Abstract
Previously, we reported that overexpression of AtRH17, an Arabidopsis DEAD-box RNA helicase gene, confers salt stress-tolerance via a pathway other than the well-known salt stress-responsive pathways. To decipher the salt stress-responsive pathway in AtRH17-overexpressing transgenic plants (OXs), we performed RNA-Sequencing and identified 397 differentially expressed genes between wild type (WT) and AtRH17 OXs. Among them, 286 genes were upregulated and 111 genes were downregulated in AtRH17 OXs relative to WT. Gene ontology annotation enrichment and KEGG pathway analysis showed that the 397 upregulated and downregulated genes are involved in various biological functions including secretion, signaling, detoxification, metabolic pathways, catabolic pathways, and biosynthesis of secondary metabolites as well as in stress responses. Genevestigator analysis of the upregulated genes showed that nine genes, namely, LEA4-5, GSTF6, DIN2/BGLU30, TSPO, GSTF7, LEA18, HAI1, ABR, and LTI30, were upregulated in Arabidopsis under salt, osmotic, and drought stress conditions. In particular, the expression levels of LEA4-5, TSPO, and ABR were higher in AtRH17 OXs than in WT under salt stress condition. Taken together, our results suggest that a high AtRH17 expression confers salt stress-tolerance through a novel salt stress-responsive pathway involving nine genes, other than the well-known ABA-dependent and ABA-independent pathways.
1. Introduction
RNA helicases (RHs) are present in most prokaryotic and eukaryotic organisms, which catalyze the unwinding of DNA or the secondary structure of RNA, and thus, play essential roles in almost every aspect of genetic processes such as replication, transcription, translation, repair, and recombination [1,2]. RHs have been classified into six superfamilies (SF1–SF6) based on specific motif sequences and domain structures. Superfamily II (SF2), the largest helicase family, mainly consists of the DEAD-box RHs, which are named after the strictly conserved sequence “Asp-Glu-Ala-Asp” (D-E-A-D) [1,2]. Fifty-eight DEAD-box RHs have been identified in Arabidopsis [1,3].
DEAD-box RH has nine well-defined motifs (Q, I, Ia, Ib, and II–VI) divided into two helicase domains: domain 1 and domain 2 [1,3,4,5,6]. Each motif has been proposed to have specific helicase functions. The newly identified motif Q of DEAD-box RHs is supposed to control hydrolysis and ATP binding; motif I (popularly known as the Walker A motif) is associated with the interaction between ATP and Mg2+; motif Ia forms a groove binding to single-stranded DNA/RNA; motif II (also known as the Walker B motif) interacts with Mg2+; motif III is responsible for helicase and NTPase activities to perform RNA unwinding, and motif VI is a part of the ATP-binding cleft that is related to helicase and NTPase activities. The molecular functions of the remaining motifs (Ib, IV, and V) are still unclear [2,3]. In addition to these conserved motifs, the N-terminal and C-terminal extended regions have also been found in each DEAD-box RH protein, which vary greatly in terms of their size and composition; it has been proposed that they function in determining the substrate-binding specificities as subcellular localization signals or possibly interact with accessory components [7,8,9].
Recent studies have indicated the important roles of DEAD-box RHs in RNA biogenesis, pre-mRNA splicing, RNA export and storage, transcription, translation, and RNA decay as well as in organelle-specific RNA metabolism [2,3]. Furthermore, multiple studies have suggested that DEAD-box RHs also play essential roles in abiotic stress responses in plants through their functions in specific RNA processing events [10,11]. AtRH7 participates in rRNA biogenesis and is involved in cold-tolerance in Arabidopsis [12]. AtRH38/LOS4 has important roles in mRNA export and in cold and heat stress responses in Arabidopsis [13]. AtRH47/RCF1 is also involved in cold stress-tolerance via proper splicing of pre-mRNAs of cold stress-responsive genes [14]. STRS1 and STRS2 reduce the expression of stress-responsive genes through RdDM-mediated gene silencing and function as negative regulators of the response to multiple abiotic stresses in Arabidopsis [15,16]. Despite these studies, the roles of DEAD-box RHs in abiotic stress response and/or abiotic stress-responsive pathways mediated by DEAD-box RHs are not well known, as yet.
Previously, we identified an Arabidopsis DEAD-box RH gene, AtRH17, using the activation tagging system. The activation tagging line, in which AtRH17 was activated and AtRH17-overexpressing transgenic plants (OXs) showed the salt-tolerant phenotype at the seedling and mature plant stages [17]. However, transcript levels of the well-known salt stress-responsive genes such as RD29A, RAB18, RD29B, RD22, COR47, DREB2A, and DREB2B, were not higher in AtRH17 OXs than in the wild type (WT) under salt stress conditions, suggesting that AtRH17 is involved in salt stress-tolerance via a pathway other than the well-known salt stress-responsive pathway.
In the present study, to elucidate the salt stress-responsive pathway in AtRH17 OXs, we performed RNA-Sequencing (RNA-Seq) and analyzed the expression of Arabidopsis genes in WT and AtRH17 OXs. RNA-Seq analysis showed 286 upregulated and 111 downregulated genes in AtRH17 OXs compared to WT. Gene Ontology (GO) annotation enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that the upregulated and downregulated genes are involved in various biological functions including stress responses. Further analysis of the upregulated genes suggested that overexpression of AtRH17 confers salt stress-tolerance via pathway(s) involving LEA4-5, GSTF6, DIN2/BGLU30, TSPO, GSTF7, LEA18, HAI1, ABR, and LTI30, other than the well-known ABA-dependent and ABA-independent stress-responsive pathways in Arabidopsis.
2. Results
2.1. Transcriptomic Profiling of AtRH17 OXs
In a previous study, we reported that overexpression of AtRH17 confers salt stress-tolerance at the seedling and mature plant stages. Interestingly, the well-known ABA-dependent and ABA-independent salt stress-responsive genes such as RD29A, RAB18, RD29B, RD22, COR47, DREB2A, and DREB2B showed similar or lower expression levels in AtRH17 OXs than in WT under salt stress conditions [17], implying that salt stress-tolerance of AtRH17 OXs is mediated by an uncharacterized pathway or mechanism other than the well-known stress-responsive pathways. In this study, to clarify the regulatory mechanism of salt stress-tolerance of AtRH17 OXs, RNA-Seq was performed using 10-day-old WT and AtRH17 OX whole seedlings. The mapping of RNA-Seq reads to the Col-0 genome was successful, with a mapping rate of 97.47% to 98.01% (Table 1). The number of mapped reads ranged from 26.4 to 30.3 million (Table 1). Genes having very low abundance were removed from the analysis, leaving 22,884 genes for further analysis.

Table 1.
Summary of mapping transcriptome reads to reference sequence.
2.2. Analysis of Up- and Downregulated Genes in AtRH17 OXs
A total of 397 differentially expressed genes (DEGs) between WT and AtRH17 OX plants were identified with >3-fold differences in expression and a p-value < 0.05. Among these, 286 genes were upregulated and 111 genes were downregulated in AtRH17 OXs (Figure 1a). The distribution of up- and downregulated genes is shown using volcano plot (Figure 1b).
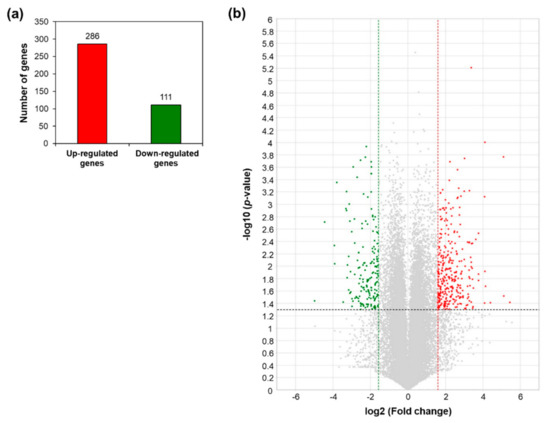
Figure 1.
Differentially expressed genes in AtRH17-overexpressing transgenic plants (OXs) compared to wild type (WT) from the RNA-Sequencing (RNA-Seq) analysis. (a) Numbers of genes more than 3-fold up- and downregulated in AtRH17 OXs compared to WT. (b) Volcano plot of differentially expressed genes (DEGs) identified between WT and AtRH17 OXs (p-value < 0.05 and log2 ratio ≥ 1). The upregulated genes are represented by a red dot and downregulated genes by a green dot, grey dots indicate genes that are not differentially expressed.
The analysis of GO terms with three categories, namely, biological processes, molecular functions, and cellular components, using the selected genes is useful in predicting the altered biological and molecular processes. Therefore, 397 DEGs were subjected to GO enrichment analysis to determine their functional significance in AtRH17 OXs. The upregulated genes were enriched in response to toxic substance, toxin catabolic process, glutathione metabolic process, and oxidation–reduction process in the biological process categories of GO annotation; glutathione transferase activity, lipid binding, heme binding, flavin adenine dinucleotide binding, and electron carrier activity in the molecular function categories; and extracellular region, apoplast, extracellular space, RNA polymerase II transcription factor complex, and cell wall in the cellular component categories (Supplementary Figure S1). The downregulated genes were enriched for plant-type cell wall organization, hydrogen peroxide catabolic process, and response to brassinosteroid in the biological process categories of GO annotation; structural constituent of cell wall, transcription factor activity, peroxidase activity in the molecular function categories; and cell wall, extracellular region, and plant-type cell wall in the cellular component categories (Supplementary Figure S2). GO enrichment analysis showed that many genes, which are involved in various biological and molecular processes such as signaling, secretion, detoxification, transcription, and stress responses, were up- and/or downregulated in AtRH17 OXs.
We further analyzed the biological process categories of GO annotation to decipher the biological functions of the DEGs in AtRH17 OXs. The upregulated genes were highly enriched in the biological process of oxidation–reduction (34 genes), defense response to fungus (14 genes), defense response (14 genes), response to oxidative stress (12 genes), response to salt stress (11 genes), response to toxic substance (10 genes), and response to water deprivation (10 genes) (Table 2), demonstrating that AtRH17 might be involved in abiotic and biotic stress responses. In addition, AtRH17 might be involved in the metabolic processes of sugar compounds and homeostasis (Table 2). The GO analysis of the downregulated genes revealed that the biological processes in which the downregulated genes are involved were mostly associated with oxidation–reduction (11 genes), defense response (seven genes), plant-type cell wall organization (six genes), response to oxidative stress (five genes), and hydrogen peroxide catabolic process (four genes) (Table 3), indicating that AtRH17 might negatively regulate oxidative stress-related genes. Moreover, AtRH17 can have a minor role in negative regulation of plant growth-related genes based on the enriched GO terms such as response to brassinosteroid, response to gibberellin, unidimensional cell growth, and response to ethylene (Table 3).
Table 2.
Biological process categories of Gene Ontology (GO) annotation of upregulated genes in AtRH17 OXs.
Table 3.
Biological process categories of GO annotation of downregulated genes in AtRH17 OXs.
To identify the functional pathways in which AtRH17 is involved, KEGG pathway analysis was performed. Both the up- and downregulated genes were involved in metabolic pathways, biosynthesis of secondary metabolites, and phenylpropanoid biosynthesis (Figure 2 and Figure 3). In contrast, only the upregulated genes were involved in glutathione metabolism and flavonoid biosynthesis, whereas the downregulated genes were involved in plant hormone signal transduction and MAPK signaling pathway (Figure 2 and Figure 3), indicating that AtRH17 might be involved in several functional pathways through the up- and/or downregulation of various related genes. We also performed hierarchical clustering to classify and identify the relationship among DEGs. The up- and downregulated genes were classified into individual hierarchies. The upregulated genes were classified into six groups, whereas the downregulated genes were classified into four groups (Figure 4).
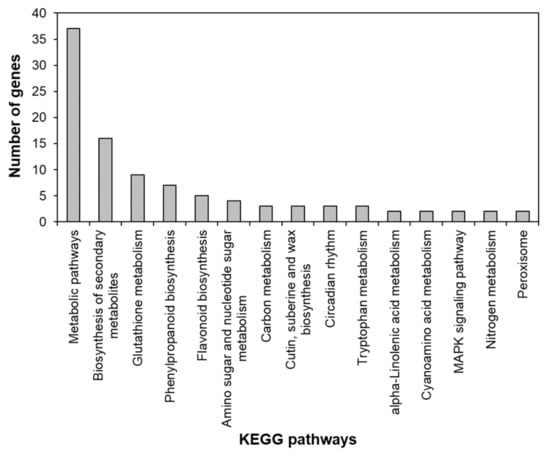
Figure 2.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway of upregulated genes in AtRH17 OXs. KEGG pathway was analyzed using KEGG Mapper.
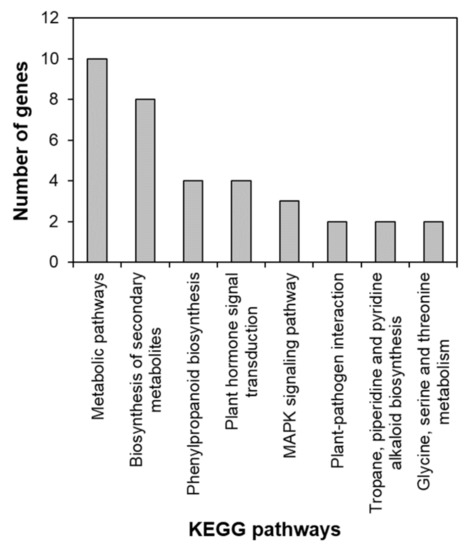
Figure 3.
KEGG pathway of downregulated genes in AtRH17 OXs. KEGG pathway was analyzed using KEGG Mapper.
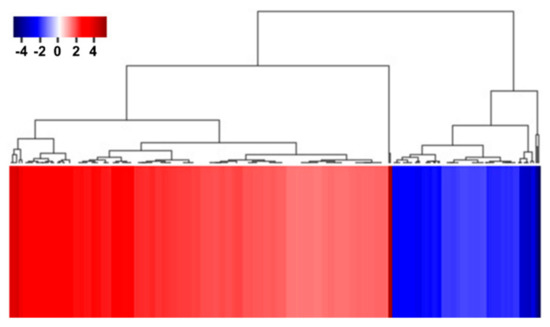
Figure 4.
Hierarchical clustering of up- and downregulated genes in AtRH17 OXs. Hierarchical clustering of up- and downregulated genes were performed using MultiExperiment Viewer (MeV).
To validate the RNA-Seq results by quantitative RT-PCR, we selected five upregulated genes, namely, TSPO, LEA4-5, ABR, LEA18, and DIN2/BGLU30 and four downregulated genes, namely, PIL1, FRF4, MYB108, and NAC019. Thereafter, we analyzed the expression of the selected genes in 10-day-old WT and AtRH17 OX seedlings. The expression of all five upregulated genes was significantly higher in AtRH17 OXs than in WT (Figure 5a–e). Especially, TSPO expression was more than 5-fold higher in AtRH17 OXs than in WT, whereas LEA4-5 expression was 2.3-fold higher in AtRH17 OXs than in WT (Figure 5a,b). The expression levels of ABR, LEA18, and DIN2/BGLU30 were more than 3-fold higher in AtRH17 OXs than in WT (Figure 5c–e). In contrast, the four downregulated genes showed lower expression in AtRH17 OXs than in WT (Figure 5f–i). PIL1 expression in AtRH17 OXs was only almost half of that in WT (Figure 5f). FRF4 and MYB108 expression in AtRH17 OXs were only one-fifth of that in WT (Figure 5g,h). Moreover, NAC019 expression in AtRH17 OXs was one-tenth of that in WT (Figure 5i). These results are consistent with the RNA-Seq results.
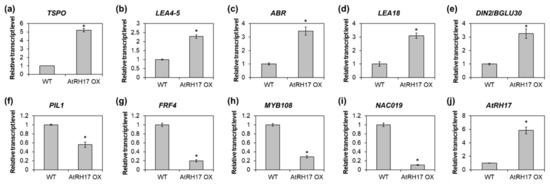
Figure 5.
Verification of up- and downregulated genes in AtRH17 OXs. Quantitative RT-PCR analysis of TSPO (a), LEA4-5 (b), ABR (c), LEA18 (d), DIN2/BGLU30 (e), PIL1 (f), FRF4 (g), MYB108 (h), NAC019 (i), and AtRH17 (j) in 10-day-old WT and AtRH17 OX seedlings. GAPc was used as an internal control. Transcript levels in WT were set as 1. Three independent reactions were performed for each technical replicate. Two technical replicates were performed for each biological replicate. At least two biological replicates showed similar results, with one shown here. Error bars represent standard deviation (n = 6 reactions) and * indicates t-test p < 0.05.
2.3. Identification of the Salt Stress-Tolerance Pathway in AtRH17 OXs
To elucidate the salt stress-tolerance mechanism in AtRH17 OXs, we isolated 19 upregulated genes enriched in the salt stress-related GO terms such as “response to salt” (GO:000965) and “response to water deprivation” (GO:0009414) (Table 4), indicating that these genes might function in salt stress-tolerance in AtRH17 OXs. To see the expression patterns of the 19 genes under stress conditions, we performed expression analysis using Genevestigator under drought, osmotic, and salt stress conditions. Five experiments were used for each stress condition. We divided the 19 genes into three groups depending on their expression patterns. In the first group, eight genes, namely, CHIA, CSD1, PER23, MYB12, ZFHD1, ANNAT7, GLP9, and MYB29, did not respond to any of the stresses (Figure 6). In the second group, LOX2 and GSTU17 showed high expression levels under drought condition, whereas their levels did not increase under osmotic and salt stress conditions (Figure 6). In the third group, nine genes, namely, DIN2/BGLU30, LEA18, GSTF6, GSTF7, LTI30, TSPO, HAI1, LEA4-5, and ABR, showed high expression levels under all three examined stress conditions (Figure 6), demonstrating that they might be involved in salt stress-tolerance in AtRH17 OXs (Table 5).
Table 4.
List of 19 genes upregulated in AtRH17 OXs, enriched in “response to salt” (GO:000965) and “response to water deprivation” (GO:0009414).
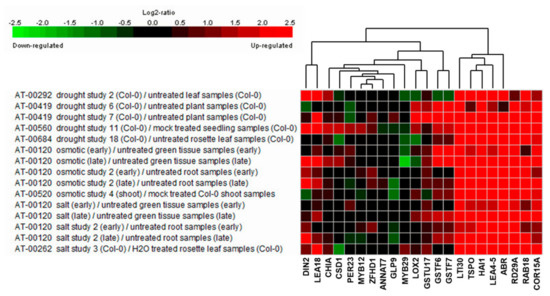
Figure 6.
Expression analysis of 19 stress-responsive genes under drought, osmotic, and salt stress conditions using Genevestigator. RD29A, RAB18, and COR15A were used as marker genes for stress conditions.
Table 5.
List of nine stress-responsive genes selected from Genevestigator analysis.
Next, we analyzed the expression of genes selected based on the Genevestigator analysis in WT and AtRH17 OXs under salt stress condition to verify whether these genes are involved in the salt stress response of AtRH17 OXs. LEA4-5 and ABR, late embryogenesis abundant (LEA) protein genes, and TSPO were selected for expression analysis among the above nine genes. The biological functions of LEA proteins in osmotic stress-tolerance are well known [18,19]. TSPOs have also been shown to be responsive to salt and osmotic stresses and ABA [20,21]. Quantitative RT-PCR analysis showed that the expression levels of LEA4-5, ABR, and TSPO were higher in AtRH17 OXs than in WT under salt stress conditions (Figure 7). Interestingly, the differences in expression levels of LEA4-5 and TSPO between WT and AtRH17 OXs were the highest under early salt stress conditions such as after 1 and 2 h of NaCl-treatment. Subsequently, the difference decreased gradually, and no difference was observed at 8 h after the NaCl-treatment (Figure 7a,b). However, the expression of ABR was almost similar in WT and AtRH17 OXs until 2 h after the NaCl-treatment. Thereafter, the expression in AtRH17 OXs significantly increased to more than that in WT until 8 h after the NaCl-treatment (Figure 7c), indicating that LEA4-5 and TSPO might function in the early salt stress response, whereas ABR might be involved in the late salt stress response.
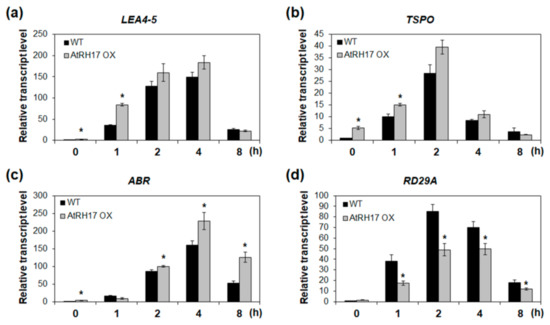
Figure 7.
Expression analysis of three stress-responsive genes in AtRH17 OXs under salt stress conditions. Quantitative RT-PCR analysis of LEA4-5 (a), TSPO (b), ABR (c), and RD29A (d) in WT and AtRH17 OX seedlings under 150 mM NaCl treatment for 0, 1, 2, 4, and 8 h. GAPc was used as an internal control. Transcript levels at 0 h in WT were set as 1. Three independent reactions were performed for each technical replicate. Two technical replicates were performed for each biological replicate. At least two biological replicates showed similar results, with one shown here. Error bars represent standard deviation (n = 6 reactions) and * indicates t-test * p < 0.05.
Protein network analysis of the nine salt stress-responsive genes using Cytoscape String Apps revealed that the nine genes were tightly connected and interacted with each other (Figure 8), indicating that the salt stress-tolerance in AtRH17 OXs might be the result of precise upregulation of these nine genes.
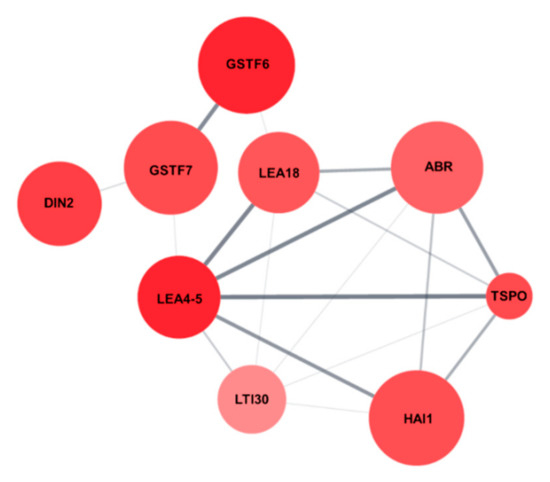
Figure 8.
Protein network of the nine salt stress-responsive genes. Protein network interaction of the nine salt stress-responsive genes were analyzed using String Apps of Cytoscape.
3. Discussion
In this study, we investigated the regulatory mechanism of the salt stress-tolerance of AtRH17 OXs using RNA-Seq analysis. AtRH17 is a member of the DEAD-box RHs, which function in RNA metabolism, ribosome biogenesis, and transcriptional and translational regulation [1,2,17]. Previous studies have shown that some DEAD-box RHs play important roles in the response of plants to abiotic stress such as cold, salt, and osmotic stresses as well as in the development of plants [11,12,13,14,15,16,22,23,24,25]. However, abiotic stress-responsive pathways, in which DEAD-box RH genes are involved, have not been well identified and are limited to a few stress-responsive DEAD-box RH genes such as AtRH38/LOS4, AtRH42/RCF1, STRS1, and STRS2 [13,14,15,16]. In addition, the identified abiotic stress-responsive pathways mediated by DEAD-box RH genes are restricted to the well-known ABA-dependent and/or ABA-independent pathways [13,14,15,16].
RNA-Seq, a high-throughput and next-generation sequencing technology, has allowed for rapid analysis of large genomic datasets and quantification of transcriptomes [26]. In addition, RNA-Seq analysis can be used to identify and quantify transcripts without prior information of genes and can provide information regarding alternative splicing and sequence variations in identified genes [26,27]. Global gene expression patterns have been determined using RNA-Seq analysis of samples at different developmental stages, in response to different stimuli or in different genotypes [28,29]. RNA-Seq analysis of AtRH17 OXs revealed that 286 genes were upregulated and 111 genes were downregulated in AtRH17 OXs relative to WT (Figure 1). The upregulated and downregulated genes were classified into six and four individual groups by hierarchy analysis, respectively (Figure 4). GO annotation enrichment analysis showed that AtRH17 might regulate genes that are involved in various functions such as signaling, secretion, detoxification, and transcription, depending on enriched GO terms such as response to toxic substances, toxin catabolic process, glutathione metabolic process, oxidation–reduction process, transcription factor activity, RNA polymerase II complex, and extracellular region (Supplementary Figures S1 and S2). KEGG pathway analysis showed that AtRH17 might be involved in metabolic pathways, biosynthesis of secondary metabolites, plant hormone signal transduction, and MAPK signaling pathway via the regulation of downstream genes functioning in these pathways (Figure 2 and Figure 3).
Previously, we reported that the expression levels of the well-known ABA-dependent and ABA-independent salt stress-responsive genes such as RD29A, RAB18, RD29B, RD22, COR47, DREB2A, and DREB2B were not higher in AtRH17 OXs than in WT under salt stress condition [17]. Using RNA-Seq analysis, we confirmed our previous results that the expression levels of these seven genes were not significantly increased in AtRH17 OXs (data not shown), indicating that the salt-tolerance in AtRH17 OXs is mediated through pathway(s) other than the well-known ABA-dependent and/or ABA-independent stress-responsive pathways.
Analysis of the upregulated genes in AtRH17 OXs using GO annotation and Genevestigator revealed that nine genes, namely, LEA4-5, GSTF6, DIN2/BGLU30, TSPO, GSTF7, LEA18, HAI1, ABR, and LTI30, are salt stress-responsive genes and might be involved in the salt-tolerance in AtRH17 OXs (Table 5). These nine genes are tightly inter-connected (Figure 8). Among these, LEA4-5, TSPO, and ABR were selected for quantitative RT-PCR analysis because their expression increased under all abiotic stress conditions examined in Genevestigator analysis and they showed high expression levels in AtRH17 OXs (Figure 6 and Table 5). Three genes showed higher expression levels in AtRH17 OXs than in WT under salt stress conditions (Figure 7). LEA4-5 is a member of LEA proteins. The C-terminal region of LEA4-5 is responsible for its antioxidant activity and in the scavenging of metal ions under stress conditions, whereas the N-terminal can function as a chaperone in the folding of enzyme proteins and in preventing their unfolding that results in protection of the enzyme activity [19,30]. TSPO, belonging to the Trp-rich sensory protein/peripheral-type benzodiazepine receptor group protein, interacts with and regulates PIP2;7, a plasma membrane aquaporin, in the endoplasmic reticulum and Golgi membrane during abiotic stress conditions [20,21]. The interaction between TSPO and PIP2;7 triggers the reduction of PIP2;7 channels present in the plasma membrane and reduces the water transport activity [20,21]. ABR is a Ser/Thr kinase and interacts with phospholipase D-derived phosphatidic acid (PA) [31,32]. ABR increases the ABR-dependent phosphorylation of PIN2, which activates auxin efflux, alters auxin accumulation, and promotes root growth under salt stress conditions [31,32]. T-DNA insertional mutants of ABR are hypersensitive to salt stress in primary root elongation and do not respond to PA [31,32]. These results suggest that the upregulated LEA4-5, TSPO, and ABR confer salt stress-tolerance in AtRH17 OXs.
Taken together, our results demonstrate that AtRH17 OXs exhibit salt stress-tolerance through a novel salt stress-responsive pathway involving LEA4-5, GSTF6, DIN2/BGLU30, TSPO, GSTF7, LEA18, HAI1, ABR, and LTI30, other than the well-known ABA-dependent and ABA-independent pathways.
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
All plant materials used in this study were of the Arabidopsis thaliana accession Col-0 background. Sterilized seeds were incubated in the dark for 2–3 days at 4 °C and then germinated, grown on half-strength Murashige and Skoog (MS) agar plates, supplemented with 1.5% (w/v) sucrose and B5 vitamin. Seedlings were grown under short-day (SD) conditions (8 h light:16 h dark photoperiod) at 22 °C.
4.2. Plant Stress Treatment for Quantitative RT-PCR
For the analysis of salt stress-responsive gene expression under salt stress conditions, 10-day-old WT and AtRH17 OX seedlings grown under SD conditions were placed on filter papers soaked in MS solution containing 150 mM NaCl. After 0, 1, 2, 4, and 8 h, the seedlings were harvested; seedlings harvested at 0 h were used as the control.
4.3. RNA Isolation and First Strand cDNA Synthesis
Total RNA was extracted using an RNAqueous RNA Isolation Kit (Invitrogen, Carlsbad, CA, USA), supplemented with Plant RNA Isolation Aid (Invitrogen, Carlsbad, CA, USA), according to the manufacturers’ instructions. Two micrograms of total RNA was reverse-transcribed in a total volume of 25 μL containing 0.5 μg of oligo dT primer, 0.5 mM dNTPs, and 200 units of Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase (Promega, Madison, WI, USA).
4.4. Quantitative RT-PCR
Quantitative RT-PCR analysis was performed using 2× POWER SYBR Green PCR Master mix (Applied Biosystems, Foster, CA, USA) with diluted cDNA as the template. The Ct (cycle at the threshold) value was set to be constant throughout the study and corresponded to the log-linear range of PCR amplification. The normalized amount of target reflected the relative amount of target transcripts with respect to that of the endogenous reference gene, GAPc. The amplification conditions included an initial denaturation at 95 °C for 10 min, followed by repeated cycles at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s. The primers used are listed in Supplementary Table S1.
4.5. Library Preparation and RNA-Sequencing
The RNA quality was assessed with Agilent 2100 bioanalyzer using the RNA 6000 Nano Chip (Agilent Technologies, Santa Clara, CA, USA), and RNA quantification was performed using a ND-2000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA). For the control and test RNAs, a library was constructed using QuantSeq 3′ mRNA-Seq Library Prep Kit (Lexogen, Inc., Vienna, Austria), according to the manufacturer’s instructions. In brief, 500 ng total RNA was hybridized to an oligo dT primer containing an Illumina-compatible sequence at its 5′-end and reverse transcription was performed. After degradation of the RNA template, second strand synthesis was initiated using a random primer containing an Illumina-compatible linker sequence at its 5′-end. The double-stranded library was purified of the reaction components by magnetic separation. The library was amplified to add the complete adapter sequences required for cluster generation, and the finished library was purified from the PCR components. High-throughput single-end 75 sequencing was performed using NextSeq 500.
4.6. RNA-Seq Data Analysis
QuantSeq 3′ mRNA-Seq reads were aligned using Bowtie2 [33]. Bowtie2 indices were either generated from the genome assembly sequence or representative transcript sequences for aligning to the genome and transcriptome. The alignment file was used for assembling the transcripts, estimating their abundance, and detecting the differential expression of genes. DEGs were determined based on the counts from unique and multiple alignments using coverage in Bedtools [34]. The Read Count data were processed based on the quantile normalization method employing EdgeR within R [35] using Bioconductor [36]. GO annotation enrichment was performed using DAVID [37] with default parameters. KEGG pathway analysis was conducted using KEGG Mapper [38] Gene clustering was performed using MeV ver. 4.9.0 [39]. Protein network analysis was performed using String Apps of Cytoscape ver. 3.7.2 [40].
4.7. Statistical Analysis
Statistical analysis was performed using one-way analysis of variance and Dunnett’s post-hoc test, as implemented in IBM SPSS v.23 (IBM Corp., Armonk, NY, USA).
Supplementary Materials
Supplementary materials can be found at https://www.mdpi.com/1422-0067/21/5/1595/s1.
Author Contributions
Conceptualization, H.-Y.S. and L.V.N.; Methodology, H.-Y.S. and L.V.N.; Software, H.-Y.S. and L.V.N.; Validation, H.-Y.S. and L.V.N.; Formal analysis, H.-Y.S. and L.V.N.; Investigation, H.-Y.S. and L.V.N.; Writing—original draft preparation, H.-Y.S. and L.V.N.; Writing—review and editing, H.-Y.S., S.-Y.L., and D.V.N.; Supervision, Y.-H.M.; Project administration, Y.-H.M.; Funding acquisition, Y.-H.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2018R1A2B6006472 and No. 2017R1D1A1B03034337).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| ABA | Abscisic acid |
| DAG | Days after germination |
| DEG | Differentially expressed gene |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LD | Long-day |
| MS | Murashige and Skoog |
| OX | Overexpressing transgenic plant |
| RH | RNA helicase |
| SD | Short-day |
References
- Aubourg, S.; Kreis, M.; Lecharny, A. The DEAD box RNA helicase family in Arabidopsis thaliana. Nucleic Acids Res. 1999, 27, 628–636. [Google Scholar] [CrossRef]
- Cordin, O.; Banroques, J.; Tanner, N.K.; Linder, P. The DEAD-box protein family of RNA helicases. Gene 2006, 367, 17–37. [Google Scholar] [CrossRef]
- Linder, P.; Rocak, S. DEAD-box proteins: The driving forces behind RNA metabolism. Nat. Rev. Mol. Cell Biol. 2004, 5, 232–241. [Google Scholar]
- Linder, P.; Jankowsky, E. From unwinding to clamping—The DEAD box RNA helicase family. Nat. Rev. Mol. Cell. Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Fuller-Pace, F.V. DExD/H box RNA helicases: Multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 2006, 34, 4206–4215. [Google Scholar] [CrossRef]
- Nawaz, G.; Kang, H. Chloroplast- or mitochondria-targeted DEAD-box RNA helicases play essential roles in organellar RNA metabolism and abiotic stress responses. Front. Plant Sci. 2017, 8, 871. [Google Scholar] [CrossRef] [PubMed]
- Iost, I.; Dreyfus, M. DEAD-box RNA helicases in Escherichia coli. Nucleic Acids Res. 2006, 34, 4189–4197. [Google Scholar] [CrossRef]
- Martin, R.; Straub, A.U.; Doebele, C.; Bohnsack, M.T. DExD/H-box RNA helicases in ribosome biogenesis. RNA Biol. 2013, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Giraud, C.; Hausmann, S.; Lemeille, S.; Prados, J.; Redder, P.; Linder, P. The C-terminal region of the RNA helicase CshA is required for the interaction with the degradosome and turnover of bulk RNA in the opportunistic pathogen Staphylococcus aureus. RNA Biol. 2015, 12, 658–674. [Google Scholar] [CrossRef]
- Liu, Y.; Imai, R. Function of plant DExD/H-box RNA helicases associated with ribosomal RNA biogenesis. Front. Plant Sci. 2018, 9, 125. [Google Scholar] [CrossRef]
- Baruah, I.; Debbarma, J.; Boruah, H.P.D.; Keshavaiah, C. The DEAD-box RNA helicases and multiple abiotic stresses in plants: A systematic review of recent advantages and challenges. Plant Omics J. 2017, 10, 252–262. [Google Scholar] [CrossRef]
- Huang, C.; Shen, Y.; Huang, L.; Wu, S.; Yeh, C.; Lu, C. The DEAD-box RNA helicase AtRH7/PRH75 participates in pre-rRNA processing, plant development and cold tolerance in Arabidopsis. Plant Cell Physiol. 2016, 57, 174–191. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Dong, C.; Lee, H.; Zhu, J.; Xiong, L.; Gong, D.; Stevenson, B.; Zhu, J.K. A DEAD box RNA helicase is essential for mRNA export and important for development and stress responses in Arabidopsis. Plant Cell 2005, 17, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Wu, J.; Zhang, Y.; Jiang, C.; Liu, R.; Chai, C.; Zhu, J. A DEAD box RNA helicase is critical for pre-mRNA splicing, cold-responsive gene regulation, and cold tolerance in Arabidopsis. Plant Cell 2013, 25, 342–356. [Google Scholar] [CrossRef]
- Kant, P.; Kant, S.; Gordon, M.; Shaked, R.; Barak, S. Stress Response Suppressor1 and Stress Response Suppressor2, two DEAD-box RNA helicases that attenuate Arabidopsis responses to multiple abiotic stresses. Plant Physiol. 2007, 145, 814–830. [Google Scholar] [CrossRef]
- Khan, A.; Garbelli, A.; Grossi, S.; Florentin, A.; Batelli, G.; Acuna, T.; Zolla, G.; Kaye, Y.; Paul, L.K.; Zhu, J.K.; et al. The Arabidopsis STRESS RESPONSE SUPPRESSOR DEAD-box RNA helicases are nucleolar- and chromocenter-localized proteins that undergo stress-mediated relocalization and are involved in epigenetic gene silencing. Plant J. 2014, 79, 28–43. [Google Scholar] [CrossRef]
- Nguyen, V.L.; Seok, H.Y.; Woo, D.H.; Lee, S.Y.; Moon, Y.H. Overexpression of the DEAD-box RNA helicase gene AtRH17 confers tolerance to salt stress in Arabidopsis. Int. J. Mol. Sci. 2018, 19, 3777. [Google Scholar] [CrossRef]
- Candat, A.; Paszkiewicz, G.; Neveu, M.; Gautier, R.; Logan, D.C.; Avelange-Macherel, M.H.; Macherel, D. The ubiquitous distribution of late embryogenesis abundant proteins across cell compartments in Arabidopsis offers tailored protection against abiotic stress. Plant Cell 2014, 26, 3148–3166. [Google Scholar] [CrossRef]
- Cuevas-Velazquez, C.L.; Saab-Rincón, G.; Reyes, J.L.; Covarrubias, A.A. The unstructured N-terminal region of Arabidopsis group 4 Late Embryogenesis Abundant (LEA) proteins is required for folding and for chaperone-like activity under water deficit. J. Biol. Chem. 2016, 291, 10893–10903. [Google Scholar] [CrossRef]
- Hachez, C.; Veljanovski, V.; Reinhardt, H.; Guillaumot, D.; Vanhee, C.; Chaumont, F.; Batoko, H. The Arabidopsis abiotic stress-induced TSPO-related protein reduces cell-surface expression of the aquaporin PIP2;7 through protein-protein interactions and autophagic degradation. Plant Cell 2014, 26, 4974–4990. [Google Scholar] [CrossRef]
- Jurkiewicz, P.; Melser, S.; Maucourt, M.; Ayeb, H.; Veljanovski, V.; Maneta-Peyret, L.; Hooks, M.; Rolin, D.; Moreau, P.; Batoko, H. The multistress-induced Translocator protein (TSPO) differentially modulates storage lipids metabolism in seeds and seedlings. Plant J. 2018, 96, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kang, H. Emerging roles of RNA-binding proteins in plant growth, development, and stress responses. Mol. Cells 2016, 39, 179–185. [Google Scholar] [PubMed]
- Stonebloom, S.; Burch-Smith, T.; Kim, I.; Meinke, D.; Mindrinoss, M.; Zambryski, P. Loss of the plant DEAD-box protein ISE1 leads to defective mitochondria and increased cell-to-cell transport via plasmodesmata. Proc. Natl. Acad. Sci. USA 2009, 106, 17229–17234. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Huang, L.; Huang, J.; Wu, S.; Yeh, C.; Lu, C. A DEAD-box protein, AtRH36, is essential for female gametophyte development and is involved in rRNA biogenesis in Arabidopsis. Plant Cell Physiol. 2010, 51, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.; Chen, Y.; Hsiao, Y.; Wang, B.; Lin, S.; Cheng, W.; Jauh, G.; Harada, J.J.; Wang, C. AtRH57, a DEAD-box RNA helicase, is involved in feedback inhibition of glucose-mediated abscisic acid accumulation during seedling development and additively affects pre-ribosomal RNA processing with high glucose. Plant J. 2014, 77, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Wold, B.; Myers, R.M. Sequence census methods for functional genomics. Nat. Methods 2008, 5, 19–21. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Miao, X.; Luo, Q. Genome-wide transcriptome analysis between small-tail Han sheep and the Surabaya fur sheep using high-throughput RNA sequencing. Reproduction 2013, 145, 587–596. [Google Scholar] [CrossRef]
- French-Pacheco, L.; Cuevas-Velazquez, C.L.; Rivillas-Acevedo, L.; Covarrubias, A.A.; Amero, C. Metal-binding polymorphism in late embryogenesis abundant protein AtLEA4-5, an intrinsically disordered protein. Peer J. 2018, 6, e4930. [Google Scholar] [CrossRef]
- Wang, P.; Shen, L.; Guo, J.; Jing, W.; Qu, Y.; Li, W.; Bi, R.; Xuan, W.; Zhang, Q.; Zhang, W. Phosphatidic acid directly regulates PINOID-dependent phosphorylation and activation of the PIN-FORMED2 auxin efflux transporter in response to salt stress. Plant Cell 2019, 31, 250–271. [Google Scholar] [CrossRef] [PubMed]
- Saini, K.; AbdElgawad, H.; Markakis, M.N.; Schoenaers, S.; Asard, H.; Prinsen, E.; Beemster, G.T.S.; Vissenberg, K. Perturbation of auxin homeostasis and signaling by PINOID overexpression induces stress responses in Arabidopsis. Front. Plant Sci. 2017, 8, 1308. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Georges, N.T.; Walter, Z.; Siméon, F. Frequentist model averaging and applications to Bernoulli trials. Open J. Stat. 2016, 6, 545–553. [Google Scholar]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef]
- Howe, E.A.; Sinha, R.; Schlauch, D.; Quackenbush, J. RNA-Seq analysis in MeV. Bioinformatics 2011, 27, 3209–3210. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).