Potential Role of Venular Amyloid in Alzheimer’s Disease Pathogenesis


{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Alzheimer’s Disease
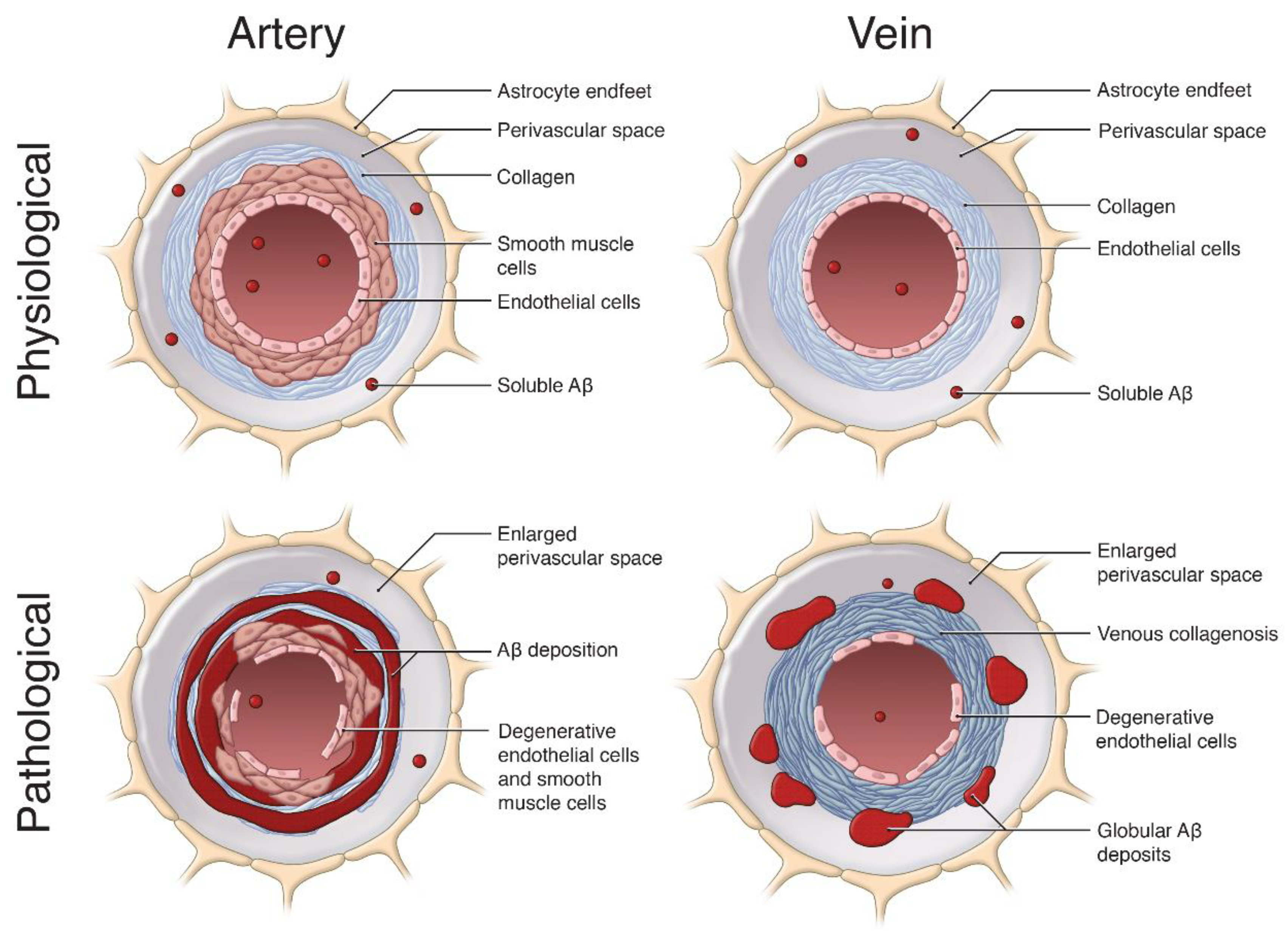
1.2. Cerebral Amyloid Angiopathy
2. Impaired Perivascular Clearance of Aβ
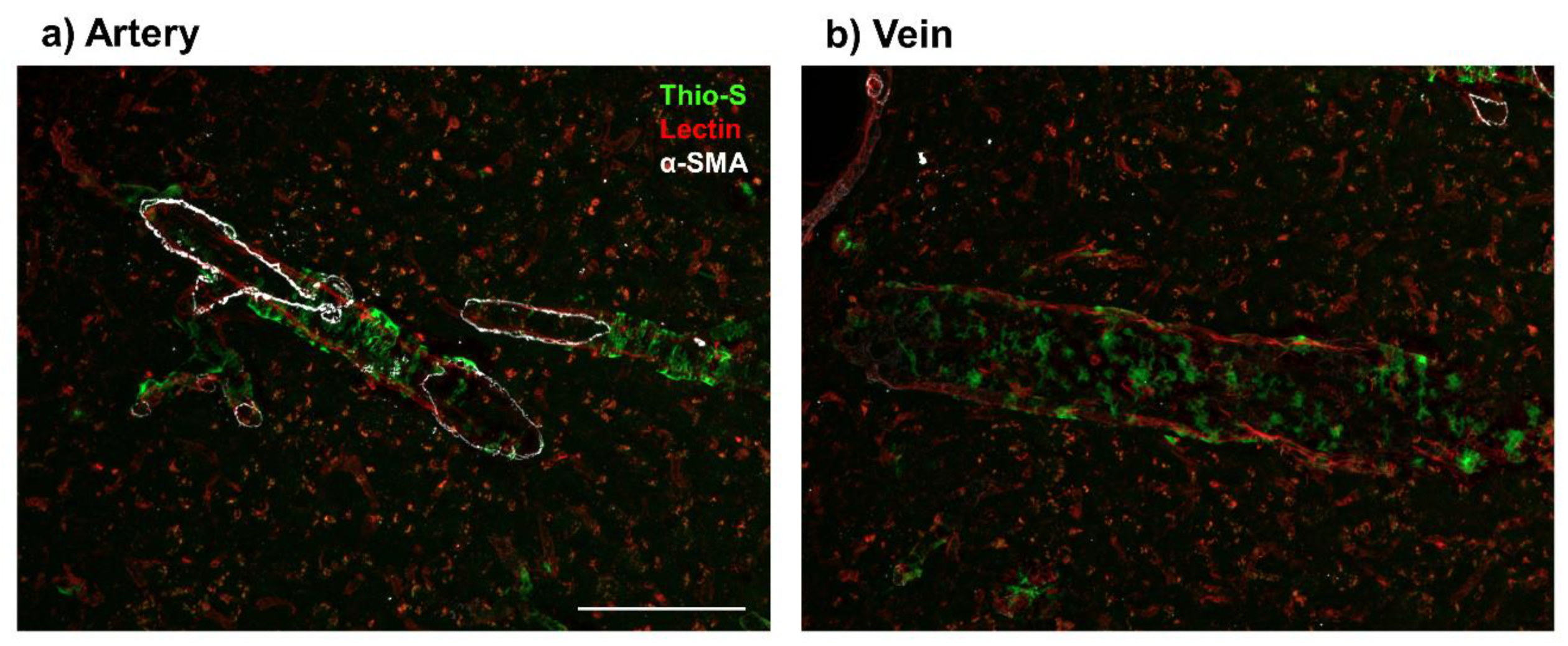
3. Evidence of Aβ in Veins and Venules
3.1. Preclinical Models
3.2. Clinical Evidence
4. Contribution of Venular Amyloid to Alzheimer’s Disease (AD) Pathogenesis
4.1. Venous Collagenosis
4.2. Cerebrovascular Pulsatility
4.3. Enlargements in the Perivascular Space
5. Limited Efficacy of Aβ-Targeted Therapeutics on Vascular Amyloid
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-SMA | alpha-smooth muscle actin |
| Aβ | amyloid-beta |
| AD | Alzheimer’s disease |
| APP | amyloid precursor protein |
| AQP4 | aquaporin 4 |
| BACE1 | beta-site amyloid precursor protein cleaving enzyme 1 |
| BBB | blood–brain barrier |
| CAA | cerebral amyloid angiopathy |
| CSF | cerebrospinal fluid |
| HCHWA | hereditary cerebral hemorrhage with amyloidosis |
| ISF | interstitial fluid |
| MRI | magnetic resonance imaging |
| PET | positron emission tomography |
| PS1/PS2 | presenilin-1/ -2 |
References
- Iadecola, C.; Gorelick, P.B. Mechanisms in Vascular and Neurodegenerative Dementia. Stroke 2003, 34, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Ihara, M. New Therapeutic Approaches for Alzhiemer’s disease and Cerebral Amyloid Angiopathy. Front. Aging Neurosci. 2014, 6, 290. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer′s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Weller, R.O.; Subash, M.; Preston, S.D.; Mazanti, I.; Carare, R.O. SYMPOSIUM: Clearance of Aβ from the Brain in Alzheimer′s Disease: Perivascular Drainage of Amyloid-β Peptides from the Brain and its Failure in Cerebral Amyloid Angiopathy and Alzheimer′s Disease. Brain Pathol. 2008, 18, 253–266. [Google Scholar] [CrossRef]
- Cai, Y.; An, S.S.; Kim, S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-Associated disorders. Clin. Interv. Aging 2015, 10, 1163–1172. [Google Scholar] [CrossRef]
- Kelleher, R.J.; Shen, J. Presenilin-I mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer′s Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef]
- Biffi, A.; Greenberg, S.M. Cerebral Amyloid Angiopathy: A Systemic Review. J. Clin. Neurol. 2011, 7, 1–9. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-Deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Hunter, J.M.; Kwan, J.; Malek-Ahmadi, M.; Maarouf, C.L.; Kokjohn, T.A.; Belden, C.; Sabbagh, M.N.; Beach, T.G.; Roher, A.E. Morphological and Pathological Evolution of the Brain Microcirculation in Aging and Alzheimer’s Disease. PLoS ONE 2012, 7, e36893. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer′s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Dominantly Inherited Alzheimer Network. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer′s Disease. N. Engl. J. Med. 2012, 367, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Morrone, C.D.; Bazzigaluppi, P.; Beckett, T.L.; Hill, M.E.; Koletar, M.M.; Stefanovic, B.; McLaurin, J. Regional differences in Alzheimer’s disease pathology confound behavioural rescue after amyloid-β attenuation. Brain 2020, 143, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.R.; Moody, D.M.; Thore, C.R.; Anstrom, J.A.; Challa, V.R. Microvascular changes in the white mater in dementia. J. Neurol. Sci. 2009, 283, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.R.; Thore, C.R. Review: Cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol. Appl. Neurobiol. 2012, 37, 56–74. [Google Scholar] [CrossRef]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Mateos-Pérez, J.M.; Evans, A.C.; The Alzheimer’s Disease Neuroimaging Initiative. Early role of vascular dysregulation on late-Onset Alzheimer’s disease based on multifactorial data-Driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef]
- Joo, I.L.; Lai, A.Y.; Bazzigaluppi, P.; Koletar, M.M.; Dorr, A.; Brown, M.E.; Thomason, L.A.M.; Sled, J.G.; McLaurin, J.; Stefanovic, B. Early neurovascular dysfunction in a transgenic rat model of Alzheimer’s disease. Sci. Rep. 2017, 7, 46427. [Google Scholar] [CrossRef]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Rostagno, A.; Ghiso, J.; Frangione, B. Sporadic and Familial Cerebral Amyloid Angiopathies. Brain Pathol. 2002, 12, 343–357. [Google Scholar] [CrossRef]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Frangione, B.; Rostagno, A.; Ghiso, J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathy. Acta Neuropathol. 2009, 118, 115–130. [Google Scholar] [CrossRef]
- Banerjee, G.; Carare, R.; Cordonnier, C.; Greenberg, S.M.; Schneider, J.A.; Smith, E.E.; Buchem, M.V.; Grond, J.V.; Verbeek, M.M.; Werring, D.J. The increasing impact of cerebral amyloid angiopathy: Essential new insights for clinical practice. J. Neurol. Neurosurg. Psychiatry 2017, 88, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.H.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease-One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 2017, 140, 1829–1850. [Google Scholar] [CrossRef] [PubMed]
- Weller, R.O.; Boche, D.; Nicoll, J.A.R. Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s disease and their potential impact on therapy. Acta Neuropathol. 2009, 118, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Scholz, W. Studien zur Pathologie der Hirngefässe II. Die drusige Entartung der Hirnarterien und Capillaren. Z. Ges. Neurol. Psychiat. 1938, 162, 694–715. [Google Scholar] [CrossRef]
- Pantelakis, S. A particular type of senile angiopathy of the central nervous system: Congophilic angiopathy, topography and frequency. Monatsschr. Psychiatr. Neurol. 1954, 128, 219–256. [Google Scholar] [CrossRef]
- Michaud, J.P.; Bellavance, M.A.; Préfontaine, P.; Rivest, S. Real-Time In Vivo Imaging Reveals the Ability of Monocytes to Clear Vascular Amyloid Beta. Cell Rep. 2013, 5, 646–653. [Google Scholar] [CrossRef]
- McGowan, E.; Pickford, F.; Kim, J.; Onstead, L.; Eriksen, J.; Yu, C.; Skipper, L.; Murphy, M.P.; Beard, J.; Das, P.; et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 2005, 47, 191–199. [Google Scholar] [CrossRef]
- Attems, J.; Lintner, F.; Jellinger, K.A. Amyloid beta peptide 1-42 highly correlates with capillary cerebral amyloid angiopathy and Alzheimer disease pathology. Acta. Neuropathol. 2004, 107, 283–291. [Google Scholar] [CrossRef]
- Black, S.; Gao, F.; Bilbao, J. Understanding White Matter Disease: Imaging-Pathological Correlations in Vascular Cognitive Impairment. Stroke 2009, 40, S48–S52. [Google Scholar] [CrossRef]
- Brown, R.; Benveniste, H.; Black, S.E.; Charpak, S.; Dichgans, M.; Joutel, A.; Nedergaard, M.; Smith, K.J.; Zlokovic, B.V.; Wardlaw, J.M. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc. Res. 2018, 114, 1462–1473. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Pantoni, L.; Love, S. The concept of sporadic cerebral small vessel disease: A road map on key definitions and current concepts. Int. J. Stroke 2016, 11, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Pantoni, L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- Weller, R.O.; Massey, A.; Newman, T.A.; Hutchings, M.; Kuo, Y.; Roher, A.E. Cerebral Amyloid Angiopathy: Amyloid β Accumulates in Putative Interstitial Fluid Drainage Pathways in Alzheimer′s Disease. Am. J. Pathol. 1998, 153, 725–733. [Google Scholar] [CrossRef]
- Yates, P.A.; Desmond, P.M.; Phal, P.M.; Steward, C.; Szoeke, C.; Salvado, O.; Ellis, K.A.; Martins, R.N.; Masters, C.L.; Ames, D.; et al. AIBL Research Group. Incidence of cerebral microbleeds in preclinical Alzheimer disease. Neurology 2014, 82, 1266–1273. [Google Scholar] [CrossRef]
- Hawkes, C.A.; Jayakody, N.; Johnston, D.A.; Bechmann, I.; Carare, R.O. Failure of Perivascular Drainage of β-amyloid in Cerebral Amyloid Angiopathy. Brain Pathol. 2014, 24, 396–403. [Google Scholar] [CrossRef]
- Hawkes, C.A.; Härtig, W.; Kacza, J.; Schliebs, R.; Weller, R.O.; Nicoll, J.A.; Carare, R.O. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011, 121, 431–443. [Google Scholar] [CrossRef]
- Peng, W.; Achariyar, T.M.; Li, B.; Liao, Y.; Mestre, H.; Hitomi, E.; Regan, S.; Kasper, T.; Peng, S.; Ding, F.; et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2016, 93, 215–225. [Google Scholar] [CrossRef]
- Morrone, C.D.; Liu, M.; Black, S.E.; McLaurin, J. Interaction between therapeutic interventions for Alzheimer’s disease and physiological Aβ clearance mechanisms. Front. Aging Neurosci. 2015, 7, 64. [Google Scholar] [CrossRef]
- Horsburgh, K.; Wardlaw, J.M.; Agtmael, T.V.; Allan, S.M.; Ashford, M.L.J.; Bath, P.M.; Brown, R.; Berwick, J.; Cader, M.Z.; Carare, R.O.; et al. Small vessels, dementia and chronic diseases-Molecular mechanisms and pathophysiology. Clin. Sci. (Lond.) 2018, 132, 851–868. [Google Scholar] [CrossRef]
- Carare, R.O.; Bernardes-Silva, M.; Newman, T.A.; Page, A.M.; Nicoll, J.A.R.; Perry, V.H.; Weller, R.O. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: Significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 2008, 34, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer’s disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- MacGregor Sharp, M.; Criswell, T.P.; Dobson, H.; Finucane, C.; Verma, A.; Carare, R.O. Solving an Old Dogma: Is it an Arteriole or a Venule? Front. Aging Neurosci. 2019, 11, 289. [Google Scholar] [CrossRef]
- Lai, A.Y.; Dorr, A.; Thomason, L.A.M.; Koletar, M.M.; Sled, J.G.; Stefanovic, B.; McLaurin, J. Venular degeneration leads to vascular dysfunction in a transgenic model of Alzheimer’s disease. Brain 2015, 138, 1046–1058. [Google Scholar] [CrossRef]
- Hartmann, D.A.; Hyacinth, H.I.; Liao, F.F.; Shih, A.Y. Does pathology of small venules contribute to cerebral infarcts and dementia? J. Neurochem. 2018, 144, 517–526. [Google Scholar] [CrossRef]
- Keith, J.; Gao, F.; Noor, R.; Kiss, A.; Balasubramaniam, G.; Au, K.; Rogaeva, E.; Masellis, M.; Black, S.E. Collagenosis of the Deep Medullary Veins: An Underrecognized Pathologic Correlate of White Matter Hyperintensities and Periventricular Infarction? J. Neuropathol. Exp. Neurol. 2017, 76, 299–312. [Google Scholar] [CrossRef]
- Simka, M.; Skula, M. Potential Involvement of Impaired Venous Outflow from the Brain in Neurodegeneration: Lessons Learned from the Research on Chronic Cerebrospinal Venous Insufficiency. Rev. Recent Clin. Trials 2019, 14, 235–236. [Google Scholar] [CrossRef]
- Mountjoy, C.Q.; Tomlinson, B.E.; Gibson, P.H. Amyloid and Senile Plaques and Cerebral Blood Vessels. A Semi-Quantitative Investigation of a Possible Relationship. J. Neurol. Sci. 1982, 57, 89–103. [Google Scholar] [CrossRef]
- Mendel, T.; Wierzba-Bobrowicz, T.; Stępień, T.; Szpak, G.M. β-Amyloid deposits in veins in patients with cerebral amyloid angiopathy and intracerebral haemorrhage. Folia Neuropathol. 2013, 51, 120–126. [Google Scholar] [CrossRef]
- Thal, D.R.; Ghebremedhin, E.; Rüb, U.; Yamaguchi, H.; Del Tredici, K.; Braak, H. Two Types of Sporadic Cerebral Amyloid Angiopathy. J. Neuropathol. Exp. Neurol. 2002, 61, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Moody, D.M.; Brown, W.R.; Challa, V.R.; Anderson, R.L. Periventricular venous collagenosis: Association with leukoaraiosis. Radiology 1995, 194, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Klakotskaia, D.; Agca, C.; Richardson, R.A.; Stopa, E.G.; Schachtman, T.R.; Agca, Y. Memory deficiency, cerebral amyloid angiopathy, and amyloid-β plaques in APP+PS1 double transgenic rat model of Alzheimer’s disease. PLoS ONE 2018, 13, e0195469. [Google Scholar] [CrossRef] [PubMed]
- Moody, D.M.; Brown, W.R.; Challa, V.R.; Ghazi-Birry, H.S.; Reboussin, D.M. Cerebral Microvascular Alterations in Aging, Leukoaraiosis, and Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 1997, 826, 103–116. [Google Scholar] [CrossRef]
- Gao, F.; van Gaal, S.; Levy-Cooperman, N.; Ramirez, J.; Scott, C.J.M.; Bilbao, J.; Black, S.E. P2-010: Does variable progression of incidental white matter hyperintensities in AD relate to venous insufficiency? In Proceedings of the Alzheimer’s Association International Conference on Alzheimer’s Disease (Alzheimers Dement), Chicago, IL, USA, 26–31 July 2008; pp. T368–T369. [Google Scholar] [CrossRef]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.J.; et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric Aβ and frank neuronal loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef]
- Dorr, A.; Sahota, B.; Chinta, L.V.; Brown, M.E.; Lai, A.Y.; Ma, K.; Hawkes, C.A.; McLaurin, J.; Stefanovic, B. Amyloid-β-Dependent compromise of microvascular structure and function in a model of Alzheimer’s disease. Brain 2012, 135, 3039–3050. [Google Scholar] [CrossRef]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef]
- Van Dorpe, J.; Smeijers, L.; Dewachter, I.; Nuyens, D.; Spittaels, K.; Van Den Haute, C.; Mercken, M.; Moechars, D.; Laenen, I.; Kuiperi, C.; et al. Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the london mutant of human APP in neurons. Am. J. Pathol. 2000, 157, 1283–1298. [Google Scholar] [CrossRef]
- Giannoni, P.; Arango-Lievano, M.; Neves, I.D.; Rousset, M.C.; Barranger, K.; Rivera, S.; Jeanneteau, F.; Claeysen, S.; Marchi, N. Cerebrovascular pathology during the pro gression of experimental Alzheimer’s disease. Neurobiol. Dis. 2016, 88, 107–117. [Google Scholar] [CrossRef]
- Heo, C.H.; Sarkar, A.R.; Baik, S.H.; Jung, T.S.; Kim, J.J.; Kang, H.; Mook-Jung, I.; Kim, H.M. A quadrupolar two-Photon fluorescent probe for in vivo imaging of amyloid-β plaques. Chem. Sci. 2016, 7, 4600–4606. [Google Scholar] [CrossRef]
- Herzig, M.C.; Winkler, D.T.; Burgermeister, P.; Pfeifer, M.; Kohler, E.; Schmidt, S.D.; Danner, S.; Abramowski, D.; Stürchler-Pierrat, C.; Bürki, K.; et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 2004, 7, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; Nostrand, W.E.V. Early-Onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-Protein precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef] [PubMed]
- El Tannir El Tayara, N.; Delatour, B.; Volk, A.; Dhenain, M. Detection of vascular alterations by in vivo magnetic resonance angiopathy and histology in APP/PS1 mouse model of Alzheimer’s disease. Magn. Reson. Mater. Phy. 2010, 23, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, M.E.; Burgermeister, P.; Phinney, A.L.; Stalder, M.; Tolnay, M.; Wiederhold, K.H.; Abramowski, D.; Stürchler-Pierrat, C.; Sommer, B.; Staufenbiel, M.; et al. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc. Natl. Acad. Sci. USA 1999, 23, 14088–14093. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Beach, T.G.; Sue, L.I.; Scott, S.; Layne, K.J.; Kokjohn, T.A.; Abramowski, D.; Stürchler-Pierrat, C.; Staufenbiel, M.; Weller, R.O.; et al. The evolution of Abeta peptide burden in the APP23 transgenic mice: Implications for Abeta deposition in Alzheimer disease. Mol. Med. 2001, 7, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Suter, O.C.; Sunthorn, T.; Kraftsik, R.; Straubel, J.; Darekar, P.; Khalili, K.; Miklossy, J. Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer’s disease. Stroke 2002, 33, 1986–1992. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Rühlmann, C.; Blinder, P.; Devor, A.; Drew, P.J.; Friedman, B.; Knutsen, P.M.; Lyden, P.D.; Matéo, C.; Mellander, L.; et al. Robust and Fragile Aspects of Cortical Blood Flow in Relation to the Underlying Angioarchitecture. Microcirculation 2015, 22, 204–218. [Google Scholar] [CrossRef]
- Nguyen, J.; Nishimura, N.; Fetcho, R.N.; Iadecola, C.; Schaffer, C.B. Occlusion of cortical ascending venules causes blood flow decreases, reversals in flow direction, and vessel dilation in upstream capillaries. J. Cereb. Blood. Flow. Metab. 2011, 31, 2243–2254. [Google Scholar] [CrossRef]
- Shih, A.Y.; Blinder, P.; Tsai, P.S.; Friedman, B.; Stanley, G.; Lyden, P.D.; Kleinfeld, D. The smallest stroke: Occlusion of one penetrating vessel leads to infarction and a cognitive deficit. Nat. Neurosci. 2013, 16, 55–63. [Google Scholar] [CrossRef]
- Summers, P.M.; Hartmann, D.A.; Hui, E.S.; Nie, X.; Deardorff, R.L.; McKinnon, E.T.; Helpern, J.A.; Jensen, J.H.; Shih, A.Y. Functional deficits induced by cortical microinfarcts. J. Cereb. Blood Flow Metab. 2017, 37, 3599–3614. [Google Scholar] [CrossRef]
- Blinder, P.; Tsai, P.S.; Kaufhold, J.P.; Knutsen, P.M.; Suhl, H.; Kleinfeld, D. The cortical angiome: An interconnected vascular network with noncolumnar patterns of blood flow. Nat. Neurosci. 2013, 16, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.R.; Moody, D.M.; Challa, V.R.; Thore, C.R.; Anstrom, J.A. Venous collagenosis and arteriolar tortuosity in leukoaraiosis. J. Neurol. Sci. 2002, 203–204, 159–163. [Google Scholar] [CrossRef]
- Yoshita, M.; Fletcher, E.; Harvey, D.; Ortega, M.; Martinez, O.; Mungas, D.M.; Reed, B.R.; DeCarli, C.S. Extent and distribution of white matter hyperintensities in normal aging, MCI, and AD. Neurology 2006, 67, 2192–2198. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Wan, J.; Zhang, X.; Tong, L.; Zhao, S.; Sun, J.; Lin, Y.; Shen, C.; Lou, M. Increased Visibility of Deep Medullary Veins in Leukoaraisos: A 3-T MRI Study. Front. Aging Neurosci. 2014, 6, 144. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, J.A.; Keith, J.; Gao, F.; Spence, J.D.; Black, S.E. CADASIL accelerated by acute hypotension: Arterial and venous contribution to leukoaraiosis. Neurology 2017, 88, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Houck, A.L.; Gutierrez, J.; Gao, F.; Igwe, K.C.; Colon, J.M.; Black, S.E.; Brickman, A.M. Increased Diameters of the Internal Cerebral Veins and the Basal Veins of Rosenthal Are Associated with White Matter Hyperintensity Volume. AJNR. Am. J. Neuroradiol. 2019, 40, 1712–1718. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Pizzo, M.E.; Preston, J.E.; Janigro, D.; Thorne, R.G. The role of brain barriers in fluid movement in the CNS: Is there a ‘glymphatic’ system? Acta Neuropathol. 2018, 135, 387–407. [Google Scholar] [CrossRef]
- Touyz, R.M.; Briones, A.M. Reactive oxygen species and vascular biology: Implications in human hypertension. Hypertens. Res. 2011, 34, 5–14. [Google Scholar] [CrossRef]
- Roher, A.E.; Garami, Z.; Alexandrov, A.V.; Kokjohn, T.A.; Esh, C.L.; Kalback, W.M.; Vedders, L.J.; Wilson, J.R.; Sabbagh, M.N.; Beach, T.G. Interaction of cardiovascular disease and neurodegeneration: Transcranial Doppler ultrasonography and Alzheimer′s disease. Neurol. Res. 2006, 28, 672–678. [Google Scholar] [CrossRef]
- Ortner, M.; Hauser, C.; Schmaderer, C.; Muggenthaler, C.; Hapfelmeier, A.; Sorg, C.; Diehl-Schmid, J.; Kurz, A.; Förstl, H.; Ikenberg, B.; et al. Decreased Vascular Pulsatility in Alzheimer’s Disease Dementia Measured by Transcranial Color-Coded Duplex Sonography. Neuropsychiatr. Dis. Treat. 2019, 15, 3487–3499. [Google Scholar] [CrossRef]
- Henry-Feugeas, M.C. Alzheimer’s disease in late life dementia: A minor toxic consequence of devastating cerebrovascular dysfunction. Med. Hypotheses 2008, 70, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Criswell, T.P.; Sharp, M.M.; Dobson, H.; Finucane, C.; Weller, R.O.; Verma, A.; Carare, R.O. The structure of the perivascular compartment in the old canine brain: A case study. Clin. Sci. (Lond.) 2017, 11, 2737–2744. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, M.M.; Van Nostrand, W.E.; Otte-Höller, I.; Wesseling, P.; De Waal, R.M. Amyloid-Beta-Induced degeneration of human brain pericytes is dependent on apolipoprotein E genotype. Ann. N. Y. Acad. Sci. 2000, 903, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, I.B.; Wilhelmus, M.M.; Kox, M.; Veerhuis, R.; de Waal, R.M.; Verbeek, M.M. Apoplipoprotein E protects cultured pericytes and astrocytes from D-Abeta(1-40)-Mediated cell death. Brain Res. 2010, 1315, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.; Brännström, K.; Byman, E.; Moussaud, S.; Nielsen, H.M.; Netherlands Brain Bank; Olofsson, A.; Wennström, M. Amyloid-Beta 1-40 is associated with alterations in NG2+ pericyte population ex vivo and in vitro. Aging Cell 2018, 17, e12728. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-Brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef]
- Bourassa, P.; Tremblay, C.; Schneider, J.A.; Bennett, D.A.; Calon, F. Brain mural cell loss in the parietal cortex in Alzheimer′s disease correlates with cognitive decline and TDP-43 pathology. Neuropathol. Appl. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Rivera-Rivera, L.A.; Turski, P.; Johnson, K.M.; Hoffman, C.; Berman, S.E.; Kilgas, P.; Rowley, H.A.; Carlsson, C.M.; Johnson, S.C.; Wieben, O. 4D flow MRI for intracranial hemodynamics assessment in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2016, 36, 1718–1730. [Google Scholar] [CrossRef]
- Rivera-Rivera, L.A.; Schubert, T.; Turski, P.; Johnson, K.M.; Berman, S.E.; Rowley, H.A.; Carlsson, C.M.; Johnson, S.C.; Wieben, O. Changes in intracranial venous blood flow and pulsatility in Alzheimer’s disease: A 4D flow MRI study. J. Cereb. Blood Flow Metab. 2017, 37, 2149–2158. [Google Scholar] [CrossRef]
- van Veluw, S.J.; Hou, S.S.; Calvo-Rodriguez, M.; Arbel-Ornath, M.; Snyder, A.C.; Frosch, M.P.; Greenberg, S.M.; Bacskai, B.J. Vasomotion as a Driving Force for Paravascular Clearance in the Awake Mouse Brain. Neuron 2020, 105, 549–561. [Google Scholar] [CrossRef]
- Drew, P.J.; Shih, A.Y.; Kleinfeld, D. Fluctuating and sensory-Induced vasodynamics in rodent cortex extend arteriole capacity. Proc. Natl. Acad. Sci. USA 2011, 108, 8473–8478. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Knutsen, P.M.; Tsai, P.S.; Shih, A.Y.; Kleinfeld, D. Entrainment of Arteriole Vasomotor Fluctuations by Neural Activity Is a Basis of Blood-Oxygenation-Level-Dependent "Resting-State" Connectivity. Neuron 2017, 96, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Abrahao, A.; Heyn, C.C.; Bethune, A.J.; Huang, Y.; Pople, C.B.; Aubert, I.; Hamani, C.; Zinman, L.; Hynynen, K.; et al. Glymphatics Visualization after Focused Ultrasound-Induced Blood-Brain Barrier Opening in Humans. Ann. Neurol. 2019, 86, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Berezuk, C.; McNeely, A.A.; Gao, F.; McLaurin, J.; Black, S.E. Imaging the Perivascular Space as a Potential Biomarker of Neurovascular and Neurodegenerative Diseases. Cell Mol. Neurobiol. 2016, 36, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Mestre, H.; Kostrikov, S.; Mehta, R.I.; Nedergaard, M. Perivascular spaces, glymphatic dysfunction, and small vessel disease. Clin. Sci. (Lond.) 2017, 131, 2257–2274. [Google Scholar] [CrossRef] [PubMed]
- Boespflug, E.L.; Simon, M.J.; Leonard, E.; Grafe, M.; Woltjer, R.; Silbert, L.C.; Kaye, J.A.; Iliff, J.J. Targeted Assessment of Enlargement of the Perivascular Space in Alzheimer’s Disease and Vascular Dementia Subtypes Implicates Astroglial Involvement Specific to Alzheimer’s Disease. J. Alzheimers Dis. 2018, 66, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Berezuk, C.; McNeely, A.A.; Scott, C.J.; Gao, F.; Black, S.E. Visible Virchow-Robin spaces on magnetic resonance imaging of Alzheimer’s disease patients and normal elderly from the Sunnybrook Dementia Study. J. Alzheimers Dis. 2015, 43, 415–424. [Google Scholar] [CrossRef]
- Banerjee, G.; Kim, H.J.; Fox, Z.; Jäger, H.R.; Wilson, D.; Charidimou, A.; Na, H.K.; Na, D.L.; Seo, S.W.; Werring, D.J. MRI-Visible perivascular space location is associated with Alzheimer’s disease independently of amyloid burden. Brain 2017, 140, 1107–1116. [Google Scholar] [CrossRef]
- Charidimou, A.; Hong, Y.T.; Jäger, H.R.; Fox, Z.; Aigbirhio, F.I.; Fryer, T.D.; Menon, D.K.; Warburton, E.A.; Werring, D.J.; Baron, J.C. White Matter Perivascular Spaces on Magnetic Resonance Imaging: Marker of Cerebrovascular Burden? Stroke 2015, 46, 1707–1709. [Google Scholar] [CrossRef]
- van Veluw, S.J.; Biessels, G.J.; Bouvy, W.H.; Spliet, W.G.; Zwanenburg, J.J.; Luijten, P.R.; Macklin, E.A.; Rozemuller, A.J.; Gurol, M.E.; Greenberg, S.M.; et al. Cerebral amyloid angiopathy severity is linked to dilation of juxtacortical perivascular spaces. J. Cereb. Blood Flow Metab. 2016, 36, 576–580. [Google Scholar] [CrossRef]
- Nicoll, J.A.; Wilkinson, D.; Holmes, C.; Steart, P.; Markham, H.; Weller, R.O. Neuropathology of human Alzheimer disease after immunization with amyloid-Beta peptide: A case report. Nat. Med. 2003, 9, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, J.A.R.; Buckland, G.R.; Harrison, C.H.; Page, A.; Harris, S.; Love, S.; Neal, J.W.; Holmes, C.; Boche, D. Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer’s disease. Brain 2019, 142, 2113–2126. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Zotova, E.; Weller, R.O.; Love, S.; Neal, J.W.; Pickering, R.M.; Wilkinson, D.; Holmes, C.; Nicoll, J.A. Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain 2008, 131, 3299–3310. [Google Scholar] [CrossRef] [PubMed]
- Gravina, S.A.; Ho, L.; Eckman, C.B.; Long, K.E.; Otvos, L., Jr.; Younkin, L.H.; Suzuki, N.; Younkin, S.G. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42 (43). J. Biol. Chem. 1995, 270, 7013–7016. [Google Scholar] [CrossRef]
- Rinne, J.O.; Brooks, D.J.; Rossor, M.N.; Fox, N.C.; Bullock, R.; Klunk, W.E.; Mathis, C.A.; Blennow, K.; Barakos, J.; Okello, A.A.; et al. 11C-PiB PET assessment of change in fibrillar amyloid-Beta load in patients with Alzheimer′s disease treated with bapineuzumab: A phase 2, double-Blind, placebo-Controlled, ascending-Dose study. Lancet Neurol. 2010, 9, 363–372. [Google Scholar] [CrossRef]
- Liu, E.; Schmidt, M.E.; Margolin, R.; Sperling, R.; Koeppe, R.; Mason, N.S.; Klunk, W.E.; Mathis, C.A.; Salloway, S.; Fox, N.C.; et al. Bapineuzumab 301 and 302 Clinical Trial Investigators. Amyloid-β 11C-PiB-PET imaging results from 2 randomized bapineuzumab phase 3 AD trials. Neurology 2015, 85, 692–700. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Bapineuzumab 301 and 302 Clinical Trial Investigators. Two phase 3 trials of bapineuzumab in mild-To-Moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef]
- Chantran, Y.; Capron, J.; Alamowitch, S.; Aucouturier, P. Anti-Aβ Antibodies and Cerebral Amyloid Angiopathy Complications. Front. Immunol. 2019, 10, 1534. [Google Scholar] [CrossRef]
- Bales, K.R.; O′Neill, S.M.; Pozdnyakov, N.; Pan, F.; Caouette, D.; Pi, Y.; Wood, K.M.; Volfson, D.; Cirrito, J.R.; Han, B.H.; et al. Passive immunotherapy targeting amyloid-β reduces cerebral amyloid angiopathy and improves vascular reactivity. Brain 2016, 139, 563–577. [Google Scholar] [CrossRef]
- Leurent, C.; Goodman, J.A.; Zhang, Y.; He, P.; Polimeni, J.R.; Gurol, M.E.; Lindsay, M.; Frattura, L.; Sohur, U.S.; Viswanathan, A.; et al. Immunotherapy with ponezumab for probable cerebral amyloid angiopathy. Ann. Clin. Transl. Neurol. 2019, 6, 795–806. [Google Scholar] [CrossRef]
- Landen, J.W.; Andreasen, N.; Cronenberger, C.L.; Schwartz, P.F.; Börjesson-Hanson, A.; Östlund, H.; Sattler, C.A.; Binneman, B.; Bednar, M.M. Ponezumab in mild-To-Moderate Alzheimer’s disease: Randomized phase II PET-PIB study. Alzheimers Dement. (N. Y). 2017, 3, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Osswald, H.L. BACE1 (β-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6765–6813. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; He, P.; Yao, H.; Dong, Q.; Li, R.; Shen, Y. Occludin deficiency with BACE1 elevation in cerebral amyloid angiopathy. Neurology 2014, 82, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Neumann, U.; Rueeger, H.; Machauer, R.; Veenstra, S.J.; Lueoend, R.M.; Tintelnot-Blomley, M.; Laue, G.; Beltz, K.; Vogg, B.; Schmid, P.; et al. A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-β and neuroinflammation in APP transgenic mice. Mol. Neurodegener. 2015, 10, 44. [Google Scholar] [CrossRef]
- Meier, S.R.; Syvänen, S.; Hultqvist, G.; Fang, X.T.; Roshanbin, S.; Lannfelt, L.; Neumann, U.; Sehlin, D. Antibody-Based In Vivo PET Imaging Detects Amyloid-β Reduction in Alzheimer Transgenic Mice After BACE-1 Inhibition. J. Nucl. Med. 2018, 59, 1885–1891. [Google Scholar] [CrossRef]
- Schelle, J.; Wegenast-Braun, B.M.; Fritschi, S.K.; Kaeser, S.A.; Jährling, N.; Eicke, D.; Skodras, A.; Beschorner, N.; Obermueller, U.; Häsler, L.M.; et al. Early Aβ reduction prevents progression of cerebral amyloid angiopathy. Ann. Neurol. 2019, 86, 561–571. [Google Scholar] [CrossRef]
- Herzig, M.C.; Paganetti, P.; Staufenbiel, M.; Jucker, M. BACE1 and mutated presenilin-1 differently modulate Abeta40 and Abeta42 levels and cerebral amyloidosis in APPDutch transgenic mice. Neurodegener. Dis. 2007, 4, 127–135. [Google Scholar] [CrossRef]
- Beckmann, N.; Doelemeyer, A.; Zurbruegg, S.; Bigot, K.; Theil, D.; Frieauff, W.; Kolly, C.; Moulin, P.; Neddermann, D.; Kreutzer, R.; et al. Longitudinal noninvasive magnetic resonance imaging of brain microhemorrhages in BACE inhibitor-treated APP transgenic mice. Neurobiol. Aging 2016, 45, 50–60. [Google Scholar] [CrossRef]
- Imbimbo, B.P.; Watling, M. Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Expert. Opin. Investig. Drugs 2019, 28, 967–975. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morrone, C.D.; Bishay, J.; McLaurin, J. Potential Role of Venular Amyloid in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2020, 21, 1985. https://doi.org/10.3390/ijms21061985
Morrone CD, Bishay J, McLaurin J. Potential Role of Venular Amyloid in Alzheimer’s Disease Pathogenesis. International Journal of Molecular Sciences. 2020; 21(6):1985. https://doi.org/10.3390/ijms21061985
Chicago/Turabian StyleMorrone, Christopher D., Jossana Bishay, and JoAnne McLaurin. 2020. "Potential Role of Venular Amyloid in Alzheimer’s Disease Pathogenesis" International Journal of Molecular Sciences 21, no. 6: 1985. https://doi.org/10.3390/ijms21061985
APA StyleMorrone, C. D., Bishay, J., & McLaurin, J. (2020). Potential Role of Venular Amyloid in Alzheimer’s Disease Pathogenesis. International Journal of Molecular Sciences, 21(6), 1985. https://doi.org/10.3390/ijms21061985