The Impact of Natural Compounds on S-Shaped Aβ42 Fibril: From Molecular Docking to Biophysical Characterization

 , , , ,
, , , ,
Abstract
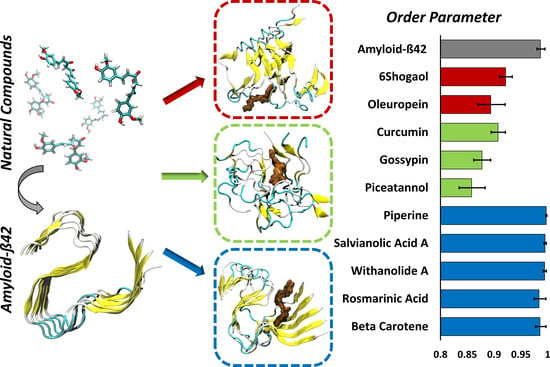
{kind=link}
{kind=link}
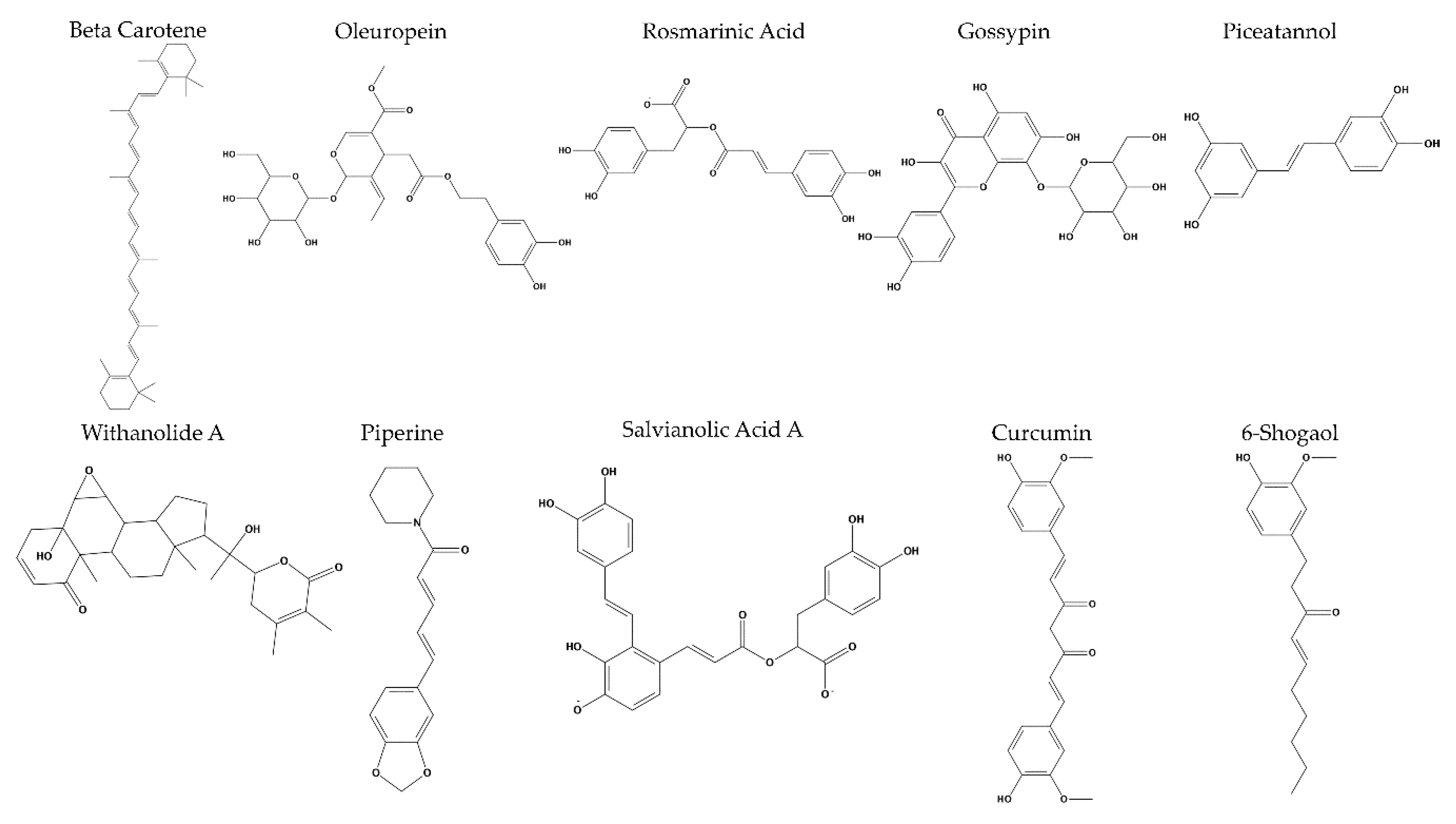
{kind=link}
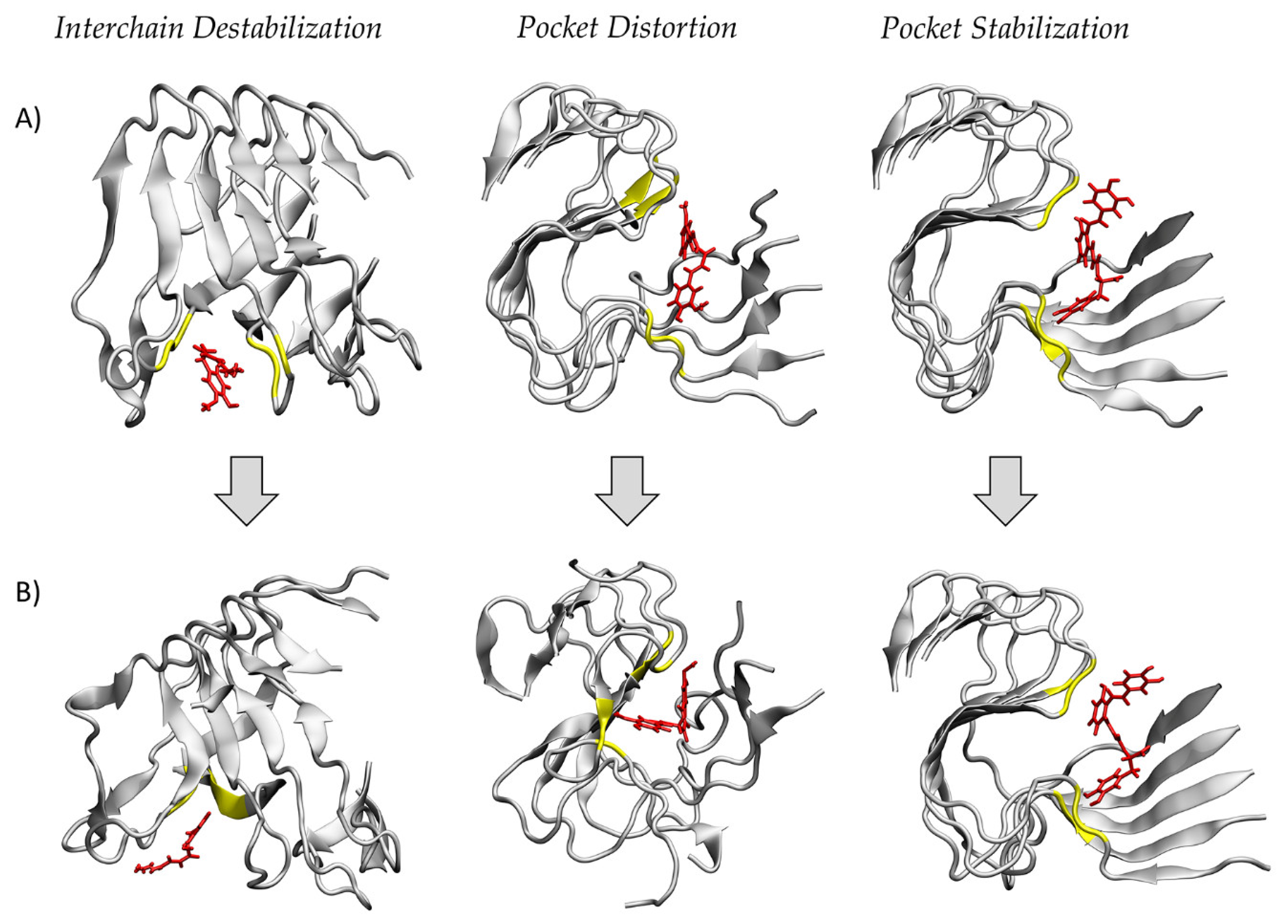
{kind=link}
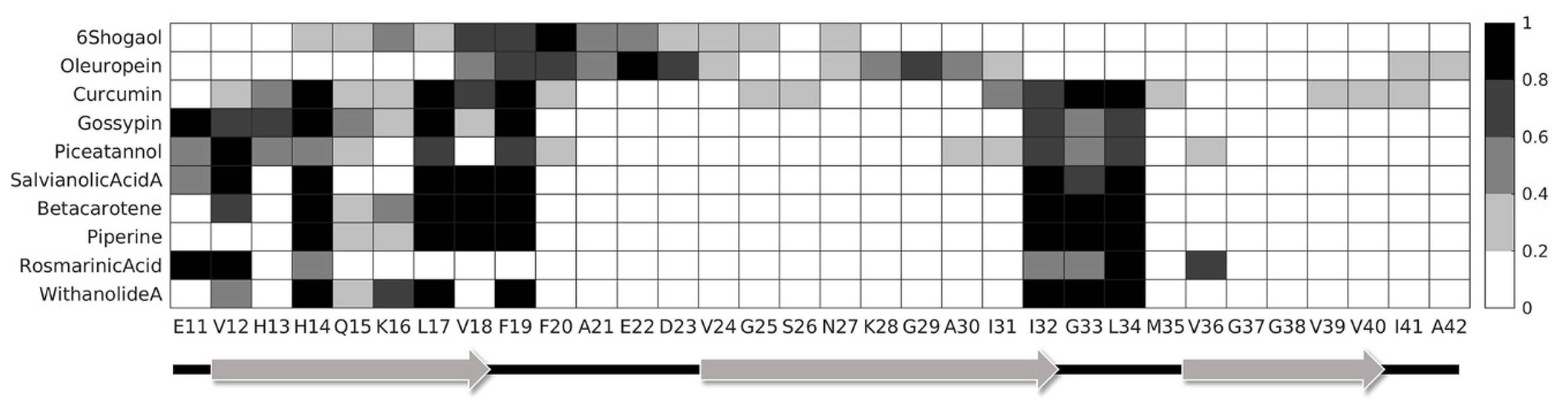{kind=link}
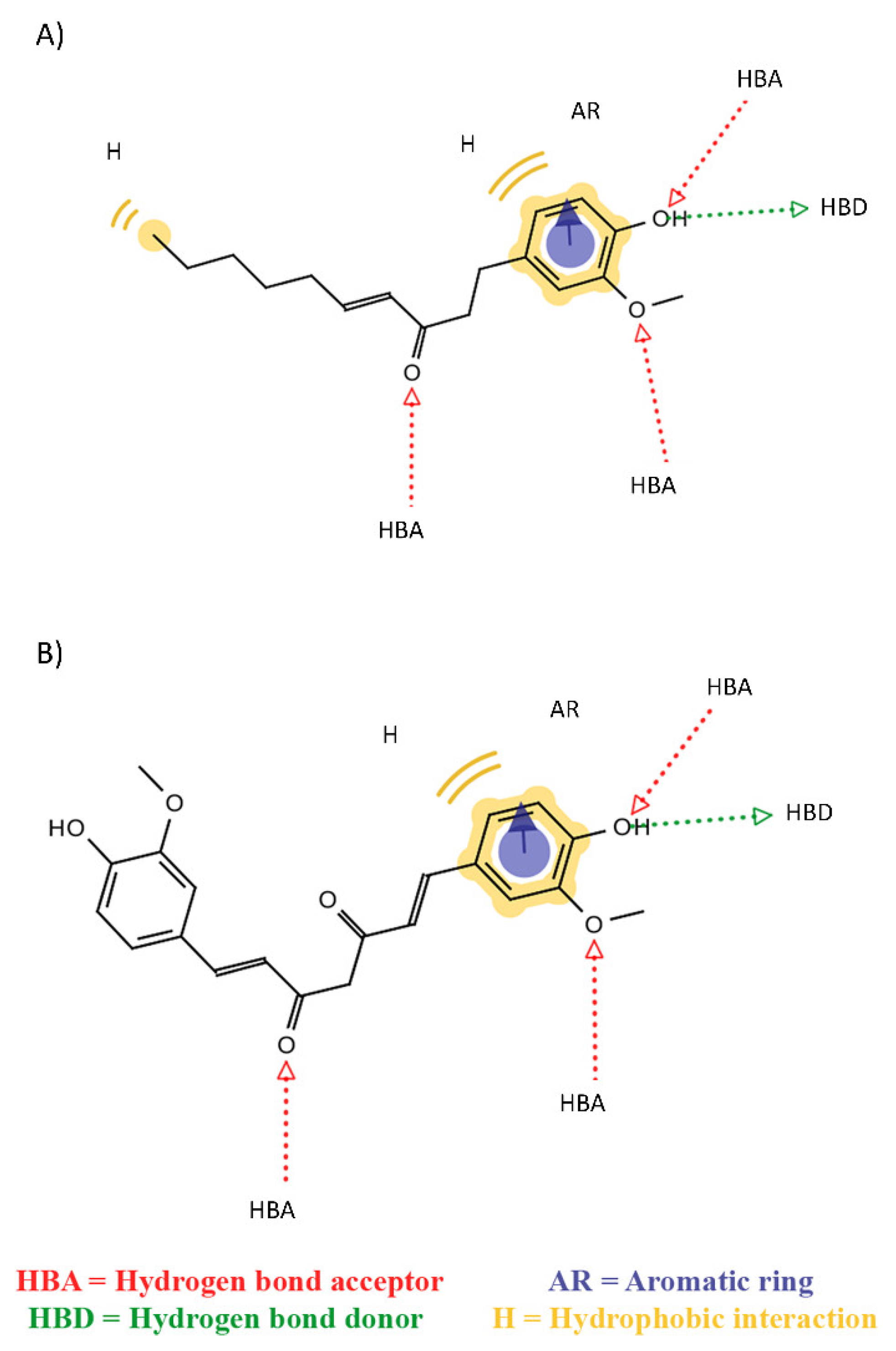{kind=link}
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Molecular Dynamics Setup
4.2. Molecular Docking Protocol
4.3. Binding Energy Estimation and Protein-Compounds Conformational Dynamics
4.4. Order Parameter
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| BBB | Blood Brain Barrier |
| APP | Amyloid Precursor Protein |
| MM–GBSA | Molecular Mechanics–Generalized Born Surface Area |
| VDW | Van der Waals |
| PME | Particle Mesh Ewald |
| GAFF | General Amber Force Field |
| RMSD | Root Mean Square Deviations |
| ordP | Order Parameter |
References
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Cummings, J.L. Alzheimer’s Disease. N. Engl. J. Med. 2004, 351, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Andrade, S.; Ramalho, M.J.; Loureiro, J.A.; Do Carmo Pereira, M. Natural compounds for alzheimer’s disease therapy: A systematic review of preclinical and clinical studies. Int. J. Mol. Sci. 2019, 20, 2313. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid- 42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [PubMed]
- Schütz, A.K.; Vagt, T.; Huber, M.; Ovchinnikova, O.Y.; Cadalbert, R.; Wall, J.; Güntert, P.; Böckmann, A.; Glockshuber, R.; Meier, B.H. Atomic-Resolution Three-Dimensional Structure of Amyloid $β$ Fibrils Bearing the Osaka Mutation. Angew. Chemie Int. Ed. 2015, 54, 331–335. [Google Scholar] [CrossRef]
- Guo, S.; Akhremitchev, B.B. Packing density and structural heterogeneity of insulin amyloid fibrils measured by AFM nanoindentation. Biomacromolecules 2006, 7, 1630–1636. [Google Scholar] [CrossRef]
- VandenAkker, C.C.; Engel, M.F.M.; Velikov, K.P.; Bonn, M.; Koenderink, G.H. Morphology and Persistence Length of Amyloid Fibrils Are Correlated to Peptide Molecular Structure. J. Am. Chem. Soc. 2011, 133, 18030–18033. [Google Scholar] [CrossRef]
- Palhano, F.L.; Lee, J.; Grimster, N.P.; Kelly, J.W. Toward the Molecular Mechanism(s) by Which EGCG Treatment Remodels Mature Amyloid Fibrils. J. Am. Chem. Soc. 2013, 135, 7503–7510. [Google Scholar] [CrossRef]
- Wang, T.; Jo, H.; DeGrado, W.F.; Hong, M. Water Distribution, Dynamics, and Interactions with Alzheimer’s β-Amyloid Fibrils Investigated by Solid-State NMR. J. Am. Chem. Soc. 2017, 139, 6242–6252. [Google Scholar] [CrossRef]
- Grasso, G.; Rebella, M.; Muscat, S.; Morbiducci, U.; Tuszynski, J.; Danani, A.; Deriu, M. Conformational Dynamics and Stability of U-Shaped and S-Shaped Amyloid β Assemblies. Int. J. Mol. Sci. 2018, 19, 571. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Rebella, M.; Morbiducci, U.; Tuszynski, J.A.; Danani, A.; Deriu, M.A. The Role of Structural Polymorphism in Driving the Mechanical Performance of the Alzheimer’s Beta Amyloid Fibrils. Front. Bioeng. Biotechnol. 2019, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Miceli, M.; Muscat, S.; Morbiducci, U.; Cavaglià, M.; Deriu, M.A. Ultrasonic waves effect on S-shaped β-amyloids conformational dynamics by non-equilibrium molecular dynamics. J. Mol. Graph. Model. 2020, 96, 107518. [Google Scholar] [CrossRef] [PubMed]
- Muscat, S.; Stojceski, F.; Danani, A. Elucidating the Effect of Static Electric Field on Amyloid Beta 1–42 Supramolecular Assembly. J. Mol. Graph. Model. 2020, 96, 107535. [Google Scholar] [CrossRef]
- Lambracht-Washington, D.; Rosenberg, R.N. Advances in the development of vaccines for Alzheimer’s disease. Discov. Med. 2013, 15, 319–326. [Google Scholar]
- Wang, C.Y.; Wang, P.-N.; Chiu, M.-J.; Finstad, C.L.; Lin, F.; Lynn, S.; Tai, Y.-H.; De Fang, X.; Zhao, K.; Hung, C.-H.; et al. UB-311, a novel UBITh ® amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 262–272. [Google Scholar] [CrossRef]
- Gold, M. Phase II clinical trials of anti–amyloid β antibodies: When is enough, enough? Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 402–409. [Google Scholar] [CrossRef]
- van Dyck, C.H. Anti-Amyloid-β Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef]
- Rajasekhar, K.; Madhu, C.; Govindaraju, T. Natural Tripeptide-Based Inhibitor of Multifaceted Amyloid β Toxicity. ACS Chem. Neurosci. 2016, 7, 1300–1310. [Google Scholar] [CrossRef]
- Viet, M.H.; Ngo, S.T.; Lam, N.S.; Li, M.S. Inhibition of Aggregation of Amyloid Peptides by Beta-Sheet Breaker Peptides and Their Binding Affinity. J. Phys. Chem. B 2011, 115, 7433–7446. [Google Scholar] [CrossRef]
- Lin, D.; Qi, R.; Li, S.; He, R.; Li, P.; Wei, G.; Yang, X. Interaction Dynamics in Inhibiting the Aggregation of Aβ Peptides by SWCNTs: A Combined Experimental and Coarse-Grained Molecular Dynamic Simulation Study. ACS Chem. Neurosci. 2016, 7, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wang, W.; Sang, J.; Jia, L.; Lu, F. Hydroxylated Single-Walled Carbon Nanotubes Inhibit Aβ 42 Fibrillogenesis, Disaggregate Mature Fibrils, and Protect against Aβ 42 -Induced Cytotoxicity. ACS Chem. Neurosci. 2019, 10, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Xiong, N.; Dong, X.Y.; Zheng, J.; Liu, F.F.; Sun, Y. Design of LVFFARK and LVFFARK-Functionalized Nanoparticles for Inhibiting Amyloid β-Protein Fibrillation and Cytotoxicity. ACS Appl. Mater. Interfaces 2015, 7, 5650–5662. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, R.; Hillert, E.-K.; Cameron, R.T.; Baillie, G.S. The role and therapeutic targeting of α-, β- and γ-secretase in Alzheimer’s disease. Futur. Sci. OA 2015, 1, fso.15.9. [Google Scholar] [CrossRef]
- Cui, J.; Wang, X.; Li, X.; Wang, X.; Zhang, C.; Li, W.; Zhang, Y.; Gu, H.; Xie, X.; Nan, F.; et al. Targeting the γ-/β-secretase interaction reduces β-amyloid generation and ameliorates Alzheimer’s disease-related pathogenesis. Cell Discov. 2015, 1, 15021. [Google Scholar] [CrossRef]
- Nie, Q.; Du, X.G.; Geng, M.Y. Small molecule inhibitors of amyloid β peptide aggregation as a potential therapeutic strategy for Alzheimer’s disease. Acta Pharmacol. Sin. 2011, 32, 545–551. [Google Scholar] [CrossRef]
- Zhu, M.; De Simone, A.; Schenk, D.; Toth, G.; Dobson, C.M.; Vendruscolo, M. Identification of small-molecule binding pockets in the soluble monomeric form of the Aβ42 peptide. J. Chem. Phys. 2013, 139. [Google Scholar] [CrossRef]
- Doig, A.J.; Derreumaux, P. Inhibition of protein aggregation and amyloid formation by small molecules. Curr. Opin. Struct. Biol. 2015, 30, 50–56. [Google Scholar] [CrossRef]
- Habchi, J.; Chia, S.; Limbocker, R.; Mannini, B.; Ahn, M.; Perni, M.; Hansson, O.; Arosio, P.; Kumita, J.R.; Challa, P.K.; et al. Systematic development of small molecules to inhibit specific microscopic steps of Aβ42 aggregation in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E200–E208. [Google Scholar] [CrossRef]
- Liu, F.; Ma, Z.; Sang, J.; Lu, F. Edaravone inhibits the conformational transition of amyloid-β42: Insights from molecular dynamics simulations. J. Biomol. Struct. Dyn. 2019, 1–12. [Google Scholar] [CrossRef]
- Liang, C.; Savinov, S.N.; Fejzo, J.; Eyles, S.J.; Chen, J. Modulation of Amyloid-β42 Conformation by Small Molecules Through Nonspecific Binding. J. Chem. Theory Comput. 2019, 15, 5169–5174. [Google Scholar] [CrossRef] [PubMed]
- Orgogozo, J.M.; Gilman, S.; Dartigues, J.F.; Laurent, B.; Puel, M.; Kirby, L.C.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S.; et al. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology 2003, 61, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Frid, P.; Anisimov, S.V.; Popovic, N. Congo red and protein aggregation in neurodegenerative diseases. Brain Res. Rev. 2007, 53, 135–160. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.T.; Nguyen, T.H. Natural product for the treatment of Alzheimer’s disease. J. Basic Clin. Physiol. Pharmacol. 2017, 28, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Robertson, A.A.B.; Cooper, M.A. Natural product and natural product derived drugs in clinical trials. Nat. Prod. Rep. 2014, 31, 1612–1661. [Google Scholar] [CrossRef]
- Andrade, S.; Ramalho, M.J.; Loureiro, J.A.; Pereira, M.C. Interaction of natural compounds with biomembrane models: A biophysical approach for the Alzheimer’s disease therapy. Colloids Surfaces B Biointerfaces 2019, 180, 83–92. [Google Scholar] [CrossRef]
- Rasool, M.; Malik, A.; Qureshi, M.S.; Manan, A.; Pushparaj, P.N.; Asif, M.; Qazi, M.H.; Qazi, A.M.; Kamal, M.A.; Gan, S.H.; et al. Recent Updates in the Treatment of Neurodegenerative Disorders Using Natural Compounds. Evidence-Based Complement. Altern. Med. 2014, 2014, 1–7. [Google Scholar] [CrossRef]
- Hiremathad, A. A Review: Natural Compounds as Anti-Alzheimer’s Disease Agents. Curr. Nutr. Food Sci. 2017, 13. [Google Scholar] [CrossRef]
- Deb, S.; Mazumder, M.K.; Dutta, A.; Phukan, B.C.; Bhattacharya, P.; Paul, R.; Borah, A. Therapeutic implications of anti-inflammatory natural products in Alzheimer’s disease. In Discovery and Development of Anti-Inflammatory Agents from Natural Products; Elsevier: Amsterdam, The Netherlands, 2019; pp. 241–258. ISBN 9780128169926. [Google Scholar]
- Mourtas, S.; Canovi, M.; Zona, C.; Aurilia, D.; Niarakis, A.; La Ferla, B.; Salmona, M.; Nicotra, F.; Gobbi, M.; Antimisiaris, S.G. Curcumin-decorated nanoliposomes with very high affinity for amyloid-β1-42 peptide. Biomaterials 2011, 32, 1635–1645. [Google Scholar] [CrossRef]
- Ringman, J.; Frautschy, S.; Cole, G.; Masterman, D.; Cummings, J. A Potential Role of the Curry Spice Curcumin in Alzheimers Disease. Curr. Alzheimer Res. 2005, 2, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Fitzpatrick, A.W.; Meehan, S.; Mott, H.R.; Vendruscolo, M.; Dobson, C.M.; Welland, M.E. Role of intermolecular forces in defining material properties of protein nanofibrils. Science 2007, 318, 1900–1903. [Google Scholar] [CrossRef] [PubMed]
- Bidone, T.C.; Kim, T.; Deriu, M.A.; Morbiducci, U.; Kamm, R.D. Multiscale impact of nucleotides and cations on the conformational equilibrium, elasticity and rheology of actin filaments and crosslinked networks. Biomech. Model. Mechanobiol. 2015, 14, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.-M.; Xu, Q.; Wei, D.-Q. Recent Studies on Mechanisms of New Drug Candidates for Alzheimer’s Disease Interacting with Amyloid-β Protofibrils Using Molecular Dynamics Simulations. In Translational Bioinformatics and Its Application; Wei, D.-Q., Ma, Y., Cho, W.C., Xu, Q., Zhou, F., Eds.; Springer: Dordrecht, The Netherlands, 2017; pp. 135–151. [Google Scholar]
- Tang, M.; Wang, Z.; Zhou, Y.; Xu, W.; Li, S.; Wang, L.; Wei, D.; Qiao, Z. A Novel Drug Candidate for Alzheimer’s Disease Treatment: Gx-50 Derived from Zanthoxylum Bungeanum. J. Alzheimer’s Dis. 2013, 34, 203–213. [Google Scholar] [CrossRef]
- Hou, S.; Gu, R.-X.; Wei, D.-Q. Inhibition of β-Amyloid Channels with a Drug Candidate wgx-50 Revealed by Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 2811–2821. [Google Scholar] [CrossRef]
- Fan, H.-M.; Gu, R.-X.; Wang, Y.-J.; Pi, Y.-L.; Zhang, Y.-H.; Xu, Q.; Wei, D.-Q. Destabilization of Alzheimer’s Aβ42 Protofibrils with a Novel Drug Candidate wgx-50 by Molecular Dynamics Simulations. J. Phys. Chem. B 2015, 119, 11196–11202. [Google Scholar] [CrossRef]
- Kanchi, P.K.; Dasmahapatra, A.K. Polyproline chains destabilize the Alzheimer’s amyloid-β protofibrils: A molecular dynamics simulation study. J. Mol. Graph. Model. 2019, 93, 107456. [Google Scholar] [CrossRef]
- Sharma, B.; Kalita, S.; Paul, A.; Mandal, B.; Paul, S. The role of caffeine as an inhibitor in the aggregation of amyloid forming peptides: A unified molecular dynamics simulation and experimental study. RSC Adv. 2016, 6, 78548–78558. [Google Scholar] [CrossRef]
- Battisti, A.; Palumbo Piccionello, A.; Sgarbossa, A.; Vilasi, S.; Ricci, C.; Ghetti, F.; Spinozzi, F.; Marino Gammazza, A.; Giacalone, V.; Martorana, A.; et al. Curcumin-like compounds designed to modify amyloid beta peptide aggregation patterns. RSC Adv. 2017, 7, 31714–31724. [Google Scholar] [CrossRef]
- Masuda, Y.; Fukuchi, M.; Yatagawa, T.; Tada, M.; Takeda, K.; Irie, K.; Akagi, K.; Monobe, Y.; Imazawa, T.; Takegoshi, K. Solid-state NMR analysis of interaction sites of curcumin and 42-residue amyloid β-protein fibrils. Bioorg. Med. Chem. 2011, 19, 5967–5974. [Google Scholar] [CrossRef]
- Rao, P.P.N.; Mohamed, T.; Teckwani, K.; Tin, G. Curcumin Binding to Beta Amyloid: A Computational Study. Chem. Biol. Drug Des. 2015, 86, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Fändrich, M.; Nyström, S.; Nilsson, K.P.R.; Böckmann, A.; LeVine, H.; Hammarström, P. Amyloid fibril polymorphism: A challenge for molecular imaging and therapy. J. Intern. Med. 2018, 283, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Acosta, D.M.Á.V.; Vega, B.C.; Basurto, J.C.; Morales, L.G.F.; Rosales Hernández, M.C. Recent Advances by In Silico and In Vitro Studies of Amyloid-β 1-42 Fibril Depicted a S-Shape Conformation. Int. J. Mol. Sci. 2018, 19, 2415. [Google Scholar] [CrossRef] [PubMed]
- Su, P.C.; Tsai, C.C.; Mehboob, S.; Hevener, K.E.; Johnson, M.E. Comparison of radii sets, entropy, QM methods, and sampling on MM-PBSA, MM-GBSA, and QM/MM-GBSA ligand binding energies of F. tularensis enoyl-ACP reductase (FabI). J. Comput. Chem. 2015, 36, 1859–1873. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A. Ekavali A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Auld, D.S.; Kornecook, T.J.; Bastianetto, S.; Quirion, R. Alzheimer’s disease and the basal forebrain cholinergic system: Relations to β-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 2002, 68, 209–245. [Google Scholar] [CrossRef]
- Farlow, M.R.; Miller, M.L.; Pejovic, V. Treatment Options in Alzheimer’s Disease: Maximizing Benefit, Managing Expectations. Dement. Geriatr. Cogn. Disord. 2008, 25, 408–422. [Google Scholar] [CrossRef]
- Du, W.J.; Guo, J.J.; Gao, M.T.; Hu, S.Q.; Dong, X.Y.; Han, Y.F.; Liu, F.F.; Jiang, S.; Sun, Y. Brazilin inhibits amyloid β-protein fibrillogenesis, remodels amyloid fibrils and reduces amyloid cytotoxicity. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Tavanti, F.; Pedone, A.; Menziani, M. Computational Insight into the Effect of Natural Compounds on the Destabilization of Preformed Amyloid-β(1–40) Fibrils. Molecules 2018, 23, 1320. [Google Scholar] [CrossRef] [PubMed]
- Pourkhodadad, S.; Alirezaei, M.; Moghaddasi, M.; Ahmadvand, H.; Karami, M.; Delfan, B.; Khanipour, Z. Neuroprotective effects of oleuropein against cognitive dysfunction induced by colchicine in hippocampal CA1 area in rats. J. Physiol. Sci. 2016, 66, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Luccarini, I.; Ed Dami, T.; Grossi, C.; Rigacci, S.; Stefani, M.; Casamenti, F. Oleuropein aglycone counteracts Aβ42 toxicity in the rat brain. Neurosci. Lett. 2014, 558, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Ferrándiz, M.L.; Alcaraz, M.J. Anti-inflammatory activity and inhibition of arachidonic acid metabolism by flavonoids. Agents Actions 1991, 32, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Thamizhiniyan, V.; Vijayaraghavan, K.; Subramanian, S.P. Gossypin, a flavonol glucoside protects pancreatic beta-cells from glucotoxicity in streptozotocin-induced experimental diabetes in rats. Biomed. Prev. Nutr. 2012, 2, 239–245. [Google Scholar] [CrossRef]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin Has Potent Anti-Amyloidogenic Effects for Alzheimer’s β-Amyloid Fibrils In Vitro. J. Neurosci. Res. 2004. [Google Scholar] [CrossRef]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005. [Google Scholar] [CrossRef]
- Diomede, L.; Rigacci, S.; Romeo, M.; Stefani, M.; Salmona, M. Oleuropein Aglycone Protects Transgenic C. elegans Strains Expressing Aβ42 by Reducing Plaque Load and Motor Deficit. PLoS ONE 2013, 8, e58893. [Google Scholar] [CrossRef]
- Rigacci, S.; Guidotti, V.; Bucciantini, M.; Nichino, D.; Relini, A.; Berti, A.; Stefani, M. Aβ(1-42) aggregates into non-toxic amyloid assemblies in the presence of the natural polyphenol oleuropein aglycon. Curr. Alzheimer Res. 2011, 8, 841–852. [Google Scholar] [CrossRef]
- Pantano, D.; Luccarini, I.; Nardiello, P.; Servili, M.; Stefani, M.; Casamenti, F. Oleuropein aglycone and polyphenols from olive mill waste water ameliorate cognitive deficits and neuropathology. Br. J. Clin. Pharmacol. 2017, 83, 54–62. [Google Scholar] [CrossRef]
- Kumar, J.; Namsechi, R.; Sim, V.L. Structure-based peptide design to modulate amyloid beta aggregation and reduce cytotoxicity. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Leri, M.; Natalello, A.; Bruzzone, E.; Stefani, M.; Bucciantini, M. Oleuropein aglycone and hydroxytyrosol interfere differently with toxic Aβ 1-42 aggregation. Food Chem. Toxicol. 2019, 129, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Hess, B.; Hess, B.; Bekker, H.; Berendsen, H.J.C.C.; Fraaije, J.G.E.M.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 27–28, 33–38. [Google Scholar] [CrossRef]
- Grasso, G.; Muscat, S.; Rebella, M.; Morbiducci, U.; Audenino, A.; Danani, A.; Deriu, M.A. Cell penetrating peptide modulation of membrane biomechanics by Molecular dynamics. J. Biomech. 2018, 73, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Klejborowska, G.; Urbaniak, A.; Preto, J.; Maj, E.; Moshari, M.; Wietrzyk, J.; Tuszynski, J.A.; Chambers, T.C.; Huczyński, A. Synthesis, biological evaluation and molecular docking studies of new amides of 4-bromothiocolchicine as anticancer agents. Bioorg. Med. Chem. 2019, 76, 115144. [Google Scholar] [CrossRef]
- Sahakyan, H.; Abelyan, N.; Arakelov, V.; Arakelov, G.; Nazaryan, K. In silico study of colchicine resistance molecular mechanisms caused by tubulin structural polymorphism. PLoS ONE 2019, 14, e0221532. [Google Scholar] [CrossRef]
- Kumbhar, B.V.; Borogaon, A.; Panda, D.; Kunwar, A. Exploring the Origin of Differential Binding Affinities of Human Tubulin Isotypes αβII, αβIII and αβIV for DAMA-Colchicine Using Homology Modelling, Molecular Docking and Molecular Dynamics Simulations. PLoS ONE 2016, 11, e0156048. [Google Scholar] [CrossRef]
- Gajewski, M.M.; Tuszynski, J.A.; Barakat, K.; Huzil, J.T.; Klobukowski, M. Interactions of laulimalide, peloruside, and their derivatives with the isoforms of β-tubulin. Can. J. Chem. 2013, 91, 511–517. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef]
- Grasso, G.; Leanza, L.; Morbiducci, U.; Danani, A.; Deriu, M.A. Aminoacid Substitutions in the Glycine Zipper Affect the Conformational Stability of Amyloid Beta Fibrils. J. Biomol. Struct. Dyn. 2019, 1–13. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muscat, S.; Pallante, L.; Stojceski, F.; Danani, A.; Grasso, G.; Deriu, M.A. The Impact of Natural Compounds on S-Shaped Aβ42 Fibril: From Molecular Docking to Biophysical Characterization. Int. J. Mol. Sci. 2020, 21, 2017. https://doi.org/10.3390/ijms21062017
Muscat S, Pallante L, Stojceski F, Danani A, Grasso G, Deriu MA. The Impact of Natural Compounds on S-Shaped Aβ42 Fibril: From Molecular Docking to Biophysical Characterization. International Journal of Molecular Sciences. 2020; 21(6):2017. https://doi.org/10.3390/ijms21062017
Chicago/Turabian StyleMuscat, Stefano, Lorenzo Pallante, Filip Stojceski, Andrea Danani, Gianvito Grasso, and Marco Agostino Deriu. 2020. "The Impact of Natural Compounds on S-Shaped Aβ42 Fibril: From Molecular Docking to Biophysical Characterization" International Journal of Molecular Sciences 21, no. 6: 2017. https://doi.org/10.3390/ijms21062017
APA StyleMuscat, S., Pallante, L., Stojceski, F., Danani, A., Grasso, G., & Deriu, M. A. (2020). The Impact of Natural Compounds on S-Shaped Aβ42 Fibril: From Molecular Docking to Biophysical Characterization. International Journal of Molecular Sciences, 21(6), 2017. https://doi.org/10.3390/ijms21062017