TLR4 Stimulation Promotes Human AVIC Fibrogenic Activity through Upregulation of Neurotrophin 3 Production

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
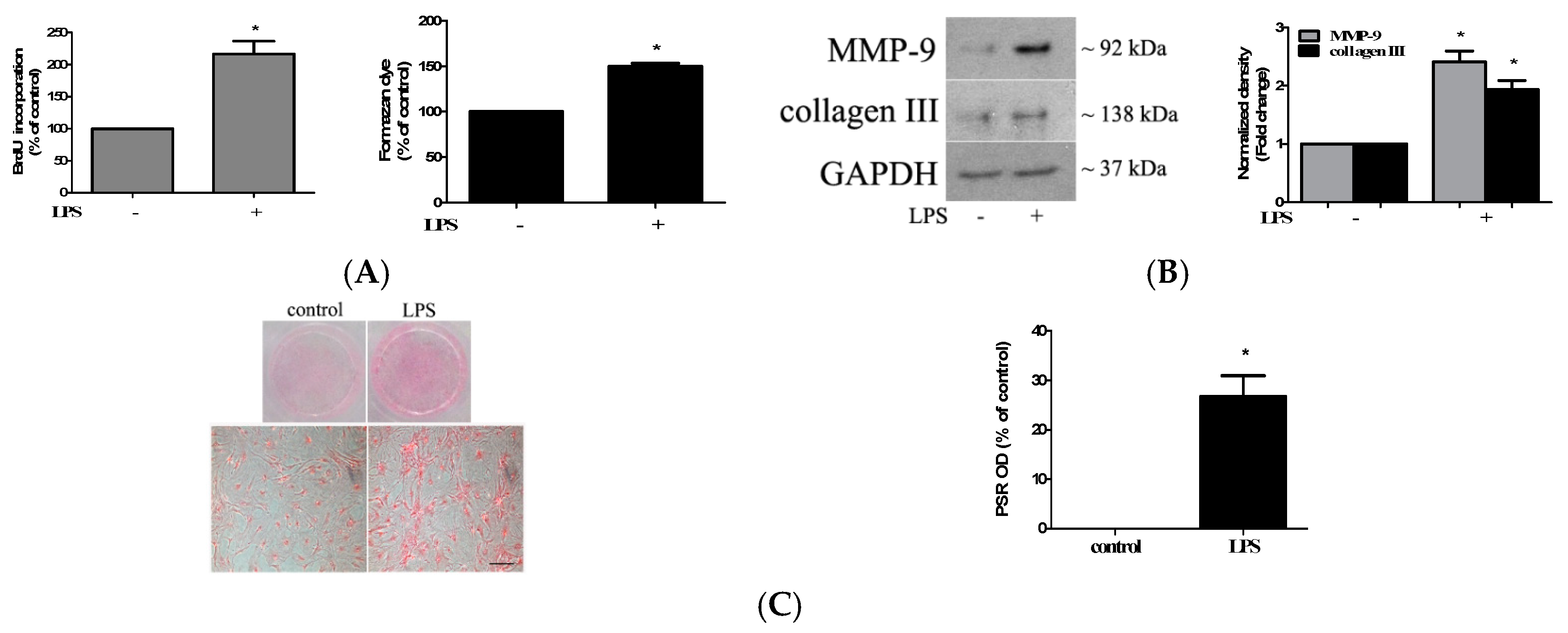
2.1. TLR4 Activation Induces Stronger Fibrogenic Activity in Human AVICs
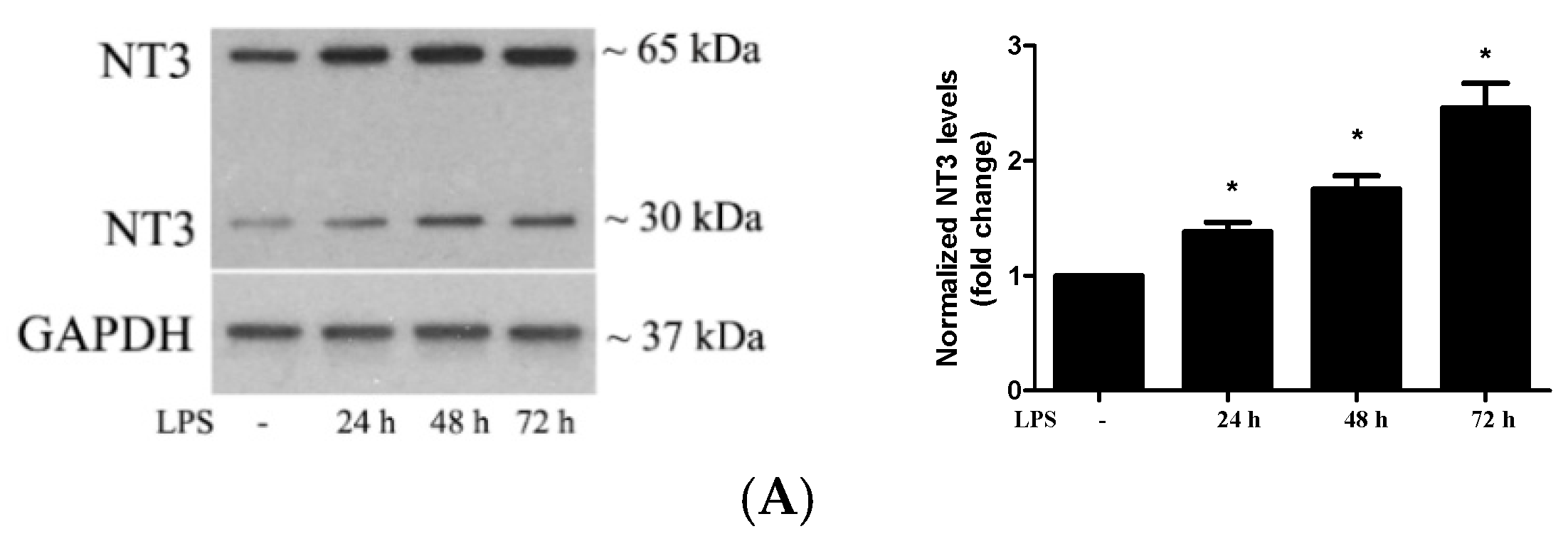
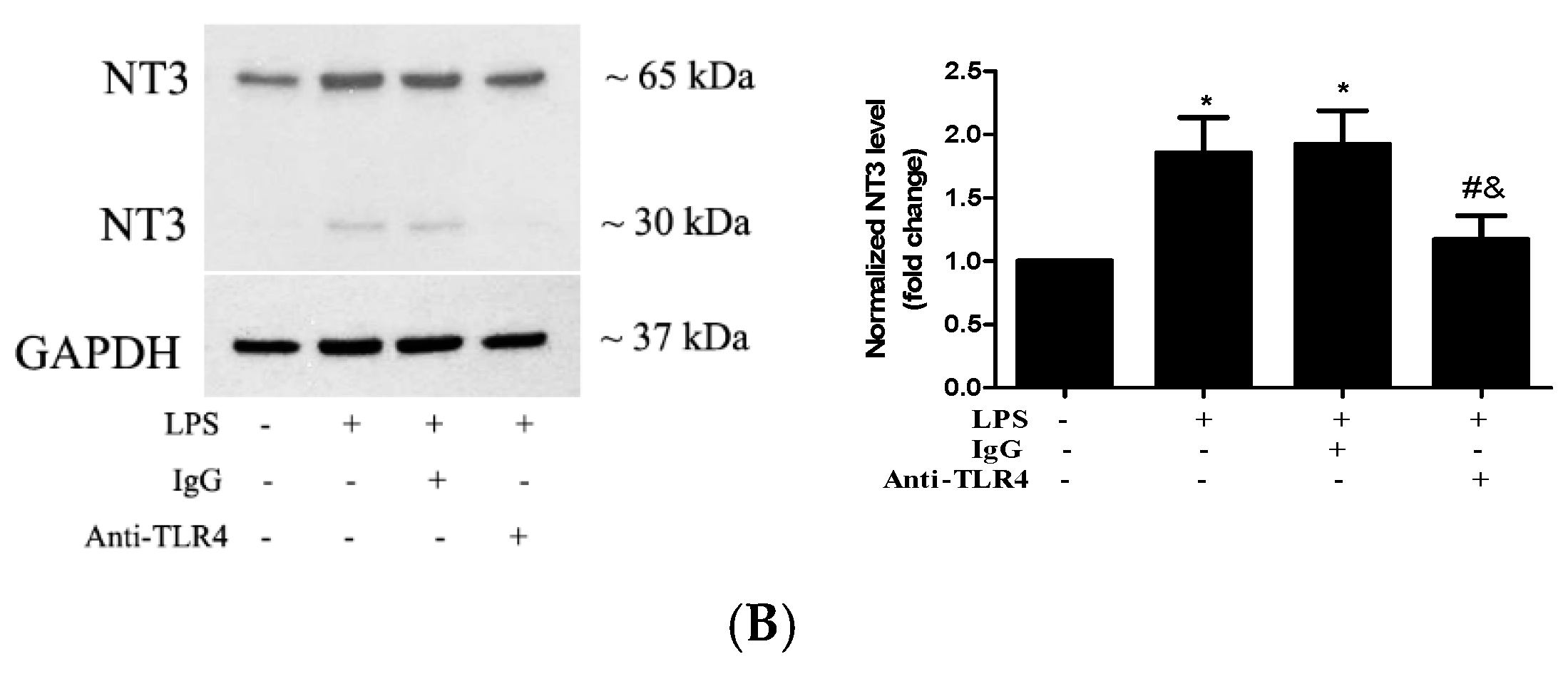
2.2. Stimulation of TLR4 in Human AVICs with LPS Upregulates NT3 Production
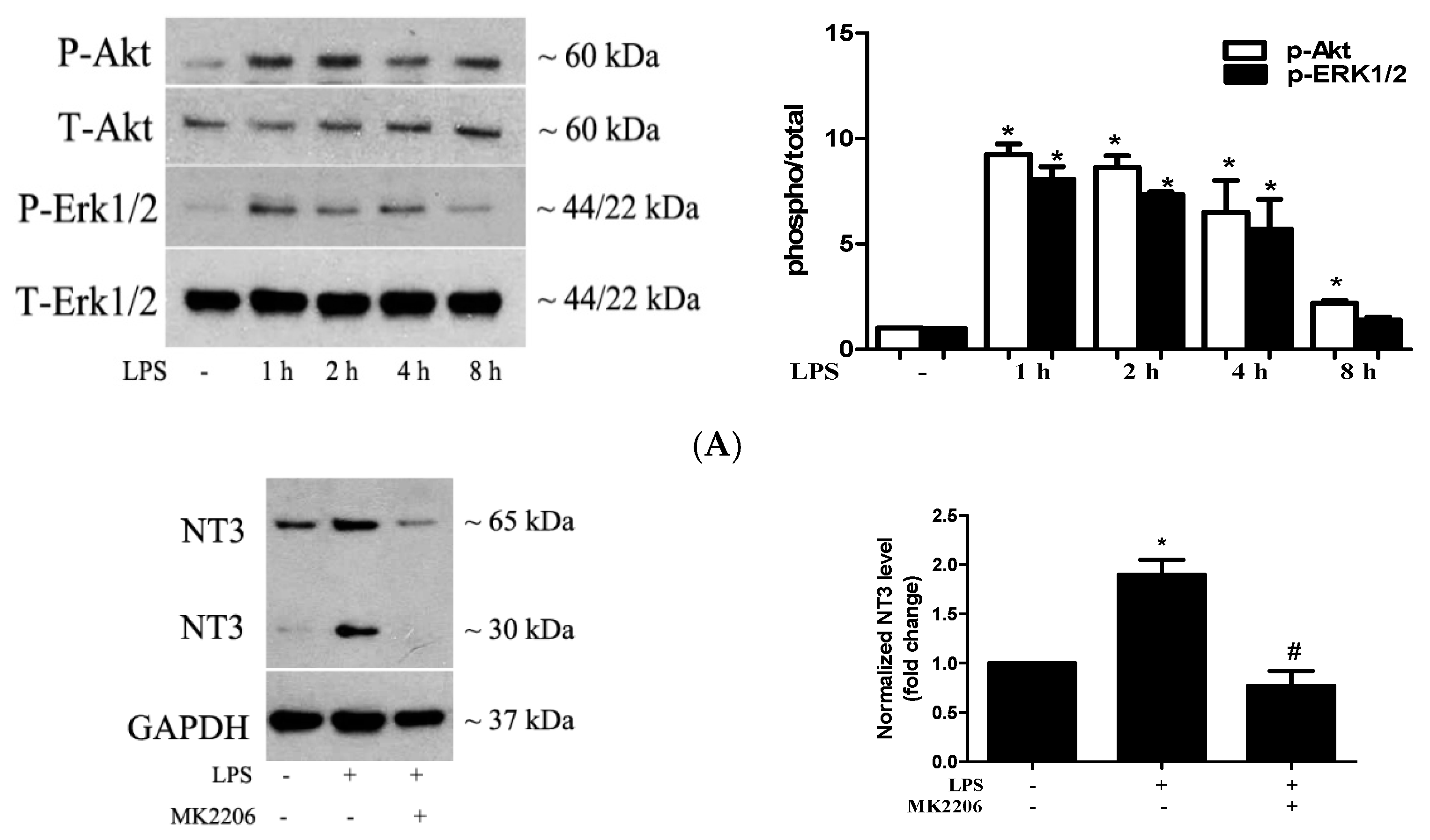
2.3. Akt and ERK1/2 are Involved in the Mechanism by which TLR4 Activation Induces NT3 Production in Human AVICs
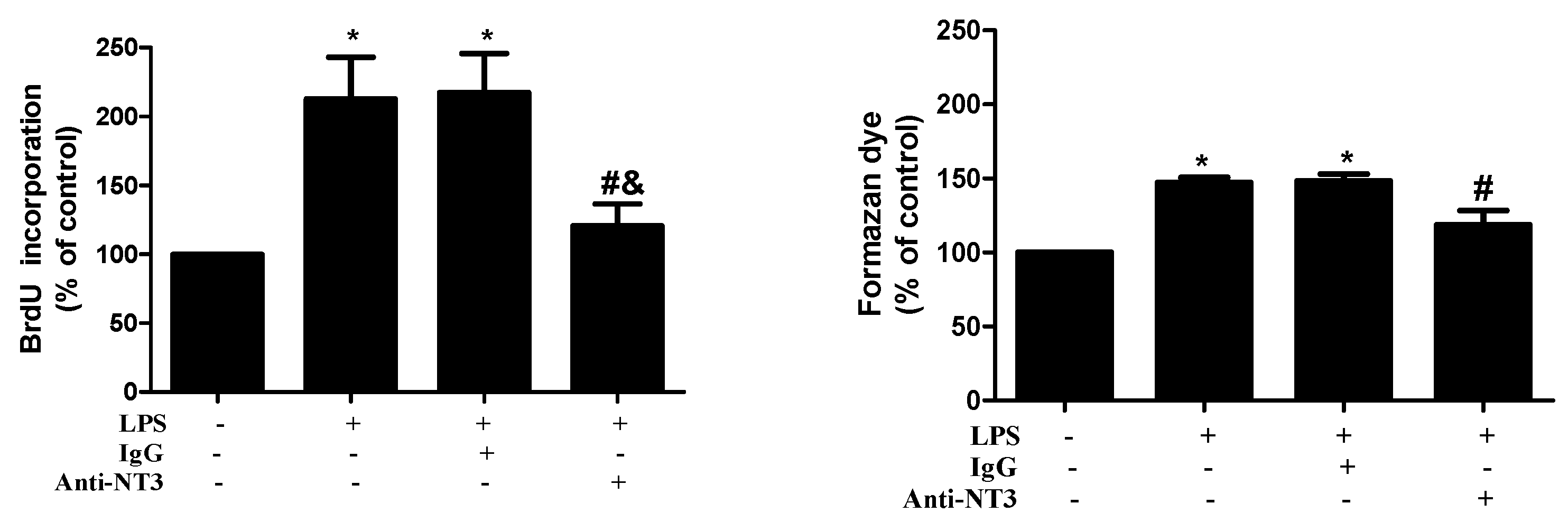
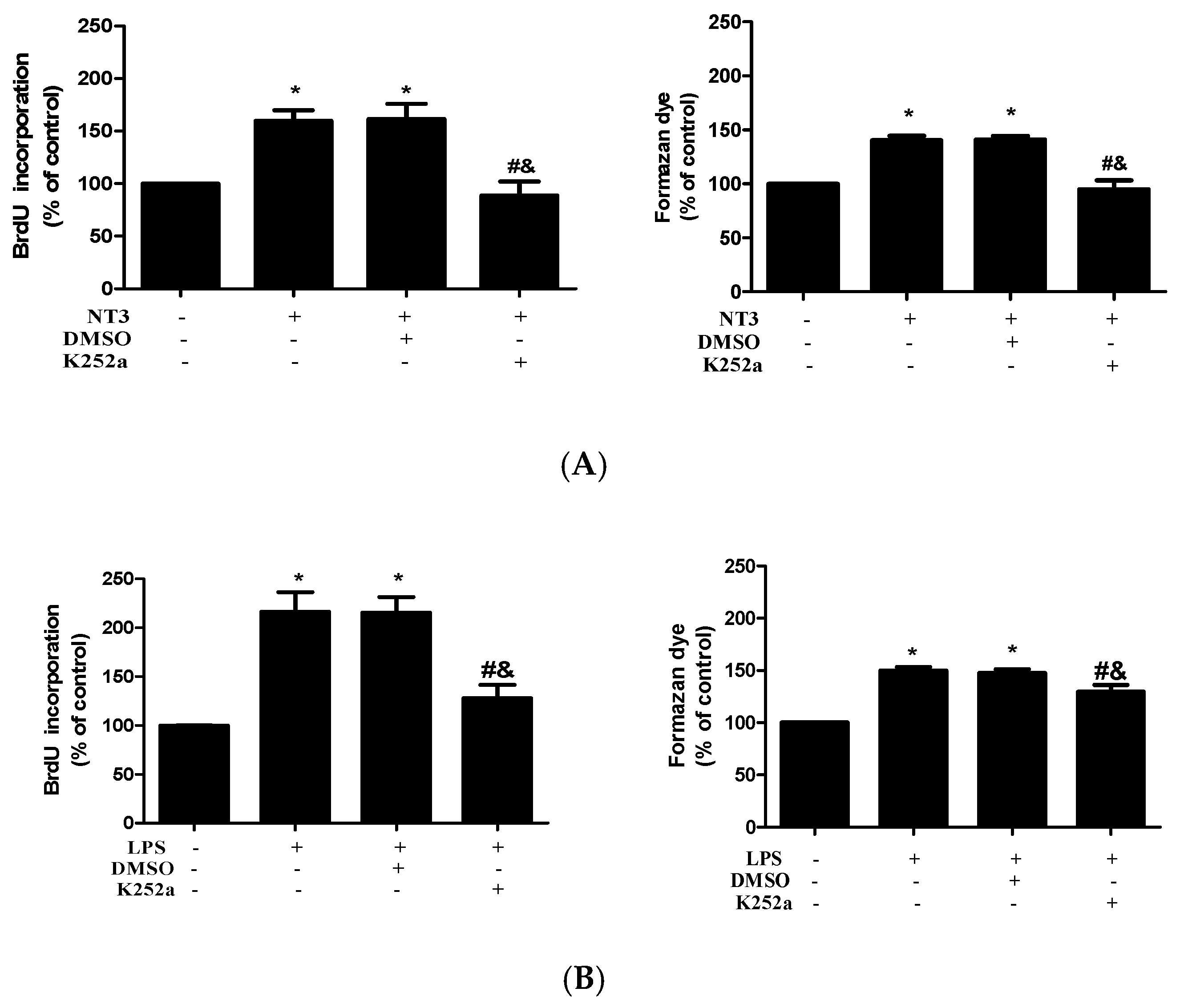
2.4. NT3 Mediates Human AVIC Proliferation Induced by TLR4 Stimulation
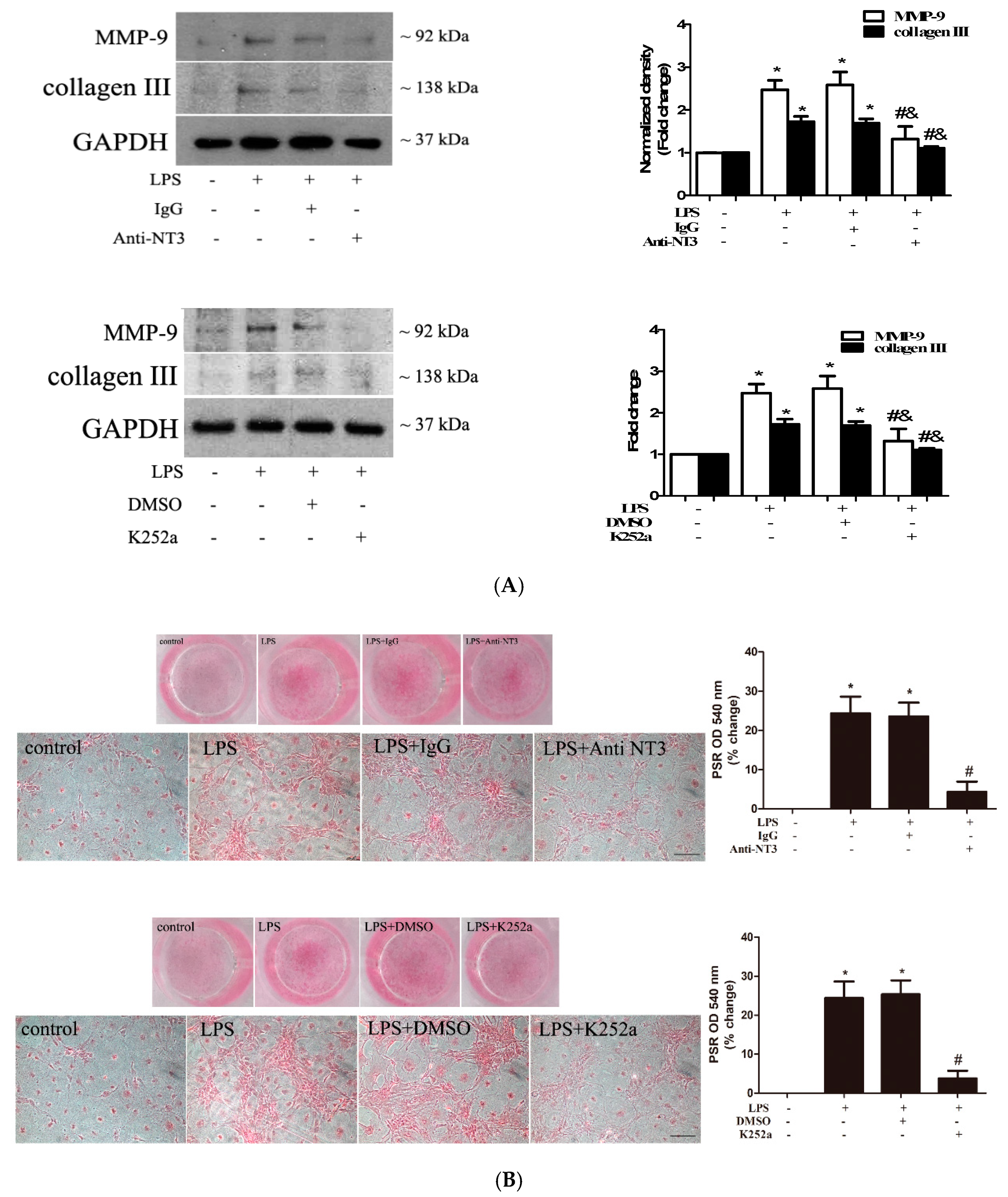
2.5. The NT3-Trk Cascade is Involved in the Mechanism by which TLR4 Stimulation Elevates Fibrogenic Activity in Human AVICs
3. Discussion
4. Materials and Methods
4.1. Material
4.2. Cell Isolation and Culture
4.3. Immunoblotting
4.4. BrdU Proliferation Assay
4.5. CCK-8 Assay
4.6. Picro-Sirius Red Staining
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAVD | calcific aortic valve disease |
| AVIC | aortic valve interstitial cell |
| ECM | extracellular matrix |
| NT3 | neurotrophin 3 |
| LPS | lipopolysaccharide |
| Trk | tropomyosin receptor kinase |
| TLR | toll-like receptor |
| α-SMA | α-smooth muscle action |
| MMP | matrix metallopeptidase |
| ERK1/2 | extracellular signal-regulated protein kinases 1 and 2 |
| DAMPs | damage-associated molecular patterns |
| Ox-LDL | oxidized low-density lipoprotein |
| TIMPs | tissue inhibitors of MMPs |
| BrdU | bromodeoxyuridine/5-bromo-2’-deoxyuridine |
| CCK-8 | cell counting kit-8 |
| PSR | picro-Sirius red |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| SDS | sodium dodecyl sulfate |
| DMSO | dimethyl sulfoxide |
| IgG | immunoglobulin G |
References
- Myasoedova, V.A.; Ravani, A.L.; Frigerio, B.; Valerio, V.; Moschetta, D.; Songia, P.; Poggio, P. Novel pharmacological targets for calcific aortic valve disease: Prevention and treatments. Pharmacol. Res. 2018, 136, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; Howard, V.J.; et al. American Heart Association Statistics, C.; Stroke Statistics, S., Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, e29–e322. [Google Scholar] [PubMed]
- Kwiecinski, J.; Chin, C.W.L.; Everett, R.J.; White, A.C.; Semple, S.; Yeung, E.; Jenkins, W.J.; Shah, A.S.V.; Koo, M.; Mirsadraee, S.; et al. Adverse prognosis associated with asymmetric myocardial thickening in aortic stenosis. Eur. Heart J. Cardiovasc. Imaging 2018, 19, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Kostyunin, A.E.; Yuzhalin, A.E.; Ovcharenko, E.A.; Kutikhin, A.G. Development of calcific aortic valve disease: Do we know enough for new clinical trials? J. Mol. Cell. Cardiol. 2019, 132, 189–209. [Google Scholar] [CrossRef] [PubMed]
- Ohukainen, P.; Ruskoaho, H.; Rysa, J. Cellular mechanisms of valvular thickening in early and intermediate calcific aortic alve disease. Curr. Cardiol. Rev. 2018, 14, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, M.A.; Madhur, M.S.; Merryman, W.D. Adaptive immune cells in calcific aortic valve disease. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H141–H155. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Holmes, A.; Murphy, G.A.; Mishra, K.; Rosenkranz, A.C.; Horowitz, J.D.; Kennedy, J.A. TGF-beta1-Induced MAPK activation promotes collagen synthesis, nodule formation, redox stress and cellular senescence in porcine aortic valve interstitial cells. J. Heart Valve Dis. 2013, 22, 621–630. [Google Scholar]
- Lim, J.; Ehsanipour, A.; Hsu, J.J.; Lu, J.; Pedego, T.; Wu, A.; Walthers, C.M.; Demer, L.L.; Seidlits, S.K.; Tintut, Y. Inflammation drives retraction, stiffening, and nodule formation via cytoskeletal machinery in a three-dimensional culture model of aortic stenosis. Am. J. Pathol. 2016, 186, 2378–2389. [Google Scholar] [CrossRef]
- Babu, A.N.; Meng, X.; Zou, N.; Yang, X.; Wang, M.; Song, Y.; Cleveland, J.C.; Weyant, M.; Banerjee, A.; Fullerton, D.A. Lipopolysaccharide stimulation of human aortic valve interstitial cells activates inflammation and osteogenesis. Ann. Thorac. Surg. 2008, 86, 71–76. [Google Scholar] [CrossRef]
- Meng, X.; Ao, L.; Song, Y.; Babu, A.; Yang, X.; Wang, M.; Weyant, M.J.; Dinarello, C.A.; Cleveland, J.C., Jr.; Fullerton, D.A. Expression of functional toll-like receptors 2 and 4 in human aortic valve interstitial cells: Potential roles in aortic valve inflammation and stenosis. Am. J. Physiol. Cell Physiol. 2008, 294, C29–C35. [Google Scholar] [CrossRef]
- Gonzalez-Guerrero, C.; Cannata-Ortiz, P.; Guerri, C.; Egido, J.; Ortiz, A.; Ramos, A.M. TLR4-mediated inflammation is a key pathogenic event leading to kidney damage and fibrosis in cyclosporine nephrotoxicity. Arch. Toxicol. 2017, 91, 1925–1939. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Z.; Wang, J.P.; Mi, S.; Liu, H.Z.; Cui, B.; Yan, H.M.; Yan, J.; Li, Z.; Liu, H.; Hua, F.; et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am. J. Pathol. 2012, 180, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Chadban, S.J.; Zhao, C.Y.; Chen, X.; Kwan, T.; Panchapakesan, U.; Pollock, C.A.; Wu, H. TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PLoS ONE 2014, 9, e97985. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wei, F.; Wang, J.; Chen, R.; Zhang, J.; Li, D.; Gan, D.; Yang, X.; Zou, Y. Manganese modifies Neurotrophin-3 (NT3) and its tropomyosin receptor kinase C (TrkC) in the cortex: Implications for manganese-induced neurotoxicity. Food Chem. Toxicol. 2019, 135, 110925. [Google Scholar] [CrossRef]
- Su, Y.W.; Zhou, X.F.; Foster, B.K.; Grills, B.L.; Xu, J.; Xian, C.J. Roles of neurotrophins in skeletal tissue formation and healing. J. Cell. Physiol. 2018, 233, 2133–2145. [Google Scholar] [CrossRef]
- Dagnell, C.; Kemi, C.; Klominek, J.; Eriksson, P.; Skold, C.M.; Eklund, A.; Grunewald, J.; Olgart Hoglund, C. Effects of neurotrophins on human bronchial smooth muscle cell migration and matrix metalloproteinase-9 secretion. Transl. Res. 2007, 150, 303–310. [Google Scholar] [CrossRef]
- Palazzo, E.; Marconi, A.; Truzzi, F.; Dallaglio, K.; Petrachi, T.; Humbert, P.; Schnebert, S.; Perrier, E.; Dumas, M.; Pincelli, C. Role of neurotrophins on dermal fibroblast survival and differentiation. J. Cell. Physiol. 2011, 227, 1017–1025. [Google Scholar] [CrossRef]
- Yao, Q.; Song, R.; Ao, L.; Zhan, Q.; Cleveland, J.C., Jr.; Yu, X.; Fullerton, D.A.; Meng, X. Over-expression of neurotrophin 3 in human aortic valves affected by calcific disease induces the osteogenic responses via the Trk-Akt pathway. Biochim. Biophys. Acta 2015, 1852, 1940–1949. [Google Scholar] [CrossRef][Green Version]
- Yao, Q.; Song, R.; Ao, L.; Cleveland, J.C., Jr.; Fullerton, D.A.; Meng, X. Neurotrophin 3 upregulates proliferation and collagen production in human aortic valve interstitial cells: A potential role in aortic valve sclerosis. Am. J. Physiol. Cell Physiol. 2017, 312, C697–C706. [Google Scholar] [CrossRef]
- Rost, B.; Hanf, G.; Ohnemus, U.; Otto-Knapp, R.; Groneberg, D.A.; Kunkel, G.; Noga, O. Monocytes of allergics and non-allergics produce, store and release the neurotrophins NGF, BDNF and NT-3. Regul. Pept. 2005, 124, 19–25. [Google Scholar] [CrossRef]
- Kolbeck, R.; Jungbluth, S.; Barde, Y.A. Characterisation of neurotrophin dimers and monomers. Eur. J. Biochem. 1994, 225, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.C.; Radziejewski, C.; Stuart, D.I.; Jones, E.Y. Structure of the brain-derived neurotrophic factor/neurotrophin 3 heterodimer. Biochemistry 1995, 34, 4139–4146. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Song, R.; Ao, L.; Weyant, M.J.; Lee, J.; Xu, D.; Fullerton, D.A.; Meng, X. Notch1 promotes the pro-osteogenic response of human aortic valve interstitial cells via modulation of ERK1/2 and nuclear factor-kappaB activation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1580–1590. [Google Scholar] [CrossRef]
- Park, S.S.; Lee, Y.J.; Han, H.J.; Kweon, O.K. Role of laminin-111 in neurotrophin-3 production of canine adipose-derived stem cells: Involvement of Akt, mTOR, and p70S6K. J. Cell. Physiol. 2011, 226, 3251–3260. [Google Scholar] [CrossRef] [PubMed]
- Podolec, J.; Baran, J.; Siedlinski, M.; Urbanczyk, M.; Krupinski, M.; Bartus, K.; Niewiara, L.; Podolec, M.; Guzik, T.; Tomkiewicz-Pajak, L.; et al. Serum rantes, transforming growth factor-beta1 and interleukin-6 levels correlate with cardiac muscle fibrosis in patients with aortic valve stenosis. J. Physiol. Pharmacol. 2018, 69, 615–623. [Google Scholar]
- Song, R.; Zeng, Q.; Ao, L.; Yu, J.A.; Cleveland, J.C.; Zhao, K.S.; Fullerton, D.A.; Meng, X. Biglycan induces the expression of osteogenic factors in human aortic valve interstitial cells via Toll-like receptor-2. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Q.; Song, R.; Li, F.; Ao, L.; Zeng, Q.; Xu, D.; Fullerton, D.A.; Meng, X. Double-stranded RNA upregulates the expression of inflammatory mediators in human aortic valve cells through the TLR3-TRIF-noncanonical NF-kappaB pathway. Am. J. Physiol. Cell Physiol. 2017, 312, C407–C417. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Fullerton, D.A.; Su, X.; Ao, L.; Cleveland, J.C., Jr.; Meng, X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J. Am. Coll. Cardiol. 2009, 53, 491–500. [Google Scholar] [CrossRef]
- Klee, S.; Lehmann, M.; Wagner, D.E.; Baarsma, H.A.; Konigshoff, M. WISP1 mediates IL-6-dependent proliferation in primary human lung fibroblasts. Sci. Rep. 2016, 6, 20547. [Google Scholar] [CrossRef]
- Calvier, L.; Chouvarine, P.; Legchenko, E.; Hoffmann, N.; Geldner, J.; Borchert, P.; Jonigk, D.; Mozes, M.M.; Hansmann, G. PPARgamma links BMP2 and TGFbeta1 pathways in vascular smooth muscle cells, regulating cell proliferation and glucose metabolism. Cell Metab. 2017, 25, 1118–1134. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.J.; Piechura, L.M.; Porras, A.M.; Masters, K.S. Manipulation of valve composition to elucidate the role of collagen in aortic valve calcification. BMC Cardiovasc. Disord. 2014, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Mohler, E.R., 3rd; Gannon, F.; Reynolds, C.; Zimmerman, R.; Keane, M.G.; Kaplan, F.S. Bone formation and inflammation in cardiac valves. Circulation 2001, 103, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Karin, D.; Koyama, Y.; Brenner, D.; Kisseleva, T. The characteristics of activated portal fibroblasts/myofibroblasts in liver fibrosis. Differentiation 2016, 92, 84–92. [Google Scholar] [CrossRef]
- Li, F.; Song, R.; Ao, L.; Reece, T.B.; Cleveland, J.C., Jr.; Dong, N.; Fullerton, D.A.; Meng, X. ADAMTS5 deficiency in calcified aortic alves is associated with elevated pro-osteogenic activity in valvular interstitial cells. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1339–1351. [Google Scholar] [CrossRef]
- Cheng, H.; Yao, Q.; Song, R.; Zhai, Y.; Wang, W.; Fullerton, D.A.; Meng, X. Lysophosphatidylcholine activates the Akt pathway to upregulate extracellular matrix protein production in human aortic valve cells. J. Surg. Res. 2017, 213, 243–250. [Google Scholar] [CrossRef]
- Rankin, A.C.; Hendry, B.M.; Corcoran, J.P.; Xu, Q. An in vitro model for the pro-fibrotic effects of retinoids: Mechanisms of action. Br. J. Pharmacol. 2013, 170, 1177–1189. [Google Scholar] [CrossRef]
- Xue, M.; McKelvey, K.; Shen, K.; Minhas, N.; March, L.; Park, S.Y.; Jackson, C.J. Endogenous MMP-9 and not MMP-2 promotes rheumatoid synovial fibroblast survival, inflammation and cartilage degradation. Rheumatology 2014, 53, 2270–2279. [Google Scholar] [CrossRef]
- Perrotta, I.; Sciangula, A.; Aquila, S.; Mazzulla, S. Matrix metalloproteinase-9 expression in calcified human aortic alves: A histopathologic, immunohistochemical, and ultrastructural study. Appl. Immunohistochem. Mol. Morphol. 2016, 24, 128–137. [Google Scholar] [CrossRef]
- Schenke-Layland, K.; Stock, U.A.; Nsair, A.; Xie, J.; Angelis, E.; Fonseca, C.G.; Larbig, R.; Mahajan, A.; Shivkumar, K.; Fishbein, M.C.; et al. Cardiomyopathy is associated with structural remodelling of heart valve extracellular matrix. Eur. Heart J. 2009, 30, 2254–2265. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Q.; The, E.; Ao, L.; Zhai, Y.; Osterholt, M.K.; Fullerton, D.A.; Meng, X. TLR4 Stimulation Promotes Human AVIC Fibrogenic Activity through Upregulation of Neurotrophin 3 Production. Int. J. Mol. Sci. 2020, 21, 1276. https://doi.org/10.3390/ijms21041276
Yao Q, The E, Ao L, Zhai Y, Osterholt MK, Fullerton DA, Meng X. TLR4 Stimulation Promotes Human AVIC Fibrogenic Activity through Upregulation of Neurotrophin 3 Production. International Journal of Molecular Sciences. 2020; 21(4):1276. https://doi.org/10.3390/ijms21041276
Chicago/Turabian StyleYao, Qingzhou, Erlinda The, Lihua Ao, Yufeng Zhai, Maren K. Osterholt, David A. Fullerton, and Xianzhong Meng. 2020. "TLR4 Stimulation Promotes Human AVIC Fibrogenic Activity through Upregulation of Neurotrophin 3 Production" International Journal of Molecular Sciences 21, no. 4: 1276. https://doi.org/10.3390/ijms21041276
APA StyleYao, Q., The, E., Ao, L., Zhai, Y., Osterholt, M. K., Fullerton, D. A., & Meng, X. (2020). TLR4 Stimulation Promotes Human AVIC Fibrogenic Activity through Upregulation of Neurotrophin 3 Production. International Journal of Molecular Sciences, 21(4), 1276. https://doi.org/10.3390/ijms21041276