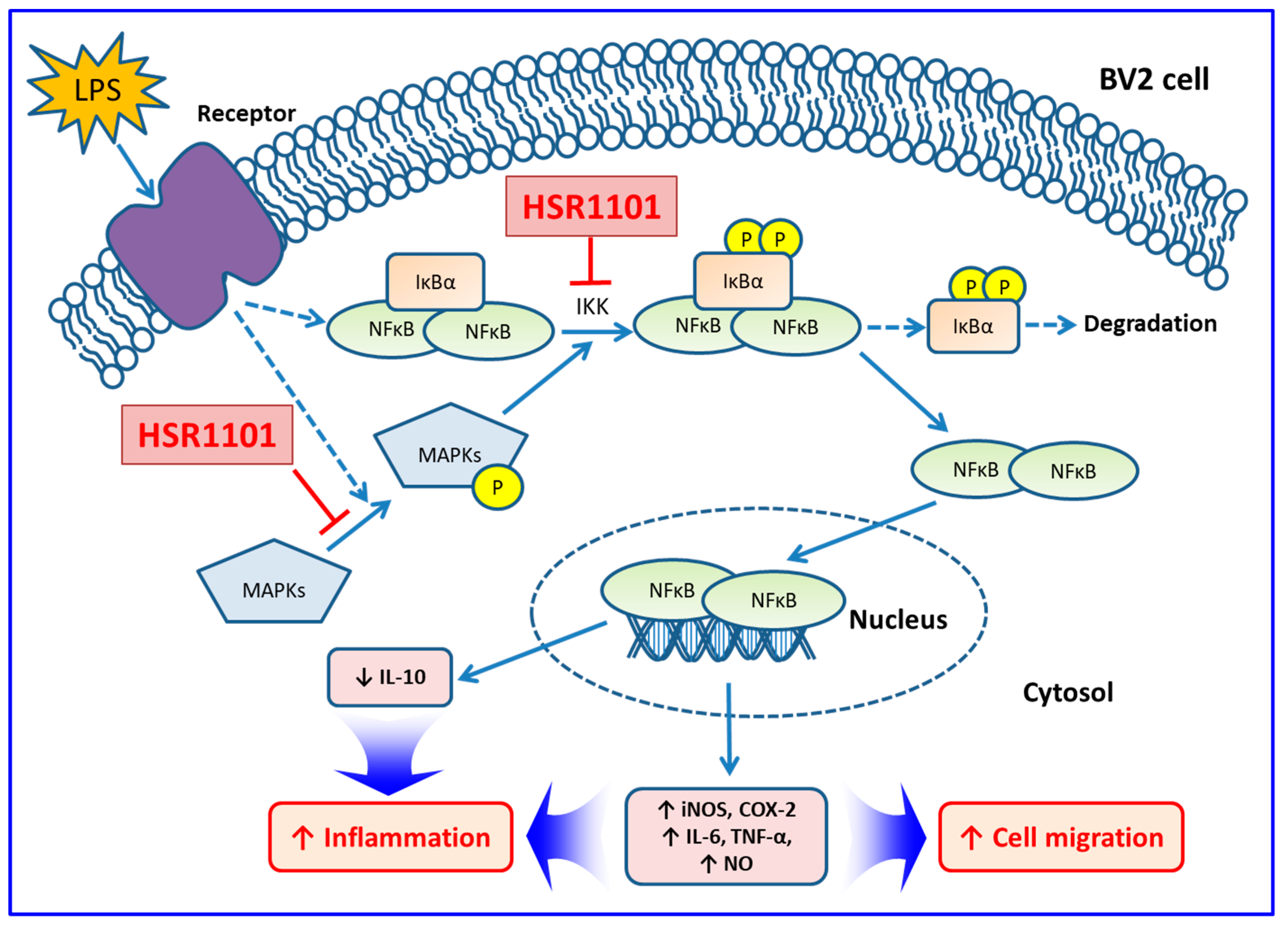
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
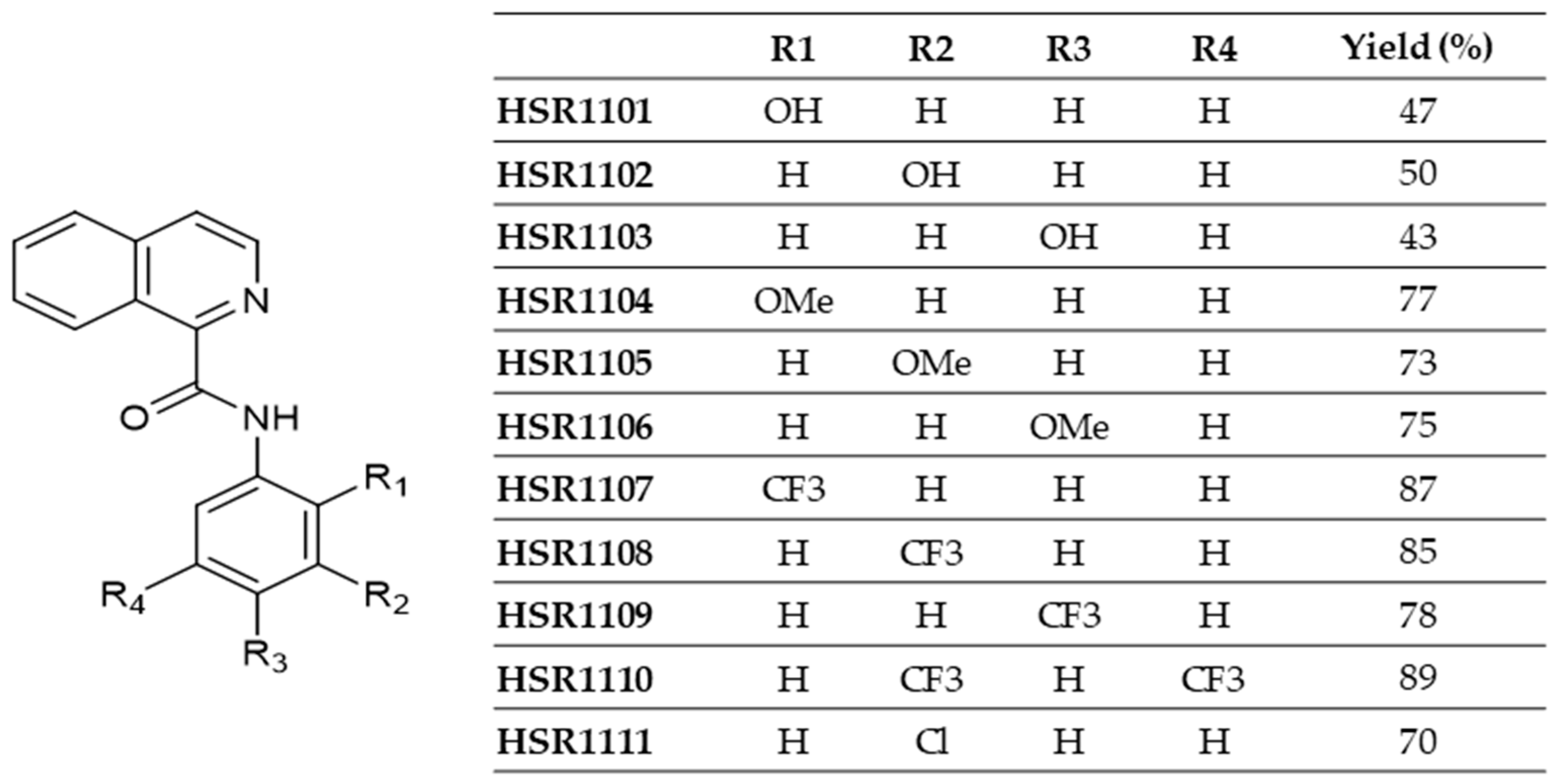
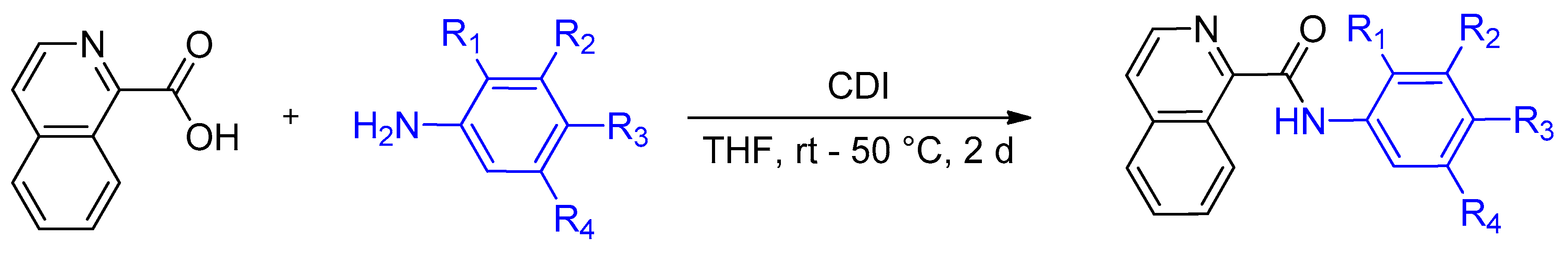
4.2. Synthesis of Isoquinoline-1-carboxamide Derivatives
4.3. Cell Culture, Treatments, and Measurements of Cell Viability
4.4. Measurements of IL-6, TNF-α, NO, and IL-10
4.5. Western Blotting
4.6. Immunocytochemistry
4.7. Cell Migration Assays
4.7.1. Wound Healing Assay
4.7.2. Transwell Migration Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| CNS | Central nervous system |
| COX-2 | Cyclooxygenase-2 |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| IκBs | Inhibitors of kappa B |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| JNK | C-Jun N-terminal kinase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| NF-κB | Nuclear factor-kappa B |
| NO | Nitric oxide |
| PD | Parkinson’s disease |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor-alpha |
References
- Amor, S.; Puentes, F.; Baker, D.; Van Der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Barron, K.D. The microglial cell. A historical review. J. Neurol. Sci. 1995, 134, 57–68. [Google Scholar] [CrossRef]
- Nakajima, K.; Kohsaka, S. Functional roles of microglia in the central nervous system. Hum. Cell 1998, 11, 141–155. [Google Scholar]
- Neumann, H.; Kotter, M.R.; Franklin, R.J. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 2009, 132, 288–295. [Google Scholar] [CrossRef]
- Zhang, Q.; Lu, Y.; Bian, H.; Guo, L.; Zhu, H. Activation of the α7 nicotinic receptor promotes lipopolysaccharide-induced conversion of M1 microglia to M2. Am. J. Transl. Res. 2017, 9, 971. [Google Scholar]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Liu, B.; Hong, J.-S. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Bose, S.; Kim, Y.M.; Chin, Y.W.; Cho, J. The ethyl acetate fraction from Physalis alkekengi inhibits LPS-induced pro-inflammatory mediators in BV2 cells and inflammatory pain in mice. J. Ethnopharmacol. 2016, 181, 26–36. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Lee, G.; Bose, S.; Choi, M.; Jung, J.K.; Lee, H.; Cho, J. Antioxidant and Anti-inflammatory Activities of N-((3,4-Dihydro-2H-benzo[h]chromene-2-yl)methyl)-4-methoxyaniline in LPS-Induced BV2 Microglial Cells. Biol. Pharm. Bull. 2015, 38, 1831–1835. [Google Scholar] [CrossRef]
- Dou, Y.; Wu, H.J.; Li, H.Q.; Qin, S.; Wang, Y.E.; Li, J.; Lou, H.F.; Chen, Z.; Li, X.M.; Luo, Q.M.; et al. Microglial migration mediated by ATP-induced ATP release from lysosomes. Cell Res. 2012, 22, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Lykhmus, O.; Mishra, N.; Koval, L.; Kalashnyk, O.; Gergalova, G.; Uspenska, K.; Komisarenko, S.; Soreq, H.; Skok, M. Molecular mechanisms regulating LPS-induced inflammation in the brain. Front. Mol. Neurosci. 2016, 9, 19. [Google Scholar] [CrossRef]
- Gao, H.M.; Jiang, J.; Wilson, B.; Zhang, W.; Hong, J.S.; Liu, B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: Relevance to Parkinson’s disease. J. Neurochem. 2002, 81, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, H.M.; Dragunow, M. Microglia induce neural cell death via a proximity-dependent mechanism involving nitric oxide. Brain Res. 2006, 1084, 1–15. [Google Scholar] [CrossRef]
- Ling, Z.; Zhu, Y.; Tong, C.; Snyder, J.A.; Lipton, J.W.; Carvey, P.M. Progressive dopamine neuron loss following supra-nigral lipopolysaccharide (LPS) infusion into rats exposed to LPS prenatally. Exp. Neurol. 2006, 199, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Jia, J.; Sun, J.; Xie, Q.; Zhang, X.; Deng, Y.; Yi, L. LXW7 attenuates inflammation via suppressing Akt/nuclear factor kappa B and mitogen-activated protein kinases signaling pathways in lipopolysaccharide-stimulated BV2 microglial cells. Int. Immunopharmacol. 2019, 77, 105963. [Google Scholar] [CrossRef]
- Subedi, L.; Lee, J.H.; Yumnam, S.; Ji, E.; Kim, S.Y. Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-kappaB Inhibition and Nrf2/HO-1 Activation. Cells 2019, 8, 194. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Invest. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Bui, B.P.; Oh, Y.; Lee, H.; Cho, J. Inhibition of inflammatory mediators and cell migration by 1,2,3,4-tetrahydroquinoline derivatives in LPS-stimulated BV2 microglial cells via suppression of NF-kappaB and JNK pathway. Int. Immunopharmacol. 2020, 80, 106231. [Google Scholar] [CrossRef]
- Pocivavsek, A.; Burns, M.P.; Rebeck, G.W. Low-density lipoprotein receptors regulate microglial inflammation through c-Jun N-terminal kinase. Glia 2009, 57, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Koppula, S.; Hong, S.S.; Jeon, S.B.; Kwon, J.H.; Hwang, B.Y.; Park, E.J.; Choi, D.K. Regulation of microglia activity by glaucocalyxin-A: Attenuation of lipopolysaccharide-stimulated neuroinflammation through NF-kappaB and p38 MAPK signaling pathways. PLoS ONE 2013, 8, e55792. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Sui, G.; Rosa, P.M.; Zhao, W. Radiation-induced c-Jun activation depends on MEK1-ERK1/2 signaling pathway in microglial cells. PLoS ONE 2012, 7, e36739. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Xiong, Z.; Chen, X.; Peng, F.; Hu, X.; Chen, Y.; Wang, Q. Artemisinin attenuates lipopolysaccharide-stimulated proinflammatory responses by inhibiting NF-κB pathway in microglia cells. PLoS ONE 2012, 7, e35125. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.Y.; Nam, J.H.; Yoon, G.; Lee, J.-Y.; Nam, Y.; Kang, H.-J.; Cho, H.-J.; Kim, J.; Hoe, H.-S. Ibrutinib suppresses LPS-induced neuroinflammatory responses in BV2 microglial cells and wild-type mice. J. Neuroinflammation 2018, 15, 271. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Li, Z.; Zhu, X.; Lin, R.; Chen, L. Salidroside reduces cell mobility via NF-κB and MAPK signaling in LPS-induced BV2 microglial cells. Evid. Based Complementary Altern. Med. 2014, 2014. [Google Scholar] [CrossRef]
- Cai, B.; Seong, K.-J.; Bae, S.-W.; Chun, C.; Kim, W.-J.; Jung, J.-Y. A synthetic diosgenin primary amine derivative attenuates LPS-stimulated inflammation via inhibition of NF-κB and JNK MAPK signaling in microglial BV2 cells. Int. Immunopharmacol. 2018, 61, 204–214. [Google Scholar] [CrossRef]
- Choi, Y.; Moon, A.; Kim, Y.C. A pinusolide derivative, 15-methoxypinusolidic acid from Biota orientalis inhibits inducible nitric oxide synthase in microglial cells: Implication for a potential anti-inflammatory effect. Int. Immunopharmacol. 2008, 8, 548–555. [Google Scholar] [CrossRef]
- Gee, M.S.; Kim, S.W.; Kim, N.; Lee, S.J.; Oh, M.S.; Jin, H.K.; Bae, J.S.; Inn, K.S.; Kim, N.J.; Lee, J.K. A Novel and Selective p38 Mitogen-Activated Protein Kinase Inhibitor Attenuates LPS-Induced Neuroinflammation in BV2 Microglia and a Mouse Model. Neurochem. Res. 2018, 43, 2362–2371. [Google Scholar] [CrossRef]
- Jeong, Y.H.; Kim, Y.; Song, H.; Chung, Y.S.; Park, S.B.; Kim, H.S. Anti-inflammatory effects of alpha-galactosylceramide analogs in activated microglia: Involvement of the p38 MAPK signaling pathway. PLoS ONE 2014, 9, e87030. [Google Scholar] [CrossRef]
- Anttila, J.E.; Whitaker, K.W.; Wires, E.S.; Harvey, B.K.; Airavaara, M. Role of microglia in ischemic focal stroke and recovery: Focus on Toll-like receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2017, 79, 3–14. [Google Scholar] [CrossRef]
- Li, N.; Liu, B.W.; Ren, W.Z.; Liu, J.X.; Li, S.N.; Fu, S.P.; Zeng, Y.L.; Xu, S.Y.; Yan, X.; Gao, Y.J.; et al. GLP-2 Attenuates LPS-Induced Inflammation in BV-2 Cells by Inhibiting ERK1/2, JNK1/2 and NF-kappaB Signaling Pathways. Int. J. Mol. Sci. 2016, 17, 190. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.P.; Zou, M.; Wang, J.Y.; Zhu, J.J.; Lai, J.M.; Zhou, L.L.; Chen, S.F.; Zhang, X.; Zhu, J.H. Paroxetine ameliorates lipopolysaccharide-induced microglia activation via differential regulation of MAPK signaling. J. Neuroinflammation 2014, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: Role of TNF-alpha. FASEB J. 2006, 20, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.L.; Abraham, C.R.; Masliah, E.; Kemper, P.; Inglis, J.D.; Oldstone, M.B.; Mucke, L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 10061–10065. [Google Scholar] [CrossRef] [PubMed]
- Bruhwyler, J.; Chleide, E.; Liégeois, J.-F.; Carreer, F. Nitric oxide: A new messenger in the brain. Neurosci. Biobehav. Rev. 1993, 17, 373–384. [Google Scholar] [CrossRef]
- Nathan, C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992, 6, 3051–3064. [Google Scholar] [CrossRef]
- Park, J.-S.; Woo, M.-S.; Kim, D.-H.; Hyun, J.-W.; Kim, W.-K.; Lee, J.-C.; Kim, H.-S. Anti-inflammatory mechanisms of isoflavone metabolites in lipopolysaccharide-stimulated microglial cells. J. Pharmacol. Exp. Ther. 2007, 320, 1237–1245. [Google Scholar] [CrossRef]
- Zhu, J.; Jiang, L.; Liu, Y.; Qian, W.; Liu, J.; Zhou, J.; Gao, R.; Xiao, H.; Wang, J. MAPK and NF-kappaB pathways are involved in bisphenol A-induced TNF-alpha and IL-6 production in BV2 microglial cells. Inflammation 2015, 38, 637–648. [Google Scholar] [CrossRef]
- Lobo-Silva, D.; Carriche, G.M.; Castro, A.G.; Roque, S.; Saraiva, M. Balancing the immune response in the brain: IL-10 and its regulation. J. Neuroinflammation 2016, 13, 297. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, C.A.; Carvalho, M.D.G.; Sousa, L.P.; Caramelli, P.; Gomes, K.B. Alzheimer’s disease and cytokine IL-10 gene polymorphisms: Is there an association? Arq. Neuropsiquiatr. 2017, 75, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S. Production of nitric oxide by glial cells: Regulation and potential roles in the CNS. Glia 2000, 29, 1–13. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Nitric oxide signaling in neurodegeneration and cell death. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 82, pp. 57–83. [Google Scholar]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Van De Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. Faseb J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease—a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- Liang, X.; Wu, L.; Wang, Q.; Hand, T.; Bilak, M.; McCullough, L.; Andreasson, K. Function of COX-2 and prostaglandins in neurological disease. J. Mol. Neurosci. 2007, 33, 94–99. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Scheiblich, H.; Roloff, F.; Singh, V.; Stangel, M.; Stern, M.; Bicker, G. Nitric oxide/cyclic GMP signaling regulates motility of a microglial cell line and primary microglia in vitro. Brain Res. 2014, 1564, 9–21. [Google Scholar] [CrossRef]
- Chen, A.; Kumar, S.M.; Sahley, C.L.; Muller, K.J. Nitric oxide influences injury-induced microglial migration and accumulation in the leech CNS. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 1036–1043. [Google Scholar] [CrossRef]
- Bohush, A.; Niewiadomska, G.; Filipek, A. Role of mitogen activated protein kinase signaling in parkinson’s disease. Int. J. Mol. Sci. 2018, 19, 2973. [Google Scholar] [CrossRef]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Nan, G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target. Int. J. Mol. Med. 2017, 39, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-Y.; Yang, C.-Y.; Hsu, W.-H.; Lin, K.-H.; Wang, C.-Y.; Shen, Y.-C.; Chen, Y.-C.; Chau, S.-F.; Tsai, H.-Y.; Cheng, C.-M. Monitoring the VEGF level in aqueous humor of patients with ophthalmologically relevant diseases via ultrahigh sensitive paper-based ELISA. Biomaterials 2014, 35, 3729–3735. [Google Scholar] [CrossRef]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef]
- Carty, M.; Bowie, A.G. Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem. Pharmacol. 2011, 81, 825–837. [Google Scholar] [CrossRef]
- Luo, Q.; Yan, X.; Bobrovskaya, L.; Ji, M.; Yuan, H.; Lou, H.; Fan, P. Anti-neuroinflammatory effects of grossamide from hemp seed via suppression of TLR-4-mediated NF-kappaB signaling pathways in lipopolysaccharide-stimulated BV2 microglia cells. Mol. Cell Biochem. 2017, 428, 129–137. [Google Scholar] [CrossRef]
- Lee, K.; Park, C.; Oh, Y.; Lee, H.; Cho, J. Antioxidant and neuroprotective effects of N-((3, 4-dihydro-2H-benzo [h] chromen-2-yl) methyl)-4-methoxyaniline in primary cultured rat cortical cells: Involvement of ERK-CREB signaling. Molecules 2018, 23, 669. [Google Scholar] [CrossRef]
- Hu, H.; Li, Z.; Zhu, X.; Lin, R.; Peng, J.; Tao, J.; Chen, L. GuaLou GuiZhi decoction inhibits LPS-induced microglial cell motility through the MAPK signaling pathway. Int. J. Mol. Med. 2013, 32, 1281–1286. [Google Scholar] [CrossRef]
- Bose, S.; Kim, S.; Oh, Y.; Moniruzzaman, M.; Lee, G.; Cho, J. Effect of CCL2 on BV2 microglial cell migration: Involvement of probable signaling pathways. Cytokine 2016, 81, 39–49. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pro-Inflammatory Mediators | IC50 Value a | ||||
|---|---|---|---|---|---|
| HSR1101 | HSR1102 | HSR1103 | HSR1105 | HSR1106 | |
| IL-6 | 39.13 | 29.52 | 30.55 | 56.59 | 76.38 |
| TNF-α | 29.18 | 25.92 | 70.17 | 71.66 | 67.81 |
| NO | 22.84 | 28.52 | 22.42 | 42.27 | 75.13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do, H.T.T.; Bui, B.P.; Sim, S.; Jung, J.-K.; Lee, H.; Cho, J. Anti-Inflammatory and Anti-Migratory Activities of Isoquinoline-1-Carboxamide Derivatives in LPS-Treated BV2 Microglial Cells via Inhibition of MAPKs/NF-κB Pathway. Int. J. Mol. Sci. 2020, 21, 2319. https://doi.org/10.3390/ijms21072319
Do HTT, Bui BP, Sim S, Jung J-K, Lee H, Cho J. Anti-Inflammatory and Anti-Migratory Activities of Isoquinoline-1-Carboxamide Derivatives in LPS-Treated BV2 Microglial Cells via Inhibition of MAPKs/NF-κB Pathway. International Journal of Molecular Sciences. 2020; 21(7):2319. https://doi.org/10.3390/ijms21072319
Chicago/Turabian StyleDo, Ha Thi Thu, Bich Phuong Bui, Seongrak Sim, Jae-Kyung Jung, Heesoon Lee, and Jungsook Cho. 2020. "Anti-Inflammatory and Anti-Migratory Activities of Isoquinoline-1-Carboxamide Derivatives in LPS-Treated BV2 Microglial Cells via Inhibition of MAPKs/NF-κB Pathway" International Journal of Molecular Sciences 21, no. 7: 2319. https://doi.org/10.3390/ijms21072319
APA StyleDo, H. T. T., Bui, B. P., Sim, S., Jung, J.-K., Lee, H., & Cho, J. (2020). Anti-Inflammatory and Anti-Migratory Activities of Isoquinoline-1-Carboxamide Derivatives in LPS-Treated BV2 Microglial Cells via Inhibition of MAPKs/NF-κB Pathway. International Journal of Molecular Sciences, 21(7), 2319. https://doi.org/10.3390/ijms21072319