Does Complement-Mediated Hemostatic Disturbance Occur in Traumatic Brain Injury? A Literature Review and Observational Study Protocol


Abstract
1. Introduction
2. Complement and Coagulation
2.1. Hemostatic Disturbance in TBI
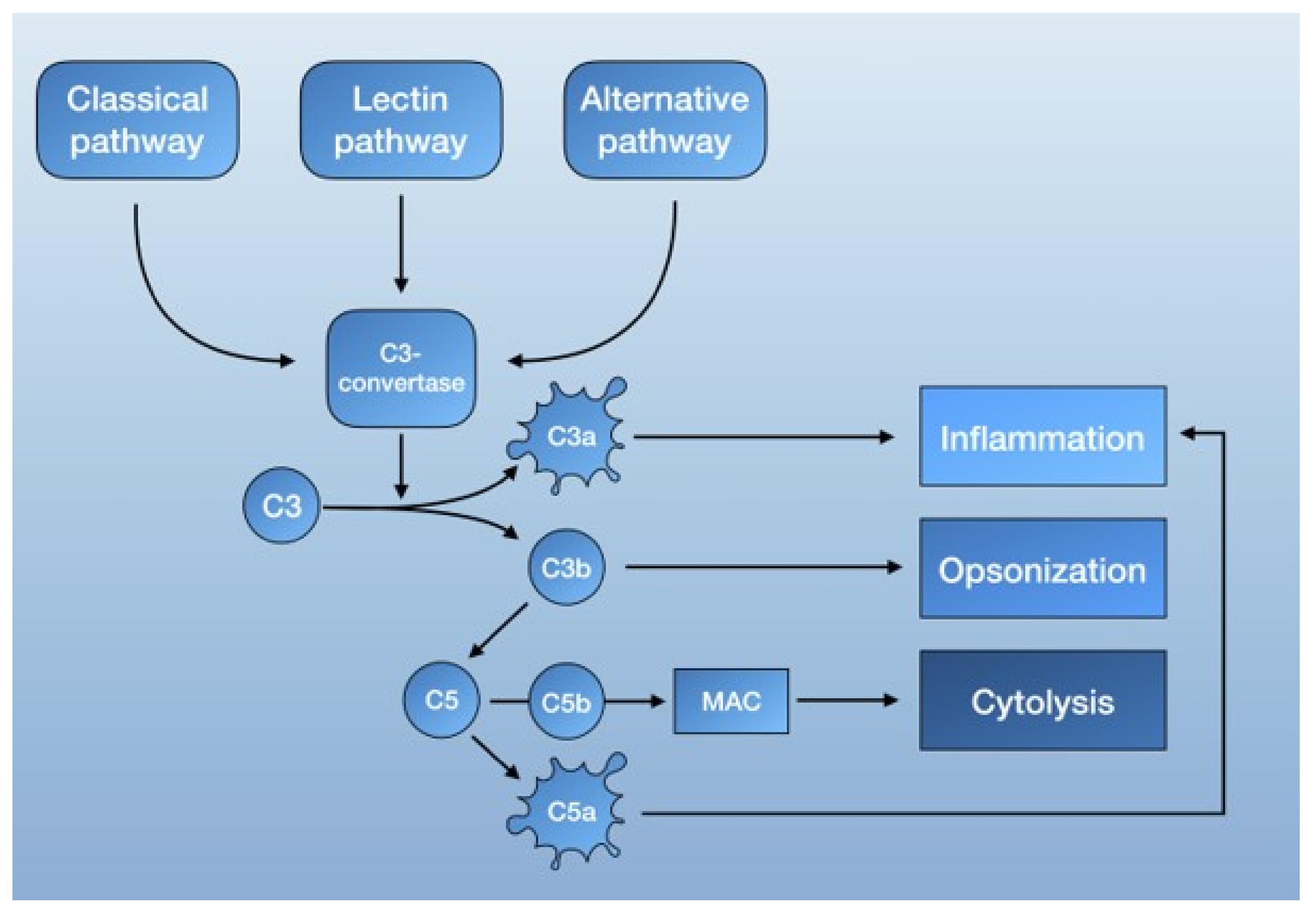
2.2. The Complement System
2.3. The Complement System in the Healthy and Injured Brain
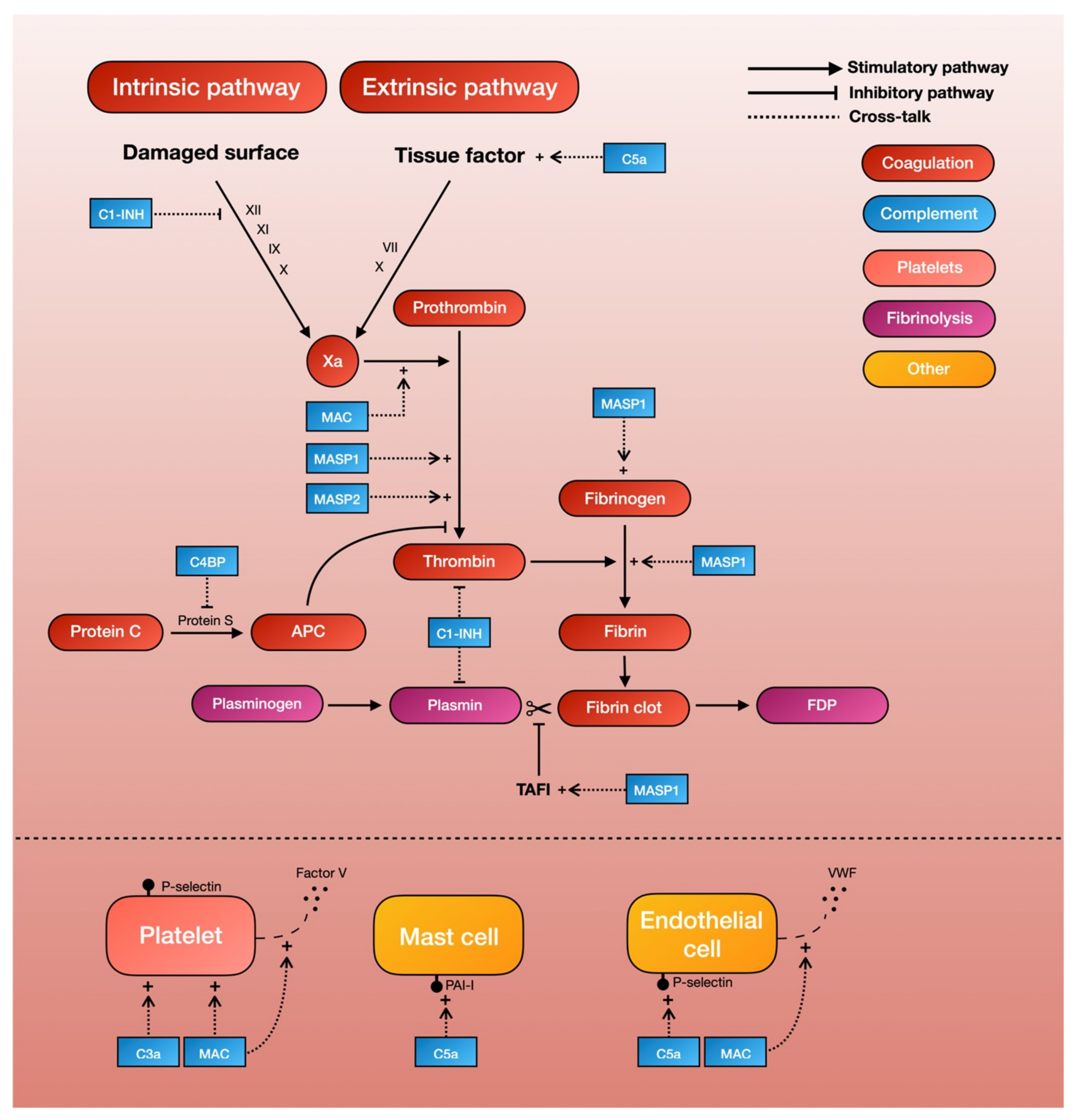
3. Crosstalk between Complement and Coagulation
3.1. Complement and the Coagulation Cascade
3.2. Complement and Platelets
3.3. Complement and Fibrinolysis
3.4. Complement and Endothelial Activation
3.5. Clinical Examples of Complement-Mediated Hemostatic Disturbance
4. Discussion
5. Observational Cohort Study: Outline
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIS | Abbreviated Injury Scale |
| aHUS BBB C1-INH | Atypical hemolytic uremic syndrome Blood-brain-barrier C1-inhibitor |
| C4BP | C4b-binding protein |
| CS-A | Chondroitin sulphate |
| CNS DAF | Central nervous system Decay-accelerating factor |
| DAI | Diffuse axonal injury |
| DIC | Disseminated intravascular coagulation |
| MAC | Membrane attack complex |
| MASP | Mannan-binding lectin serine protease |
| PAI-1 PNH | Plasminogen activator inhibitor-1 Paroxysmal nocturnal hemoglobinuria |
| SLE | Systemic lupus erythematosus |
| TAFI TAT | Thrombin activatable fibrinolysis inhibitor Thrombin-antithrombin complex |
| TBI | Traumatic brain injury |
References
- Stocchetti, N.; Zanier, E.R. Chronic impact of traumatic brain injury on outcome and quality of life: A narrative review. Crit. Care 2016, 20, 148. [Google Scholar] [CrossRef]
- Maegele, M.; Schöchl, H.; Menovsky, T.; Maréchal, H.; Marklund, N.; Buki, A.; Stanworth, S. Coagulopathy and haemorrhagic progression in traumatic brain injury: advances in mechanisms, diagnosis, and management. Lancet Neurol. 2017, 16, 630–677. [Google Scholar] [CrossRef]
- Bellander, B.-M.; Singhrao, S.K.; Ohlsson, M.; Mattsson, P.; Svensson, M. Complement Activation in the Human Brain after Traumatic Head Injury. J. Neurotrauma 2001, 18, 1295–1311. [Google Scholar] [CrossRef] [PubMed]
- Bellander, B.-M.; Bendel, O.; Euler, G. Von; Ohlsson, M.; Svensson, M. Activation of Microglial Cells and Complement following Traumatic Injury in Rat Entorhinal-Hippocampal Slice Cultures. J. Neurotrauma 2004, 21, 605–615. [Google Scholar] [CrossRef]
- Stahel, P.F.; Trentz, O.; Kossmann, T.; Morganti-Kossmann, M.C.; Perez, D.; Redaelli, C.; Gloor, B. Intrathecal levels of complement-derived soluble membrane attack complex (sc5b-9) correlate with blood-brain barrier dysfunction in patients with traumatic brain injury. J. Neurotrauma 2001, 18, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Ritis, K.; Doumas, M.; Mastellos, D.; Micheli, A.; Giaglis, S.; Magotti, P.; Rafail, S.; Kartalis, G.; Sideras, P.; Lambris, J.D. A Novel C5a Receptor-Tissue Factor Cross-Talk in Neutrophils Links Innate Immunity to Coagulation Pathways. J. Immunol. 2006, 177, 4794–4802. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Nagasawa, K.; Horiuchi, T.; Tsuru, T.; Nishizaka, H.; Niho, Y. C5a induces tissue factor activity on endothelial cells. Thromb. Haemost. 1997, 77, 394–398. [Google Scholar] [CrossRef]
- Davis, A.E. Biological effects of C1 inhibitor. Drug News Perspect. 2004, 17, 439–446. [Google Scholar] [CrossRef]
- De Agostini, A.; Lijnen, H.R.; Pixley, R.A.; Colman, R.W.; Schapira, M. Inactivation of factor XII active fragment in normal plasma. Predominant role of C1-inhibitor. J. Clin. Investig. 1984, 73, 1542–1549. [Google Scholar] [CrossRef]
- Cugno, M.; Bos, I.; Lubbers, Y.; Hack, C.E.; Agostoni, A. In vitro interaction of C1-inhibitor with thrombin. Blood Coagul. Fibrinolysis 2001, 12, 253–260. [Google Scholar] [CrossRef]
- Rezende, S.M.; Simmonds, R.E.; Lane, D.A. Coagulation, inflammation, and apoptosis: Different roles for protein S and the protein S-C4b binding protein complex. Blood 2004, 103, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Brückner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular intercommunication between the complement and coagulation systems. J. Immunol. 2010, 185, 5628–5636. [Google Scholar] [CrossRef] [PubMed]
- Amara, U.; Rittirsch, D.; Flierl, M.; Bruckner, U.; Klos, A.; Gebhard, F.; Lambris, J.D.; Huber-Lang, M. Interaction between the coagulation and complement system. Adv. Exp. Med. Biol. 2008, 632, 71–79. [Google Scholar] [PubMed]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.H.; Conway, E.M. Cross Talk Pathways Between Coagulation and Inflammation. Circ. Res. 2016, 118, 1392–1408. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.H. Examining coagulation-complement crosstalk: complement activation and thrombosis. Thromb. Res. 2016, 141, S50–S54. [Google Scholar] [CrossRef]
- Rittirsch, D.; Flierl, M.A.; Ward, P.A. Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 2008, 8, 776–787. [Google Scholar] [CrossRef]
- Landsem, A.; Fure, H.; Christiansen, D.; Nielsen, E.W.; Østerud, B.; Mollnes, T.E.; Brekke, O.L. The key roles of complement and tissue factor in Escherichia coli-induced coagulation in human whole blood. Clin. Exp. Immunol. 2015, 182, 81–89. [Google Scholar] [CrossRef]
- Gushiken, F.C.; Han, H.; Li, J.; Rumbaut, R.E.; Afshar-Kharghan, V. Abnormal platelet function in C3-deficient mice. J. Thromb. Haemost. 2009, 7, 865–870. [Google Scholar] [CrossRef]
- Polley, M.J.; Nachman, R.L. Human complement in thrombin-mediated platelet function: uptake of the C5b-9 Complex. J. Exp. Med. 1979, 150, 633–645. [Google Scholar] [CrossRef]
- Sims, P.J.; Faioni, E.M.; Wiedmer, T.; Shattil, S.J. Complement proteins C5b-9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J. Biol. Chem. 1988, 263, 18205–18212. [Google Scholar] [PubMed]
- Polley, M.J.; Nachman, R. The human complement system in thrombin-mediated platelet function. J. Exp. Med. 1978, 147, 1713–1726. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Jurk, K.; Hobohm, L.; Jackel, S.; Saffarzadeh, M.; Schwierczek, K.; Wenzel, P.; Langer, F.; Reinhardt, C.; Ruf, W. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 2017, 129, 2291–2302. [Google Scholar] [CrossRef] [PubMed]
- Sauter, R.J.; Sauter, M. Functional relevance of the Anaphylatoxin receptor C3aR for platelet function and arterial thrombus formation marks an intersection point between innate immunity and thrombosis. Circulation 2018, 138, 1720–1735. [Google Scholar] [CrossRef]
- Wiedmer, T.; Esmon, C.T.; Sims, P.J. On the mechanism by which complement proteins C5b-9 increase platelet prothrombinase activity. J. Biol. Chem. 1986, 261, 14587–14592. [Google Scholar]
- Wiedmer, T.; Esmon, C.T.; Sims, P.J. Complement proteins C5b-9 stimulate procoagulant activity through platelet prothrombinase. Blood 1986, 68, 875–880. [Google Scholar] [CrossRef]
- Huber-Lang, M.; Kovtun, A.; Ignatius, A. The role of complement in trauma and fracture healing. Semin. Immunol. 2013, 25, 73–78. [Google Scholar] [CrossRef]
- Van Griensven, M.; Ricklin, D.; Denk, S.; Halbgebauer, R.; Braun, C.K.; Schultze, A.; Hönes, F.; Koutsogiannaki, S.; Primikyri, A.; Reis, E.; et al. Protective Effects of the Complement Inhibitor Compstatin CP40 in Hemorrhagic Shock. Shock 2019, 51, 78–87. [Google Scholar] [CrossRef]
- Øvstebø, R.; Hellum, M.; Aass, H.C.D.; Trøseid, A.M.; Brandtzaeg, P.; Mollnes, T.E.; Henriksson, C.E. Microparticle-associated tissue factor activity is reduced by inhibition of the complement protein 5 in Neisseria meningitidis-exposed whole blood. Innate Immun. 2014, 20, 552–560. [Google Scholar] [CrossRef]
- Del Conde, I.; Crúz, M.A.; Zhang, H.; López, J.A.; Afshar-Kharghan, V. Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 2005, 201, 871–879. [Google Scholar] [CrossRef]
- Hamad, O.A.; Ekdahl, K.N.; Nilsson, P.H.; Andersson, J.; Magotti, P.; Lambris, J.D.; Nilsson, B. Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J. Thromb. Haemost. 2008, 6, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Devine, D.V.; Rosse, W.F. Regulation of the activity of platelet-bound C3 convertase of the alternative pathway of complement by platelet factor H. Proc. Natl. Acad. Sci. USA 1987, 84, 5873–5877. [Google Scholar] [CrossRef] [PubMed]
- Schmaier, A.H.; Smith, P.M.; Colman, R.W. Platelet C1- inhibitor. A secreted alpha-granule protein. J. Clin. Investig. 1985, 75, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Peerschke, E.I.B.; Yin, W.; Grigg, S.E.; Ghebrehiwet, B. Blood platelets activate the classical pathway of human complement. J. Thromb. Haemost. 2006, 4, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Kozarcanin, H.; Lood, C.; Munthe-Fog, L.; Sandholm, K.; Hamad, O.A.; Bengtsson, A.A.; Skjoedt, M.O.; Huber-Lang, M.; Garred, P.; Ekdahl, K.N.; et al. The lectin complement pathway serine proteases (MASPs) represent a possible crossroad between the coagulation and complement systems in thromboinflammation. J. Thromb. Haemost. 2016, 14, 531–545. [Google Scholar] [CrossRef]
- Brown, E.W.; Ravindran, S.; Patston, P.A. The reaction between plasmin and C1-inhibitor results in plasmin inhibition by the serpin mechanism. Blood Coagul. Fibrinolysis 2002, 13, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.H.; Walton, B.L.; Aleman, M.M.; O’Byrne, A.M.; Lei, V.; Harrasser, M.; Foley, K.A.; Wolberg, A.S.; Conway, E.M. Complement Activation in Arterial and Venous Thrombosis is Mediated by Plasmin. EBioMedicine 2016, 5, 175–182. [Google Scholar] [CrossRef]
- Wojta, J.; Huber, K.; Valent, P. New aspects in thrombotic research: Complement induced switch in mast cells from a profibrinolytic to a prothrombotic phenotype. Pathophysiol. Haemost. Thromb. 2003, 33, 438–441. [Google Scholar] [CrossRef]
- Krarup, A.; Wallis, R.; Presanis, J.S.; Gál, P.; Sim, R.B. Simultaneous Activation of Complement and Coagulation by MBL-Associated Serine Protease 2. PLoS ONE 2007, 2, e623. [Google Scholar] [CrossRef]
- Dobó, J.; Schroeder, V.; Jenny, L.; Cervenak, L.; Závodszky, P.; Gál, P. Multiple roles of complement MASP-1 at the interface of innate immune response and coagulation. Mol. Immunol. 2014, 61, 69–78. [Google Scholar] [CrossRef]
- Hess, K.; Ajjan, R.; Phoenix, F.; Dobó, J.; Gál, P.; Schroeder, V. Effects of MASP-1 of the Complement System on Activation of Coagulation Factors and Plasma Clot Formation. PLoS ONE 2012, 7, e35690. [Google Scholar] [CrossRef] [PubMed]
- Dzik, S. Complement and Coagulation: Cross Talk Through Time. Transfus. Med. Rev. 2019, 33, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Jenny, L.; Dobó, J.; Gál, P.; Pál, G.; Lam, W.A.; Schroeder, V. MASP-1 of the complement system enhances clot formation in a microvascular whole blood flow model. PLoS ONE 2018, 13, e0191292. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Cardenas, J.C.; Wade, C.E.; Holcomb, J.B. Advances in the understanding of trauma-induced coagulopathy. Blood 2016, 128, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Herbert, J.P.; Guillotte, A.R.; Hammer, R.D.; Scott Litofsky, N. Coagulopathy in the setting of mild traumatic brain injury: Truths and consequences. Brain Sci. 2017, 7, 92. [Google Scholar] [CrossRef] [PubMed]
- Folkerson, L.E.; Sloan, D.; Cotton, B.A.; Holcomb, J.B.; Tomasek, J.S.; Wade, C.E. Predicting progressive hemorrhagic injury from isolated traumatic brain injury and coagulation. Surgery 2015, 158, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, F.; Dong, J. fei Coagulopathy induced by traumatic brain injury: Systemic manifestation of a localized injury. Blood 2018, 131, 2001–2006. [Google Scholar] [CrossRef]
- Yuan, Q.; Sun, Y.R.; Wu, X.H.X.; Yu, J.; Li, Z.Q.; Du, Z.Y.; Wu, X.H.X.; Zhou, L.F.; Hu, J. Coagulopathy in Traumatic Brain Injury and Its Correlation with Progressive Hemorrhagic Injury: A Systematic Review and Meta-Analysis. J. Neurotrauma 2016, 33, 1279–1291. [Google Scholar] [CrossRef]
- Skrifvars, M.B.; Bailey, M.; Presneill, J.; French, C.; Nichol, A.; Little, L.; Duranteau, J.; Huet, O.; Haddad, S.; Arabi, Y.; et al. Venous thromboembolic events in critically ill traumatic brain injury patients. Intensive Care Med. 2017, 43, 419–428. [Google Scholar] [CrossRef]
- Strollo, B.P.; Bennett, G.J.; Chopko, M.S.; Guo, W.A. Timing of venous thromboembolism chemoprophylaxis after traumatic brain injury. J. Crit. Care 2018, 43, 75–80. [Google Scholar] [CrossRef]
- Glass, N.E.; Vadlamani, A.; Hwang, F.; Sifri, Z.C.; Kunac, A.; Bonne, S.; Pentakota, S.R.; Yonclas, P.; Mosenthal, A.C.; Livingston, D.H.; et al. Bleeding and Thromboembolism After TBI in the Elderly: A Real Conundrum. J. Surg. Res. 2019, 235, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Hachem, L.D.; Mansouri, A.; Scales, D.C.; Geerts, W.; Pirouzmand, F. Anticoagulant prophylaxis against venous thromboembolism following severe traumatic brain injury: A prospective observational study and systematic review of the literature. Clin. Neurol. Neurosurg. 2018, 175, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, J.S.; Liu, X.; Smith, G.S.; Baumgarten, M.; Rattinger, G.B.; Gambert, S.R.; Langenberg, P.; Zuckerman, I.H. Stroke Incidence Following Traumatic Brain Injury in Older Adults HHS Public Access. J. Head Trauma Rehabil. 2015, 30, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, R.G.; Haarbauer-Krupa, J.K.; Bell, J.M.; Corrigan, J.D.; Hammond, F.M.; Torbey, M.T.; Hofmann, M.C.; Dams-O’Connor, K.; Miller, A.C.; Whiteneck, G.G. Acute Ischemic Stroke After Moderate to Severe Traumatic Brain Injury: Incidence and Impact on Outcome. Stroke 2017, 48, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.W.; Huang, L.C.; Chung, W.F.; Chang, H.K.; Wu, J.C.; Chen, L.F.; Chen, Y.C.; Huang, W.C.; Cheng, H.; Lo, S.S. Increased risk of stroke in patients of concussion: A nationwide cohort study. Int. J. Environ. Res. Public Health 2017, 14, 230. [Google Scholar] [CrossRef]
- Eric Nyam, T.T.; Ho, C.H.; Chio, C.C.; Lim, S.W.; Wang, J.J.; Chang, C.H.; Kuo, J.R.; Wang, C.C. Traumatic Brain Injury Increases the Risk of Major Adverse Cardiovascular and Cerebrovascular Events: A 13-Year, Population-Based Study. World Neurosurg. 2019, 122, e740–e753. [Google Scholar] [CrossRef]
- McFarlane, T.D.; Love, J.; Hanley, S.; Dixon, B.E.; Hammond, F.M. Increased Risk of Stroke Among Young Adults With Serious Traumatic Brain Injury. J. Head Trauma Rehabil. 2019. [Google Scholar] [CrossRef]
- Stein, S.C.; Graham, D.I.; Chen, X.-H.; Smith, D.H. Association between intravascular microthrombosis and cerebral ischemia in traumatic brain injury. Neurosurgery 2004, 54, 687–691. [Google Scholar] [CrossRef]
- Hammad, A.; Westacott, L.; Zaben, M. The role of the complement system in traumatic brain injury: A review. J. Neuroinflammation 2018, 15, 24. [Google Scholar] [CrossRef]
- Huber-Lang, M.; Lambris, J.D.; Ward, P.A. Innate immune responses to trauma review-article. Nat. Immunol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Krem, M.M.; Di Cera, E. Evolution of enzyme cascades from embryonic development to blood coagulation. Trends Biochem. Sci. 2002, 27, 67–74. [Google Scholar] [CrossRef]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part II: Role in immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Kharghan, V. The role of the complement system in cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P.; Gasque, P. Extrahepatic complement biosynthesis: Where, when and why? Clin. Exp. Immunol. 1997, 107, 1–7. [Google Scholar] [CrossRef]
- Nataf, S.; Levison, S.W.; Barnum, S.R. Expression of the anaphylatoxin C5a receptor in the oligodendrocyte lineage. Brain Res. 2001, 894, 321–326. [Google Scholar] [CrossRef]
- Alawieh, A.; Elvington, A.; Tomlinson, S. Complement in the Homeostatic and Ischemic Brain. Front. Immunol. 2015, 6, 417. [Google Scholar] [CrossRef]
- Krukowski, K.; Chou, A.; Feng, X.; Tiret, B.; Paladini, M.-S.; Riparip, L.-K.; Chaumeil, M.; Lemere, C.; Rosi, S. Traumatic Brain Injury in Aged Mice Induces Chronic Microglia Activation, Synapse Loss, and Complement-Dependent Memory Deficits. Int. J. Mol. Sci. 2018, 19, 3753. [Google Scholar] [CrossRef]
- Sillesen, M.; Rasmussen, L.S.; Jin, G.; Jepsen, C.H.; Imam, A.; Hwabejire, J.O.; Halaweish, I.; DeMoya, M.; Velmahos, G.; Johansson, P.I.; et al. Assessment of coagulopathy, endothelial injury, and inflammation after traumatic brain injury and hemorrhage in a porcine model. J. Trauma Acute Care Surg. 2014, 76, 12–19, discussion 19–20. [Google Scholar] [CrossRef]
- Nekludov, M.; Antovic, J.; Bredbacka, S.; Blombäck, M. Coagulation Abnormalities Associated with Severe Isolated Traumatic Brain Injury: Cerebral Arterio-Venous Differences in Coagulation and Inflammatory Markers. J. Neurotrauma 2007, 24, 174–180. [Google Scholar] [CrossRef]
- Kossmann, T.; Stahel, P.F.; Morganti-Kossmann, M.C.; Jones, J.L.; Barnum, S.R. Elevated levels of the complement components C3 and factor B in ventricular cerebrospinal fluid of patients with traumatic brain injury. J. Neuroimmunol. 1997, 73, 63–69. [Google Scholar] [CrossRef]
- Kaczorowski, S.L.; Schiding, J.K.; Toth, C.A.; Kochanek, P.M. Effect of soluble complement receptor-1 on neutrophil accumulation after traumatic brain injury in rats. J. Cereb. Blood Flow Metab. 1995, 15, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Nakamura, T.; Hua, Y.; Keep, R.F.; Younger, J.G.; He, Y.; Hoff, J.T.; Xi, G. The role of complement C3 in intracerebral hemorrhage-induced brain injury. J. Cereb. Blood Flow Metab. 2006, 26, 1490–1495. [Google Scholar] [CrossRef] [PubMed]
- Sewell, D.L.; Nacewicz, B.; Liu, F.; Macvilay, S.; Erdei, A.; Lambris, J.D.; Sandor, M.; Fabry, Z. Complement C3 and C5 play critical roles in traumatic brain cryoinjury: Blocking effects on neutrophil extravasation by C5a receptor antagonist. J. Neuroimmunol. 2004, 155, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Ruseva, M.M.; Ramaglia, V.; Morgan, B.P.; Harris, C.L. An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice. Proc. Natl. Acad. Sci. USA 2015, 112, 14319–14324. [Google Scholar] [CrossRef]
- Lindblad, C.; Thelin, E.P.; Nekludov, M.; Frostell, A.; Nelson, D.W.; Svensson, M.; Bellander, B.M. Assessment of platelet function in traumatic brain injury-A retrospective observational study in the neuro-critical care setting. Front. Neurol. 2018, 9, 15. [Google Scholar] [CrossRef]
- Castellino, F.J.; Chapman, M.P.; Donahue, D.L.; Thomas, S.; Moore, E.E.; Wohlauer, M.V.; Fritz, B.; Yount, R.; Ploplis, V.; Davis, P.; et al. Traumatic brain injury causes platelet adenosine diphosphate and arachidonic acid receptor inhibition independent of hemorrhagic shock in humans and rats. J. Trauma Acute Care Surg. 2014, 76, 1169–1176. [Google Scholar] [CrossRef]
- Kutcher, M.E.; Redick, B.J.; McCreery, R.C.; Crane, I.M.; Greenberg, M.D.; Cachola, L.M.; Nelson, M.F.; Cohen, M.J. Characterization of platelet dysfunction after trauma. J. Trauma Acute Care Surg. 2012, 73, 13–19. [Google Scholar] [CrossRef]
- Nekludov, M.; Bellander, B.-M.M.; Blombäck, M.; Wallen, H.N.; Blomback, M.; Wallen, H.N. Platelet dysfunction in patients with severe traumatic brain injury. J. Neurotrauma 2007, 24, 1699–1706. [Google Scholar] [CrossRef]
- Schnüriger, B.; Inaba, K.; Abdelsayed, G.A.; Lustenberger, T.; Eberle, B.M.; Barmparas, G.; Talving, P.; Demetriades, D. The impact of platelets on the progression of traumatic intracranial hemorrhage. J. Trauma Inj. Infect. Crit. Care 2010, 68, 881–885. [Google Scholar]
- Atefi, G.; Aisiku, O.; Shapiro, N.; Hauser, C.; Lucca, D.; Flaumenhaft, R.; Tsokos, G.C.; Dalle Lucca, J.; Flaumenhaft, R.; Tsokos, G.C. Complement activation in trauma patients alters platelet function. Shock 2016, 46, 83–88. [Google Scholar] [CrossRef]
- Fair, K.; Farrell, D.; McCully, B.; Rick, E.; Dewey, E.N.; Hilliard, C.; Dean, R.; Lin, A.L.; Hinson, H.E.; Barbosa, R.R.; et al. Fibrinolytic Activation in Patients with Progressive Intracranial Hemorrhage after Traumatic Brain Injury. J. Neurotrauma 2019. [Google Scholar] [CrossRef] [PubMed]
- Nakae, R.; Takayama, Y.; Kuwamoto, K.; Naoe, Y.; Sato, H.; Yokota, H. Time Course of Coagulation and Fibrinolytic Parameters in Patients with Traumatic Brain Injury. J. Neurotrauma 2016, 33, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Leeper, C.M.; Neal, M.D.; McKenna, C.J.; Gaines, B.A. Trending Fibrinolytic Dysregulation: Fibrinolysis Shutdown in the Days after Injury Is Associated with Poor Outcome in Severely Injured Children. Ann. Surg. 2017, 266, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Leeper, C.M.; Strotmeyer, S.J.; Neal, M.D.; Gaines, B.A. Window of Opportunity to Mitigate Trauma-induced Coagulopathy: Fibrinolysis Shutdown not Prevalent Until 1 Hour Post-injury. Ann. Surg. 2019, 270, 528–534. [Google Scholar] [CrossRef]
- Moore, H.B.; Moore, E.E.; Gonzalez, E.; Chapman, M.P.; Chin, T.L.; Silliman, C.C.; Banerjee, A.; Sauaia, A. Hyperfibrinolysis, physiologic fibrinolysis, and fibrinolysis shutdown: the spectrum of postinjury fibrinolysis and relevance to antifibrinolytic therapy. J. Trauma Acute Care Surg. 2014, 77, 811–817, discussion 817. [Google Scholar] [CrossRef]
- Hattori, R.; Hamilton, K.K.; McEver, R.P.; Sims, P.J. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J. Biol. Chem. 1989, 264, 9053–9060. [Google Scholar]
- Hamilton, K.K.; Hattori, R.; Esmon, C.T.; Sims, P.J. Complement proteins C5b-9 induce vesiculation of the endothelial plasma membrane and expose catalytic surface for assembly of the prothrombinase enzyme complex. J. Biol. Chem. 1990, 265, 3809–3814. [Google Scholar]
- Foreman, K.E.; Vaporciyan, A.A.; Bonish, B.K.; Jones, M.L.; Johnson, K.J.; Glovsky, M.M.; Eddy, S.M.; Ward, P.A. C5a-induced expression of P-selectin in endothelial cells. J. Clin. Investig. 1994, 94, 1147–1155. [Google Scholar] [CrossRef]
- Merrill, S.A.; Brodsky, R.A. Complement-driven anemia: More than just paroxysmal nocturnal hemoglobinuria. Hematology 2018, 2018, 371–376. [Google Scholar] [CrossRef]
- Chapin, J.; Terry, H.S.; Kleinert, D.; Laurence, J. The role of complement activation in thrombosis and hemolytic anemias. Transfus. Apher. Sci. 2016, 54, 191–198. [Google Scholar] [CrossRef]
- Eriksson, O.; Mohlin, C.; Nilsson, B.; Ekdahl, K.N. The human platelet as an innate immune cell: Interactions between activated platelets and the complement system. Front. Immunol. 2019, 10, 1590. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.; Uchino, K.; Fakhran, S.; Sattar, M.A.; Branstetter, B.F.; Au, K.; Navratil, J.S.; Paul, B.; Lee, M.; Gallagher, K.M.; et al. Platelet C4d Is Associated With Acute Ischemic Stroke and Stroke Severity. Stroke 2008, 39, 3236–3241. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.A.; Conklin, J.; O’Malley, T.; Dervieux, T. Platelet-bound C4d, low C3 and lupus anticoagulant associate with thrombosis in SLE. Lupus Sci. Med. 2019, 6, e000318. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; Dezern, A.E.; Kinoshita, T.; Brodsky, R.A. Paroxysmal nocturnal haemoglobinuria. Nat. Rev. Dis. Prim. 2017, 3, 17028. [Google Scholar] [CrossRef]
- Zhang, K.; Lu, Y.; Harley, K.T.; Tran, M.H. Atypical hemolytic uremic syndrome: A brief review. Hematol. Rep. 2017, 9, 62–67. [Google Scholar] [CrossRef]
- Abe, T.; Sasaki, A.; Ueda, T.; Miyakawa, Y.; Ochiai, H. Complement-mediated thrombotic microangiopathy secondary to sepsis-induced disseminated intravascular coagulation successfully treated with eculizumab a case report. Medicine 2017, 96, e6056. [Google Scholar] [CrossRef]
- Keragala, C.B.; Draxler, D.F.; McQuilten, Z.K.; Medcalf, R.L. Haemostasis and innate immunity—A complementary relationship. Br. J. Haematol. 2017, 180, 782–798. [Google Scholar] [CrossRef]
- Gialeli, C.; Gungor, B.; Blom, A.M. Novel potential inhibitors of complement system and their roles in complement regulation and beyond. Mol. Immunol. 2018, 102, 73–83. [Google Scholar] [CrossRef]
- Thurman, J.M.; Le Quintrec, M. Targeting the complement cascade: novel treatments coming down the pike. Kidney Int. 2016, 90, 746–752. [Google Scholar] [CrossRef]
- Reddy, Y.N.V.; Siedlecki, A.M.; Francis, J.M. Breaking down the complement system: A review and update on novel therapies. Curr. Opin. Nephrol. Hypertens. 2017, 26, 123–128. [Google Scholar] [CrossRef]
- Andrighetto, S.; Leventhal, J.; Zaza, G.; Cravedi, P. Complement and complement targeting therapies in glomerular diseases. Int. J. Mol. Sci. 2019, 20, 6336. [Google Scholar] [CrossRef]
- Baines, A.C.; Brodsky, R.A. Complementopathies. Blood Rev. 2017, 31, 213–223. [Google Scholar] [CrossRef]
- Wada, T.; Gando, S.; Maekaw, K.; Katabami, K.; Sageshima, H.; Hayakawa, M.; Sawamura, A. Disseminated intravascular coagulation with increased fibrinolysis during the early phase of isolated traumatic brain injury. Crit. Care 2017, 21, 219. [Google Scholar] [CrossRef]
- The CRASH-3 trial collaborators. Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): a randomised, placebo-controlled trial. Lancet 2019, 394, 1713–1723. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Complement Substrate | Effect on Hemostasis | Presumed Physiological Effect |
|---|---|---|
| C3a | Activate platelets | ↑ Platelet aggregation |
| C5a | Increase tissue factor activity Increase expression of endothelial P-selectin Induce expression of PAI-1 on mast cells | ↑Fibrin formation ↑ Platelet aggregation ↓ Fibrinolysis |
| MAC (C5b-9) | Activate platelets Initiates release of factor V from platelet alpha-granules Increase binding of coagulation factors Va and Xa Induce endothelial cells to secrete von Willebrand factor | ↑ Platelet aggregation ↑ Platelet aggregation ↑Fibrin formation ↑Platelet aggregation, ↑ Fibrin formation |
| MASP 1 | Activate thrombin (by cleaving prothrombin) Activate fibrinogen Activate factor XII Activate TAFI | ↑ Fibrin formation ↑ Fibrin formation ↑ Fibrin formation ↓ Fibrinolysis |
| MASP 2 | Activate thrombin (by cleaving prothrombin) | ↑ Fibrin formation |
| C1-INH | Inhibit factor XII Inhibit thrombin Inhibit plasmin | ↓ Fibrin formation ↓ Fibrin formation ↓ Fibrinolysis |
| C4BP | Inhibit protein S | ↑ Fibrin formation |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fletcher-Sandersjöö, A.; Maegele, M.; Bellander, B.-M. Does Complement-Mediated Hemostatic Disturbance Occur in Traumatic Brain Injury? A Literature Review and Observational Study Protocol. Int. J. Mol. Sci. 2020, 21, 1596. https://doi.org/10.3390/ijms21051596
Fletcher-Sandersjöö A, Maegele M, Bellander B-M. Does Complement-Mediated Hemostatic Disturbance Occur in Traumatic Brain Injury? A Literature Review and Observational Study Protocol. International Journal of Molecular Sciences. 2020; 21(5):1596. https://doi.org/10.3390/ijms21051596
Chicago/Turabian StyleFletcher-Sandersjöö, Alexander, Marc Maegele, and Bo-Michael Bellander. 2020. "Does Complement-Mediated Hemostatic Disturbance Occur in Traumatic Brain Injury? A Literature Review and Observational Study Protocol" International Journal of Molecular Sciences 21, no. 5: 1596. https://doi.org/10.3390/ijms21051596
APA StyleFletcher-Sandersjöö, A., Maegele, M., & Bellander, B.-M. (2020). Does Complement-Mediated Hemostatic Disturbance Occur in Traumatic Brain Injury? A Literature Review and Observational Study Protocol. International Journal of Molecular Sciences, 21(5), 1596. https://doi.org/10.3390/ijms21051596