Altered Growth and Envelope Properties of Polylysogens Containing Bacteriophage Lambda N−cI− Prophages

Abstract
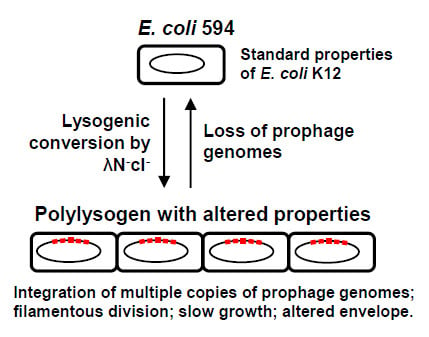
1. Introduction
2. Results
2.1. Envelope-associated Properties of the Polylysogen
2.1.1. Properties Associated with the Cytoplasmic Membrane
2.1.2. Properties Associated with the Peptidoglycan and the Outer Membrane
2.1.3. Envelope Composition
2.2. Characteristics of the Polylysogen Filaments
2.2.1. Filament Length
2.2.2. Irregular Placement and Uniqueness of Septa in the Polylysogen Filament
2.2.3. Division Status in Different Segments of the Polylysogen Filament
2.3. Deconversion of the Polylysogen
2.4. Role of Lambda Genes in Conversion
3. Summary and Discussion
4. Materials and Methods
4.1. Chemicals, Bacteria, and Phages
4.2. Methods
4.2.1. General and Common Procedures
4.2.2. Visualization and Microscopy of Bacteria
4.2.3. Isolation and Analysis of Bacterial Envelope and Its Constituents
4.2.4. Quantification of Prophage Copies in Lysogens
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| cAMP | Cyclic AMP |
| CFU | Colony-forming unit |
| DMSO | Dimethyl sulfoxide |
| DNA | Deoxyribonucleic acid |
| DOC | Deoxycholate |
| E. coli | Escherichia coli |
| EDTA | Ethylenediaminetetraacetic acid |
| E.O.P. | Efficiency of plating |
| FAD | Flavin adenine dinucleotide |
| Glc | Glucose |
| GlcN | N-acetyl glucosamine |
| Hep | Heptose |
| HPr | Histidine-containing protein (Phosphocarrier protein) |
| KDO | 2-Keto-3-deoxy-octonate |
| LDH | Lactate dehydrogenase |
| LPS | Lipopolysaccharide |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) |
| NADH | Nicotinamide adenine dinucleotide |
| OL | Operator, left |
| OR | Operator, right |
| PAGE | Polyacrylamide gel electrophoresis |
| PEP | Phosphoenolpyruvate |
| PMS | Phenazine methosulfate |
| PL | Promoter, left |
| pNPP | Para-nitrophenylphosphate |
| PR | Promoter, right |
| PTS | Phosphotransferase enzyme system |
| Rha | Rhamnose |
| RNA | Ribonucleic acid |
| SDS | Sodium dodecyl sulfate |
| TCA | Trichloroacetic acid |
| TEMED | Tetramethylethylenediamine |
References
- Casjens, S.R.; Hendrix, R.W. Bacteriophage lambda: Early pioneer and still relevant. Virology 2015, 479–480, 310–330. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W.; Roberts, J.; Stahl, F.; Weisberg, R.E. Lambda II; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1983. [Google Scholar]
- Gottesman, M.E.; Weisberg, R.A. Little lambda, who made thee? Microbiol. Mol. Biol. Rev. 2004, 68, 796–813. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, A.B.; Kobiler, O.; Stavans, J.; Court, D.L.; Adhya, S. Switches in bacteriophage lambda development. Annu. Rev. Genet. 2005, 39, 409–429. [Google Scholar] [CrossRef] [PubMed]
- Golding, I. Infection by bacteriophage lambda: An evolving paradigm for cellular individuality. Curr. Opin. Microbiol. 2018, 43, 9–13. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, H.; Gu, Z.; Roberts, J.W. High-resolution view of bacteriophage lambda gene expression by ribosome profiling. Proc. Natl. Acad. Sci. USA 2013, 110, 11928–11933. [Google Scholar] [CrossRef] [PubMed]
- Kobiler, O.; Rokney, A.; Friedman, N.; Court, D.L.; Stavans, J.; Oppenheim, A.B. Quantitative kinetic analysis of the bacteriophage lambda genetic network. Proc. Natl. Acad. Sci. USA 2005, 102, 4470–4475. [Google Scholar] [CrossRef]
- Salstrom, J.S.; Szybalski, W. Coliphage lambdanutL-: A unique class of mutants defective in the site of gene N product utilization for antitermination of leftward transcription. J. Mol. Biol. 1978, 124, 195–221. [Google Scholar] [CrossRef]
- Benzer, S. Fine structure of a genetic region in bacteriophage. Proc. Natl. Acad. Sci. USA 1955, 41, 344–354. [Google Scholar] [CrossRef]
- Shinedling, S.; Parma, D.; Gold, L. Wild-type bacteriophage T4 is restricted by the lambda rex genes. J. Virol. 1987, 61, 3790–3794. [Google Scholar] [CrossRef]
- Lieb, M. Properties of polylysogens containing depressed lambda N- prophages. II. The number of prophages affects bacterial viability. Virology 1972, 49, 818–820. [Google Scholar] [CrossRef]
- Mandal, N.C.; Crochet, M.; Lieb, M. Properties of polylysogens containing derepressed lambda N- prophages. A large number of integrated lambda prophages. Virology 1974, 57, 85–92. [Google Scholar] [CrossRef]
- Barik, S. Studies on the polylysogens of bacteriophage lambda. Ph.D. Thesis, Calcutta University, Calcutta, India, 1981. [Google Scholar]
- Chattopadhyay, D.J.; Mandal, N.C. Studies on polylysogens containing lambda N- cI- prophages. I. Control of synthesis and maintenance of a large number of integrated lambda genomes. Virology 1982, 30(118), 439–447. [Google Scholar] [CrossRef]
- Chattopadhyay, D.J.; Nag, D.K.; Mandal, N.C. Studies on polylysogens containing lambda N-cI- prophages. II. Role of high multiplicities in lysogen formation by lambda N-cI- phage. Virology 1983, 128, 265–270. [Google Scholar] [CrossRef]
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
- Postma, P.W.; Lengeler, J.W.; Jacobson, G.R. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 1993, 57, 543–594. [Google Scholar] [CrossRef] [PubMed]
- Kundig, W.; Ghosh, S.; Roseman, S. Phosphate bound to histidine in a protein as an intermediate in a novel phospho-transferase system. Proc. Natl. Acad. Sci. USA 1964, 52, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, T.J. Structures of gram-negative cell walls and their derived membrane vesicles. J. Bacteriol. 1999, 181, 4725–4733. [Google Scholar] [CrossRef]
- Sobell, H.M. Actinomycin and DNA transcription. Proc. Natl. Acad. Sci. USA 1985, 82, 5328–5331. [Google Scholar] [CrossRef]
- Wilkinson, S.G. Composition and structure of bacterial lipopolysaccharides. In Surface Carbohydrates of the Prokaryotic Cell; Sutherland, I.W., Ed.; Academic Press, Inc.: New York, NY, USA, 1977; pp. 97–175. [Google Scholar]
- Jann, B.; Reske, K.; Jann, K. Heterogeneity of lipopolysaccharides. Analysis of polysaccharide chain lengths by sodium dodecylsulfate-polyacrylamide gel electrophoresis. Eur. J. Biochem. 1975, 60, 239–246. [Google Scholar] [CrossRef]
- DiRienzo, J.M.; Nakamura, K.; Inouye, M. The outer membrane proteins of Gram-negative bacteria: Biosynthesis, assembly, and functions. Annu. Rev. Biochem. 1978, 47, 481–532. [Google Scholar] [CrossRef]
- Schnaitman, C.A. Outer membrane proteins of Escherichia coli. IV. Differences in outer membrane proteins due to strain and cultural differences. J. Bacteriol. 1974, 118, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Slater, M.; Schaechter, M. Control of cell division in bacteria. Bacteriol. Rev. 1974, 38, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.J.; Munson, R. Separation of the inner (cytoplasmic) and outer membranes of Gram-negative bacteria. Methods Enzymol. 1974, 31, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Inouye, M. Reversal by sodium chloride of envelope protein changes related to DNA replication and cell division of Escherichia coli. J. Mol. Biol. 1972, 63, 597–600. [Google Scholar] [CrossRef]
- Ingram, L.O.; Fisher, W.D. Stimulation of cell division by membrane-active agents. Biochem. Biophys. Res. Commun. 1973, 50, 200–210. [Google Scholar] [CrossRef]
- Grula, E.A.; Grula, M.M. Cell division in a species of Erwinia III. Reversal of inhibition of cell division caused by D-amino acids, penicillin, and ultraviolet light. J. Bacteriol. 1962, 83, 981–998. [Google Scholar] [CrossRef]
- Onitsuka, M.O.; Rikihisa, Y.; Maruyama, H.B. Biochemical and topographical studies on Escherichia coli cell surface. IV. Giant spheroplast formation from a filamentous cell. J. Bacteriol. 1979, 138, 567–574. [Google Scholar] [CrossRef]
- Maniatis, T.; Fritsch, E.F.; Sambrook, J. Molecular Cloning: A Laboratory Manual, 1st ed.; Cold Spring Harbor Laboratory: New York, NY, USA, 1982; pp. 1–545. [Google Scholar]
- Cahill, J.; Young, R. Phage lysis: Multiple genes for multiple barriers. Adv. Virus Res. 2019, 103, 33–70. [Google Scholar] [CrossRef]
- Barik, S.; Mandal, N.C. Role of S gene product of bacteriophage lambda in host cell lysis. J. Biosci. 1982, 4, 361–368. [Google Scholar] [CrossRef]
- Canchaya, C.; Fournous, G.; Brüssow, H. The impact of prophages on bacterial chromosomes. Mol. Microbiol. 2004, 53, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Gödeke, J.; Paul, K.; Lassak, J.; Thormann, K.M. Phage-induced lysis enhances biofilm formation in Shewanella oneidensis MR-1. ISME J. 2011, 5, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, L.; Sigal, N.; Borovok, I.; Nir-Paz, R.; Herskovits, A.A. Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell 2012, 150, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kim, Y.; Ma, Q.; Hong, S.H.; Pokusaeva, K.; Sturino, J.M.; Wood, T.K. Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 2010, 1, 147. [Google Scholar] [CrossRef]
- Bossi, L.; Fuentes, J.A.; Mora, G.; Figueroa-Bossi, N. Prophage contribution to bacterial population dynamics. J. Bacteriol. 2003, 185, 6467–6471. [Google Scholar] [CrossRef]
- Anderson, J.J.; Wilson, J.M.; Oxender, D.L. Defective transport and other phenotypes of a periplasmic “leaky” mutant of Escherichia coli K-12. J. Bacteriol. 1979, 140, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Friedman, D.I. Phage regulatory circuits and virulence gene expression. Curr. Opin. Microbiol. 2005, 8, 459–465. [Google Scholar] [CrossRef]
- Yates, S.P.; Jørgensen, R.; Andersen, G.R.; Merrill, A.R. Stealth and mimicry by deadly bacterial toxins. Trends Biochem. Sci. 2006, 31, 123–133. [Google Scholar] [CrossRef] [PubMed]
- McLeod, S.M.; Kimsey, H.H.; Davis, B.M.; Waldor, M.K. CTXphi and Vibrio cholerae: Exploring a newly recognized type of phage-host cell relationship. Mol. Microbiol. 2005, 57, 347–356. [Google Scholar] [CrossRef]
- Steinberg, K.M.; Levin, B.R. Grazing protozoa and the evolution of the Escherichia coli O157:H7 Shiga toxin-encoding prophage. Proc. Biol. Sci. 2007, 274, 1921–1929. [Google Scholar] [CrossRef]
- Henning, U.; Jann, K. Two-component nature of bacteriophage T4 receptor activity in Escherichia coli K-12. J. Bacteriol. 1979, 137, 664–666. [Google Scholar] [CrossRef]
- Mutoh, N.; Furukawa, H.; Mizushima, S. Role of lipopolysaccharide and outer membrane protein of Escherichia coli K-12 in the receptor activity for bacteriophage T4. J. Bacteriol. 1978, 136, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Greer, H. The kil gene of bacteriophage lambda. Virology 1975, 66, 589–604. [Google Scholar] [CrossRef]
- Haeusser, D.P.; Hoashi, M.; Weaver, A.; Brown, N.; Pan, J.; Sawitzke, J.A.; Thomason, L.C.; Court, D.L.; Margolin, W. The Kil peptide of bacteriophage λ blocks Escherichia coli cytokinesis via ZipA-dependent inhibition of FtsZ assembly. PLoS Genet. 2014, 10, e1004217. [Google Scholar] [CrossRef] [PubMed]
- Court, D.; Gottesman, M.; Gallo, M. Bacteriophage lambda hin function. I. Pleiotropic alteration in host physiology. J. Mol. Biol. 1980, 138, 715–729. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Galanos, C.; Lüderitz, O.; Westphal, O. A new method for the extraction of R lipopolysaccharides. Eur. J. Biochem. 1969, 9, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.; Gilles, K.; Hamilton, J.K.; Rebers, P.A.; Smith, F. A colorimetric method for the determination of sugars. Nature 1951, 168, 167. [Google Scholar] [CrossRef]
- Dische, Z.; Shettles, L.B. A new spectrophotometric test for the detection of methylpentose. J. Biol. Chem. 1951, 192, 579–582. [Google Scholar]
- Wright, B.G.; Rebers, P.A. Procedure for determining heptose and hexose in lipopolysaccharides. Modification of the cysteine-sulfuric acid method. Anal. Biochem. 1972, 49, 307–319. [Google Scholar] [CrossRef]
- Washko, M.E.; Rice, E.W. Determination of glucose by an improved enzymatic procedure. Clin. Chem. 1961, 7, 542–545. [Google Scholar] [CrossRef]
- Elbein, A.D.; Heath, E.C. The biosynthesis of cell wall lipopolysaccharide in Escherichia coli. I. The biochemical properties of a uridine diphosphate galactose 4-epimeraseless mutant. J. Biol. Chem. 1965, 240, 1919–1925. [Google Scholar] [PubMed]
- Ellwood, D.C. The distribution of 2-keto-3-deoxy-octonic acid in bacterial walls. J. Gen. Microbiol. 1970, 60, 373–380. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Garen, A.; Levinthal, C. A fine-structure genetic and chemical study of the enzyme alkaline phosphatase of E. coli. I. Purification and characterization of alkaline phosphatase. Biochim. Biophys. Acta. 1960, 38, 470–483. [Google Scholar] [CrossRef]
- Neu, H.C.; Heppel, L.A. The release of enzymes from Escherichia coli by osmotic shock and during the formation of spheroplasts. J. Biol. Chem. 1965, 240, 3685–3692. [Google Scholar]
- Chung, A.E.; Langdon, R.G. Human erythrocyte glucose 6-phosphate dehydrogenase. I. Isolation and properties of the enzyme. J. Biol. Chem. 1963, 238, 2309–2316. [Google Scholar]
- Kohn, L.D.; Kaback, H.R. Mechanisms of active transport in isolated bacterial membrane vesicles. XV. Purification and properties of the membrane-bound D-lactate dehydrogenase from Escherichia coli. J. Biol. Chem. 1973, 248, 7012–7017. [Google Scholar]
- Gutman, M.; Schejter, A.; Avi-Dor, Y. The preparation and properties of the membranal DPNH dehydrogenase from Escherichia coli. Biochim. Biophys. Acta. 1968, 162, 506–517. [Google Scholar] [CrossRef]
- Datta, A.; Franklin, R.M. Structure and synthesis of a lipid-containing bacteriophage. II. Alterations in cytoplasmic and membrane-bound enzymes during replication of bacteriophage PM2. Virology 1969, 39, 408–418. [Google Scholar] [CrossRef]
- Ghosh, S.; Ghosh, D. Probable role of a membrane-bound phosphoenolpyruvate-hexose phosphotransferase system of Escherichia coli in the permeation of sugars. Indian J. Biochem. 1968, 5, 49–52. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria | Activity / Property Assayed 1 | |||
|---|---|---|---|---|
| PTS activity a Total (in frozen-thawed cells) | Amount exposed in saline-washed cells | % of total exposed in saline-washed cells | ||
| Nonlysogen | 43.3 ± 3 | 0.53 ± 0.02 | 1.20 | |
| Polylysogen | 16.6 ± 2 | 9.70 ± 0.8 | 60.5 | |
| Uptake of: | ||||
| 14C-Glucose | 14C-Galactose | 3H-Uridine | ||
| Nonlysogen | 3.60 × 104 | 3.6 × 103 | 1.93 × 104 | |
| Polylysogen | 2.15 × 104 (60%) | 2.3 × 103 (63%) | 1.00 × 104 (52%) | |
| Enzyme activity in membrane fraction | ||||
| NADH oxidase b | FAD-linked LDH b | Dye reductase b | ATPase c | |
| Nonlysogen | 26.5 ± 2 | 8.00 ± 0.2 | 14.5 ± 1 | 28.0 ± 1 |
| Polylysogen | 19.0 (60.1%) | 4.08 (57%) | 8.5 (58%) | 14.00 (50%) |
| Enzyme | % Total Activity Released by EDTA Treatment 1 | |
|---|---|---|
| Nonlysogen | Polylysogen | |
| (a) Periplasmic: | ||
| Alkaline phosphatase | 1.8 | 32.0 |
| 5′-nucleotidase | 8.0 | 47.0 |
| Acid phosphatase | ND | 30.0 |
| (b) Cytoplasmic: | ||
| G6PD | ND | ND |
| Inorganic pyrophosphatase | 9.0 | 10.0 |
| Phage | Efficiency of Plating, Relative to E. coli 594 2 | |
|---|---|---|
| Monolysogen 594 (λ+) | Polylysogen 594 (λN-cI-) | |
| T4D | 0.8 | 0.2 |
| T3 | 0.9 | 1.0 |
| T7 | 1.0 | 1.1 |
| Bacteria | Total LPS (% of Dry Envelope, w/v) | Carbohydrate (% of dry LPS, w/w) | |||||
|---|---|---|---|---|---|---|---|
| Monosaccharides | |||||||
| Total | Rha | GlcN | Glc | Hep | KDO | ||
| Nonlysogen | 5.0 (100) | 31.3 | 0.92 | 6.5 | 11.1 | 18.6 | 4.22 |
| Polylysogen | 3.6 (72) | 32.8 | 0.97 | 6.0 | 11.9 | 18.45 | 4.58 |
| Major Peak# | ~Mr/1000 | Area of the Peak * | Ratio (b/a) | |
|---|---|---|---|---|
| Nonlysogen (a) | Polylysogen (b) | |||
| 7 | 57.0 | 210 | 235 | 1.10 |
| 9 | 39.0 | 141 | 82 | 0.58 |
| 11 | 30.0 | 214 | 360 | 1.60 |
| 14 | 18.0 | 60 | 123 | 2.05 |
| 15 | 14.5 | 435 | 99 | 0.22 |
| 17 | 8.0 | 93 | 108 | 1.16 |
| Bacteria | Colony Size | Cell Length | % AP Leakage | E.O.P. of T4 | λDNA Copy |
|---|---|---|---|---|---|
| Nonlysogen a | Large | Small rod (1×) | 2.4 | 1.0 | 0 |
| Deconverted | Large | Small rod (1×) | 3.2 | 0.9 | 0 |
| Semi-deconverted | Medium | Short filament (3–4×) | 12.0 | 0.48 | 16 ± 1 |
| Polylysogen | Small | Long filament (14–25×) | 32.0 | 0.2 | 22 ± 2 |
| Lysogen | Colony Size | Cell Morphology | PTS Activity (% of 594) | % AP Leakage | E.O.P. of T4 (Fraction of 594) | λDNA Copy |
|---|---|---|---|---|---|---|
| 594(λN-cI-) | Small a | Filament, 15× | 39.0 | 32 ± 4 | 0.20 | 24 ± 4 |
| 594(λN-cI-R-) | Small | Filament, 15× | 34.6 | 30 ± 3 | 0.12 | 22 ± 3 |
| 594(λN-cI-S-) | Small | Filament, 15× | 34.4 | 29 ± 5 | 0.10 | 24 ± 4 |
| 594(λN-cI-R-S-) | Small | Filament, 15× | 24.1 | 32 ± 2 | 0.13 | 23 ± 4 |
| 594(λN-cI-O-) | Large | Short filament, 2× | ND | 4.9 ± 1 | 0.65 | 8 ± 1 |
| 594(λN-cI-P-) | Large | Short filament, 2× | ND | 7.8 ± 0.4 | 0.60 | 8 ± 1 |
| 594 | Large | Small rod, 1× | 100 | 1.8 ± 0.1 | 1.00 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barik, S.; Mandal, N.C. Altered Growth and Envelope Properties of Polylysogens Containing Bacteriophage Lambda N−cI− Prophages. Int. J. Mol. Sci. 2020, 21, 1667. https://doi.org/10.3390/ijms21051667
Barik S, Mandal NC. Altered Growth and Envelope Properties of Polylysogens Containing Bacteriophage Lambda N−cI− Prophages. International Journal of Molecular Sciences. 2020; 21(5):1667. https://doi.org/10.3390/ijms21051667
Chicago/Turabian StyleBarik, Sailen, and Nitai C. Mandal. 2020. "Altered Growth and Envelope Properties of Polylysogens Containing Bacteriophage Lambda N−cI− Prophages" International Journal of Molecular Sciences 21, no. 5: 1667. https://doi.org/10.3390/ijms21051667
APA StyleBarik, S., & Mandal, N. C. (2020). Altered Growth and Envelope Properties of Polylysogens Containing Bacteriophage Lambda N−cI− Prophages. International Journal of Molecular Sciences, 21(5), 1667. https://doi.org/10.3390/ijms21051667