A Rare Mutation in The APOB Gene Associated with Neurological Manifestations in Familial Hypobetalipoproteinemia

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
Family Description
2. Results
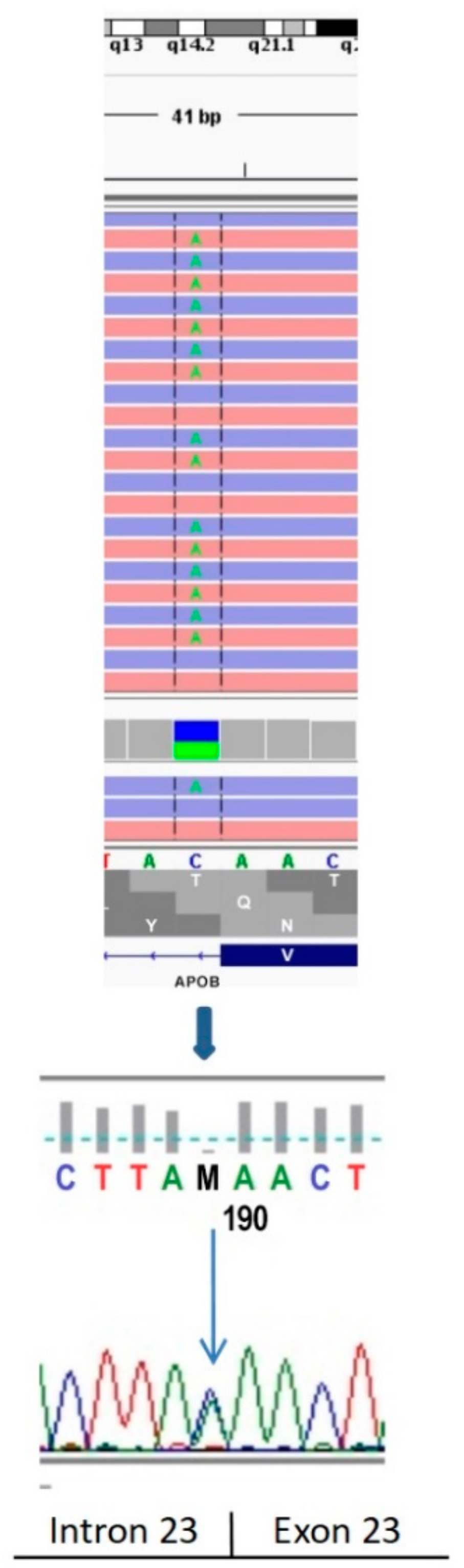
2.1. Genetic Testing
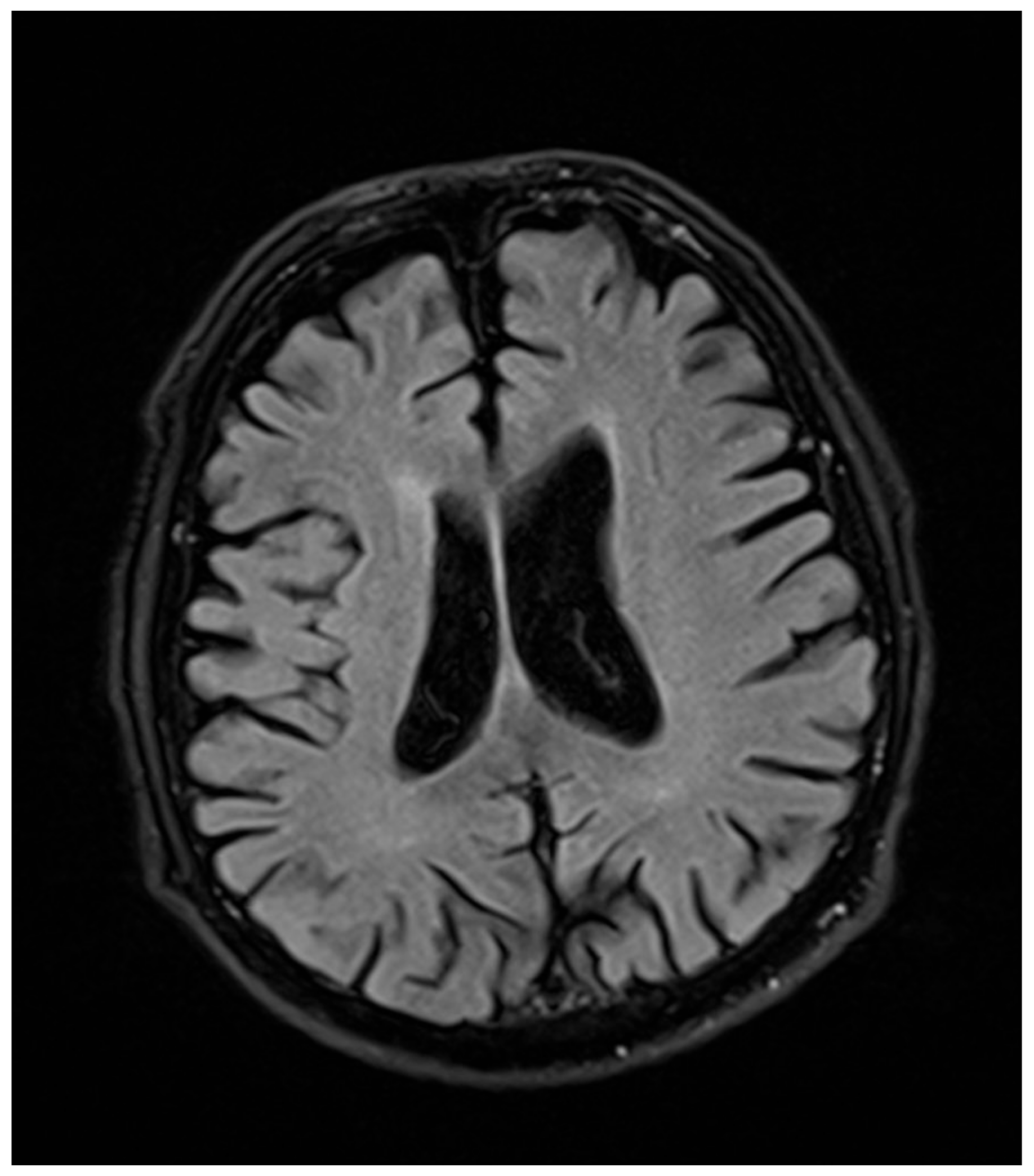
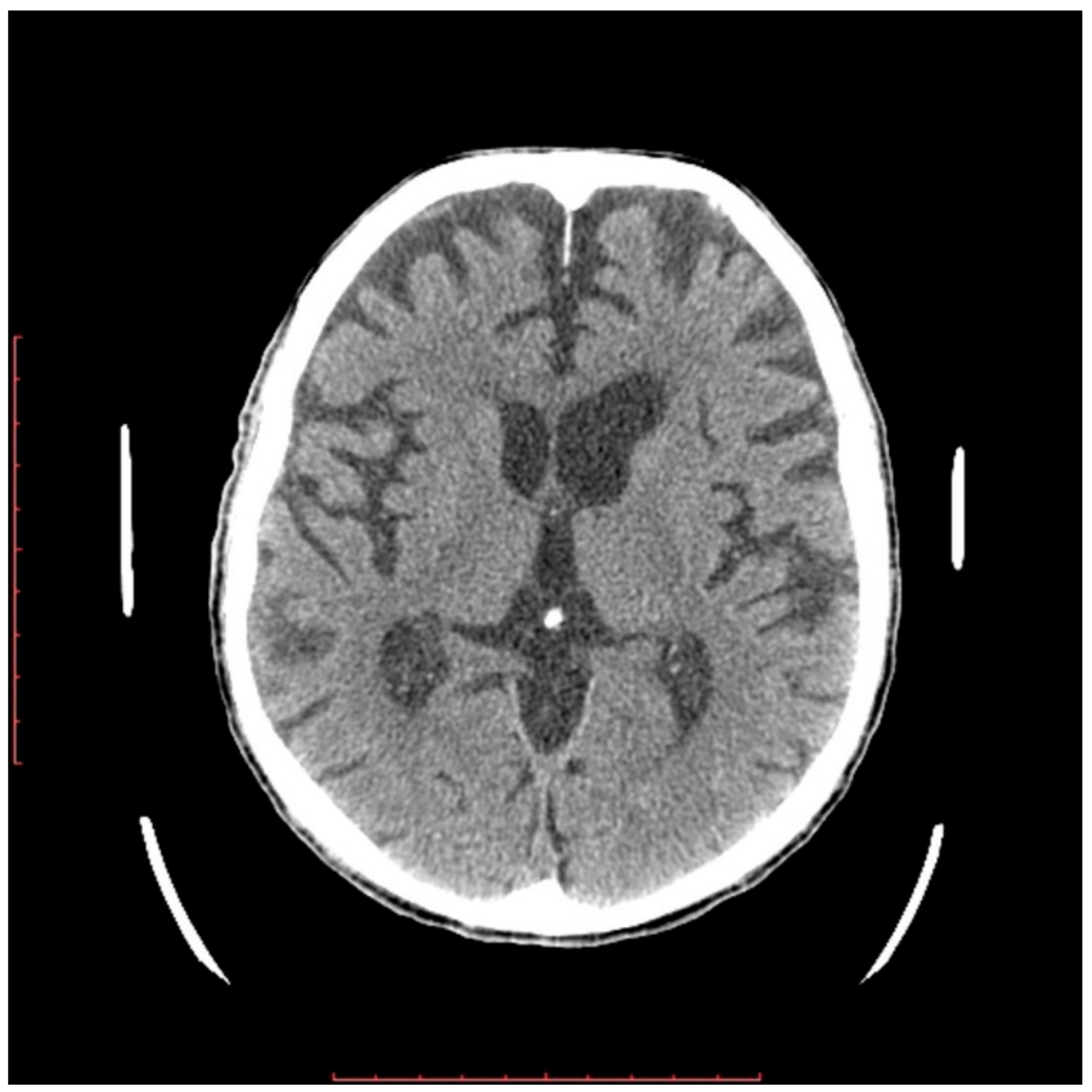
2.2. Neurological Examination
2.3. Ophthalmological Examination
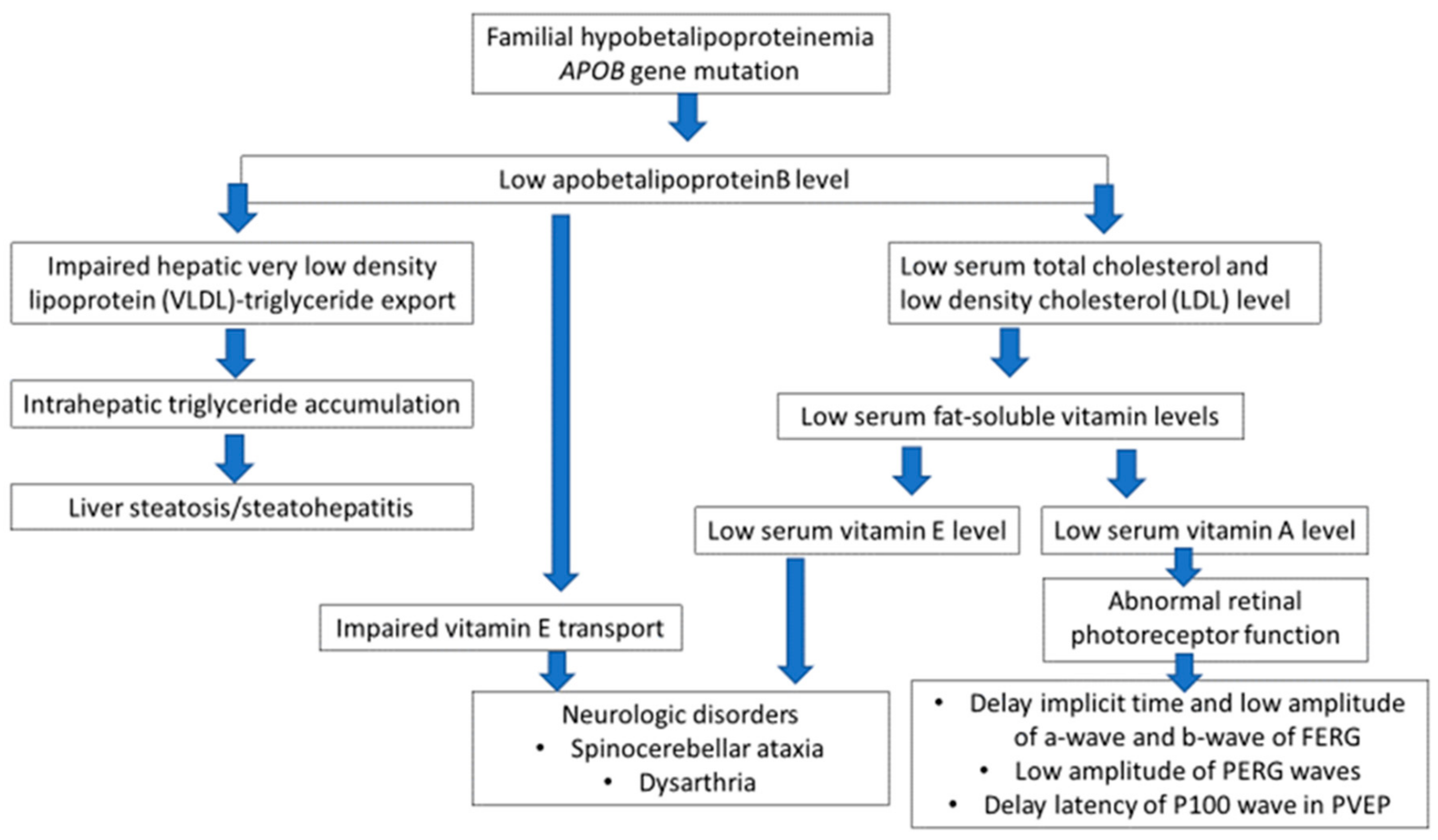
3. Discussion
4. Materials and Methods
4.1. Genetic Testing
4.2. The Neurological Conditions
4.3. The Ophthalmological Examination
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FHBL | Familial hypobetalipoproteinemia |
| APOB | Apolipoprotein B |
| NAFLD | Nonalcoholic fatty liver disease |
| ABL | Abetalipoproteinemia |
| NASH | Nonalcoholic steatohepatitis |
| MR | Magnetic resonance |
| CT | Computed tomography |
| TC | Total cholesterol |
| LDL | Low-density lipoprotein |
| TG | Triglycerides |
| DOB | Date of birth |
| NGS | Next-generation sequencing |
| HGVS | Human Genome Variation Society |
| OCT | Optical coherence tomography |
| PERG | Pattern electroretinography |
| PVEP | Pattern visual evoked potentials |
| ACMG | American College of Medical Genetics and Genomics |
| ERG | Electroretinography |
| LDLR | Low-density lipoprotein receptor |
| HGMD | Human Gene Mutation Database |
| UPDRS | Unified Parkinson’s Disease Rating Scale |
| BCVA | Best-corrected visual acuity |
| FERG | Flash full-field electroretinography |
| ISCEV | International Society for Clinical Electrophysiology of Vision |
References
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, G.; Patterson, B.W.; Yablonskiy, D.A.; Tanoli, T.S.K.; Averna, M.; Elias, N.; Yue, P.; Ackerman, J. Fatty liver in familial hypobetalipoproteinemia: Triglyceride assembly into VLDL particles is affected by the extent of hepatic steatosis. J. Lipid Res. 2003, 44, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, A.J.; Barrett, H.R.; Robertson, K.; Havlat, M.F.; van Bockxmeer, F.M.; Burnett, J.R. Liver dysfunction and steatosis in familial hypobetalipoproteinemia. Clin. Chem. 2005, 51, 266–269. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lam, M.; Singham, J.; Hegele, R.A.; Riazy, M.; Hiob, M.A.; Urs, G.F.; Steinbrecher, P. Familial hypobetalipoproteinemia-induced nonalcoholic steatohepatitis. Case Rep. Gastroenterol. 2012, 6, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Familial hypobetalipoproteinemia. Available online: https://ghr.nlm.nih.gov/condition/familial-hypobetalipoproteinemia (accessed on 3 November 2018).
- Lee, J.; Hegele, R. Abetalipoproteinemia and homozygous hypobetalipoproteinemia: A framework for diagnosis and management. J. Inherit. Metab. Dis. 2014, 3, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Welty, F.K. Hypobetalipoproteinemia and abetalipoproteinemia. Curr. Opin. Lipidol. 2014, 25, 161–168. [Google Scholar] [CrossRef]
- Tarugi, P.; Averna, M.; Di Leo, E.; Cefalù, A.B.; Noto, D.; Magnolo, L.; Cattin, L.; Bertolini, S.; Calandra, S. Molecular diagnosis of hypobetalipoproteinemia: An ENID review. Atherosclerosis 2007, 195, 19–27. [Google Scholar] [CrossRef]
- Cefalù, A.B.; Pirruccello, J.P.; Noto, D.; Gabriel, S.; Valenti, V.; Gupta, N.; Spina, R.; Tarugi, P.; Kathiresan, S.; Averna, M.R. A novel APOB mutation identified by exome sequencing cosegregates with steatosis, liver cancer, and hypocholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2013, 8, 2021–2025. [Google Scholar] [CrossRef]
- Schonfeld, G. Familial hypobetalipoproteinemia: A review. J. Lipid Res. 2003, 44, 878–883. [Google Scholar] [CrossRef]
- Fu, J.; Kwok, S.; Sinai, L.; Abdel-Razek, O.; Babula, J.; Chen, D.; Farago, E.; Fernandopulle, N.; Leith, S.; Loyzer, M.; et al. Western Database of lipid variants (WDLV): A catalogue of genetic variants in monogenic dyslipidemias. Can. J. Cardiol. 2013, 29, 934–939. [Google Scholar] [CrossRef]
- Wang, L.R.; McIntyre, A.D.; Hegele, R.A. Complex genetic architecture in severe hypobetalipoproteinemia. Lipids Health Dis. 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.; Min, J.L.; Huang, J.; Crooks, L.; Memari, Y.; McCarthy, S.; Perry, J.R.; Xu, C.; Futema, M.; Lawson, D.; et al. The UK10K project identifies rare variants in health and disease. Nature 2015, 526, 82–90. [Google Scholar] [PubMed]
- HGMD. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 30 August 2018).
- UniProtKB. Available online: https://www.uniprot.org/help/uniprotkb (accessed on 30 August 2018).
- Clinvar. Available online: https://www.clinvar.com/ (accessed on 30 August 2018).
- IGSR: The International Genome Sample Resource. Available online: https://www.internationalgenome.org/ (accessed on 30 August 2018).
- Boguszewska-Chachulska, A.; Matczynska, M.; Lyszkiewicz, E.; Krawczyk, M.; Kowalczyk, D.; Szymanczak, R.; Piotrowska, A.; Mossakowska, M.; Puzianowska-Kuznicka, M.; Zagulski, M. Whole genome sequencing of successfully aging long-lived Polish Caucasians and creation of a reference database. Eur. J. Hum. Genetics 2016, 24, 355. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 5, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Cefalù, A.B.; Norata, G.D.; Ghiglioni, D.G.; Noto, D.; Uboldi, P.; Garlaschelli, K.; Baragetti, A.; Spina, R.; Valenti, V.; Pederiva, C.; et al. Homozygous familial hypobetalipoproteinemia: Two novel mutations in the splicing sites of apolipoprotein B gene and review of the literature. Atherosclerosis 2015, 239, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Edgar, P.F.; Hooper, A.J.; Poa, N.R.; Burnett, J.R. Violent behavior associated with hypocholesterolemia due to a novel APOB gene mutation. Mol. Psychiatry 2007, 12, 258–263. [Google Scholar] [CrossRef]
- Rampoldi, L.; Danek, A.; Monaco, A.P. Clinical features and molecular bases of neuroacanthocytosis. J. Mol. Med. 2002, 80, 475–491. [Google Scholar] [CrossRef]
- Holder, G.E.; Brigell, M.G.; Hawlina, M.; Meigen, T.; Vaegan; Bach, M. ISCEV standard for clinical pattern electroretinography—2007 update. Doc. Ophthalmol. 2007, 114, 111–116. [Google Scholar]
- Halliday, A.M.; McDonald, W.I.; Mushin, J. Visual evoked response in diagnosis of multiple sclerosis. Br. Med. J. 1973, 4, 661–664. [Google Scholar] [CrossRef]
- Pojda-Wilczek, D. Retrospective analysis of pattern VEP results in different ocular and systemic diseases. Klin. Oczna 2010, 7, 205–209. [Google Scholar]
- Buonuomo, P.S.; Ruggiero, A.; Valeriani, M.; Mariotti, P. Familial hypobetalipoproteinemia: Early neurological, hematological, and ocular manifestations in two affected twins responding to vitamin supplementation. Curr. Opin. Pediatr. 2009, 21, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Odom, J.V.; Bach, M.; Brigell, M.; Holder, G.E.; McCulloch, D.L.; Tormene, A.P. ISCEV standard for clinical visual evoked potentials (2009 update). Doc. Ophthalmol. 2010, 120, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Bach, M.; Brigell, M.G.; Hawlina, M.; Holder, G.E.; Johnson, M.A.; McCulloch, D.L.; Meigen, T.; Viswanathan, S. ISCEV standard for clinical pattern electroretinography (PERG)—2012 update. Doc. Ophthalmol. 2013, 126, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Fulton, A.B.; Holder, G.E.; Miyake, Y.; Brigell, M.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc. Ophthalmol. 2009, 118, 69–77. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musialik, J.; Boguszewska-Chachulska, A.; Pojda-Wilczek, D.; Gorzkowska, A.; Szymańczak, R.; Kania, M.; Kujawa-Szewieczek, A.; Wojcieszyn, M.; Hartleb, M.; Więcek, A. A Rare Mutation in The APOB Gene Associated with Neurological Manifestations in Familial Hypobetalipoproteinemia. Int. J. Mol. Sci. 2020, 21, 1439. https://doi.org/10.3390/ijms21041439
Musialik J, Boguszewska-Chachulska A, Pojda-Wilczek D, Gorzkowska A, Szymańczak R, Kania M, Kujawa-Szewieczek A, Wojcieszyn M, Hartleb M, Więcek A. A Rare Mutation in The APOB Gene Associated with Neurological Manifestations in Familial Hypobetalipoproteinemia. International Journal of Molecular Sciences. 2020; 21(4):1439. https://doi.org/10.3390/ijms21041439
Chicago/Turabian StyleMusialik, Joanna, Anna Boguszewska-Chachulska, Dorota Pojda-Wilczek, Agnieszka Gorzkowska, Robert Szymańczak, Magdalena Kania, Agata Kujawa-Szewieczek, Małgorzata Wojcieszyn, Marek Hartleb, and Andrzej Więcek. 2020. "A Rare Mutation in The APOB Gene Associated with Neurological Manifestations in Familial Hypobetalipoproteinemia" International Journal of Molecular Sciences 21, no. 4: 1439. https://doi.org/10.3390/ijms21041439
APA StyleMusialik, J., Boguszewska-Chachulska, A., Pojda-Wilczek, D., Gorzkowska, A., Szymańczak, R., Kania, M., Kujawa-Szewieczek, A., Wojcieszyn, M., Hartleb, M., & Więcek, A. (2020). A Rare Mutation in The APOB Gene Associated with Neurological Manifestations in Familial Hypobetalipoproteinemia. International Journal of Molecular Sciences, 21(4), 1439. https://doi.org/10.3390/ijms21041439