Constitutive Differential Features of Type 2 Transglutaminase in Cells Derived from Celiac Patients and from Healthy Subjects

 ,
, 
 and
and
Abstract
1. Introduction
2. Results
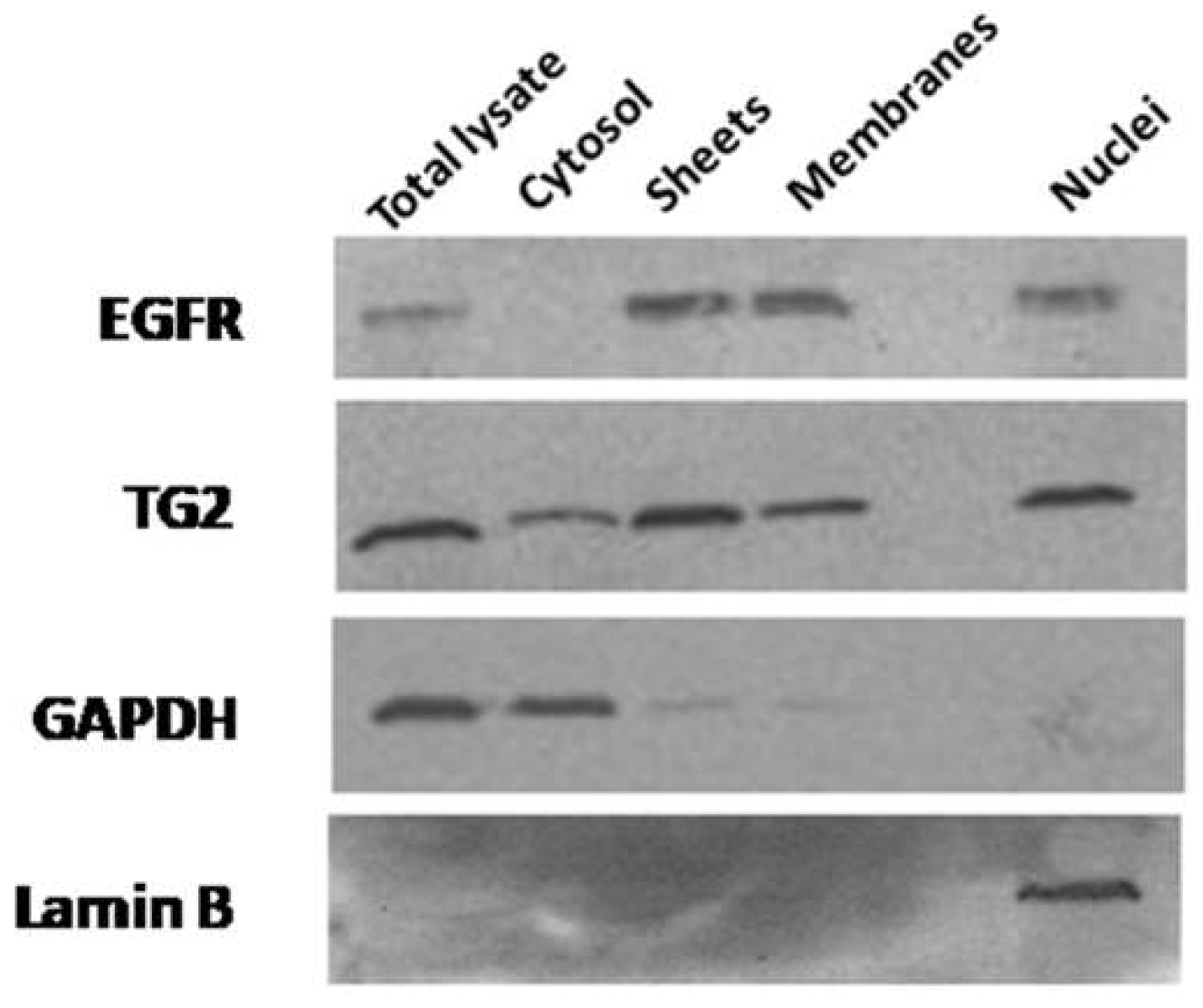
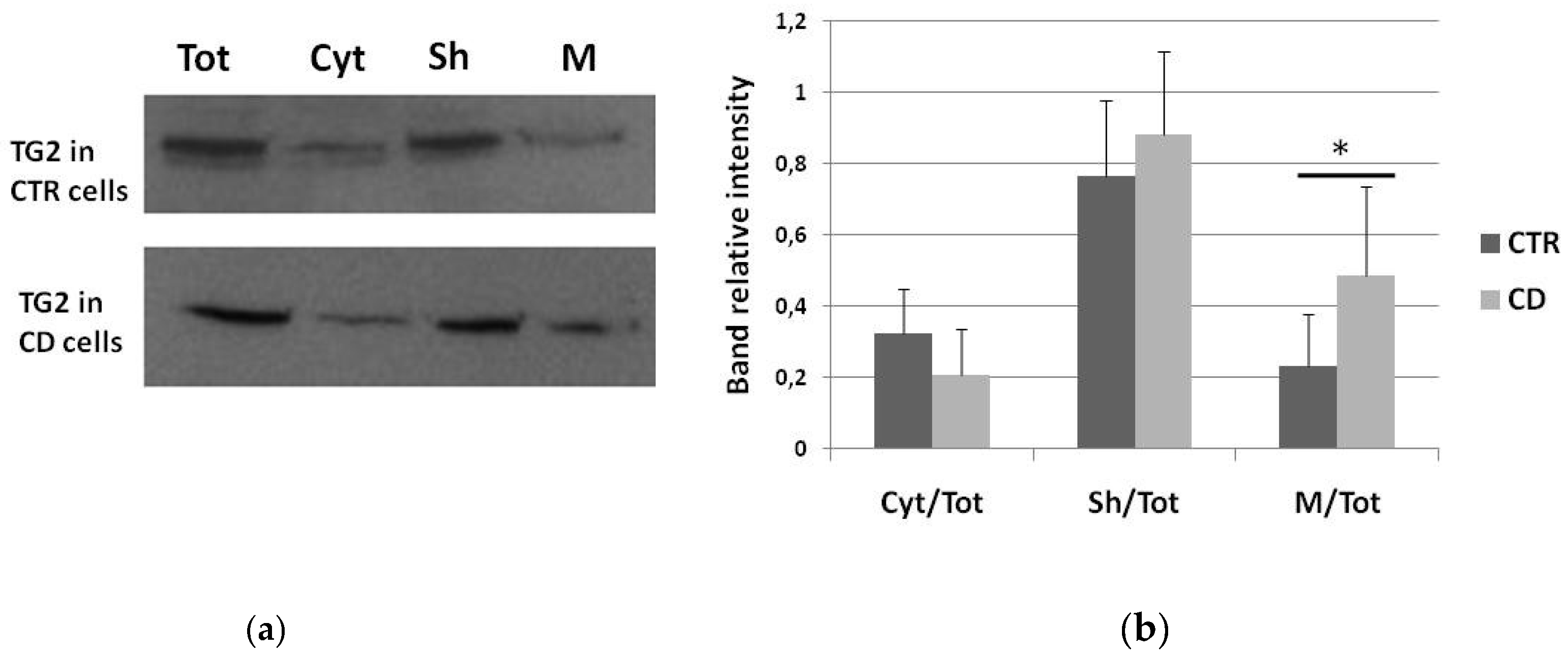
2.1. Subcellular Distribution of TG2
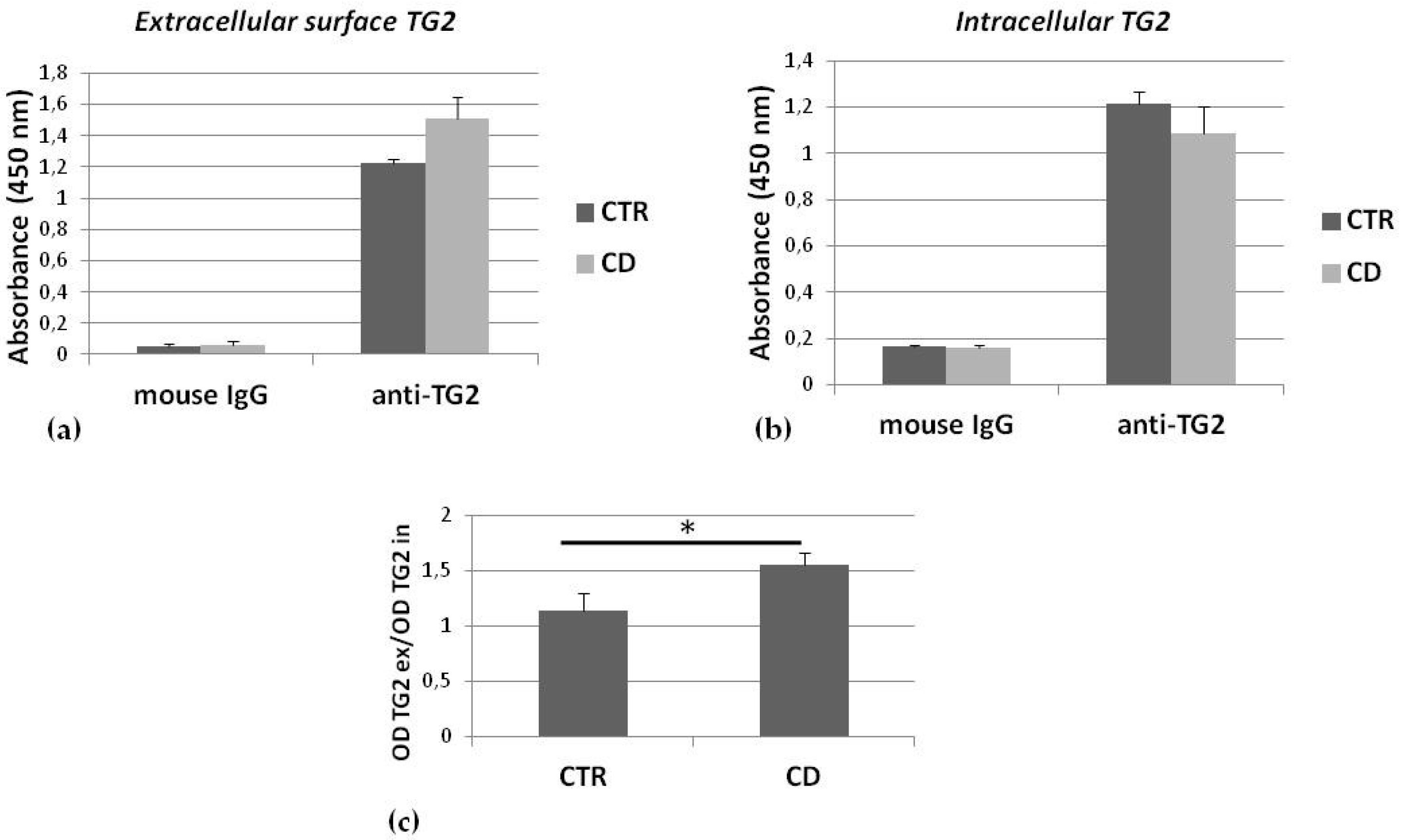
2.2. TG2 on the Membrane Surface
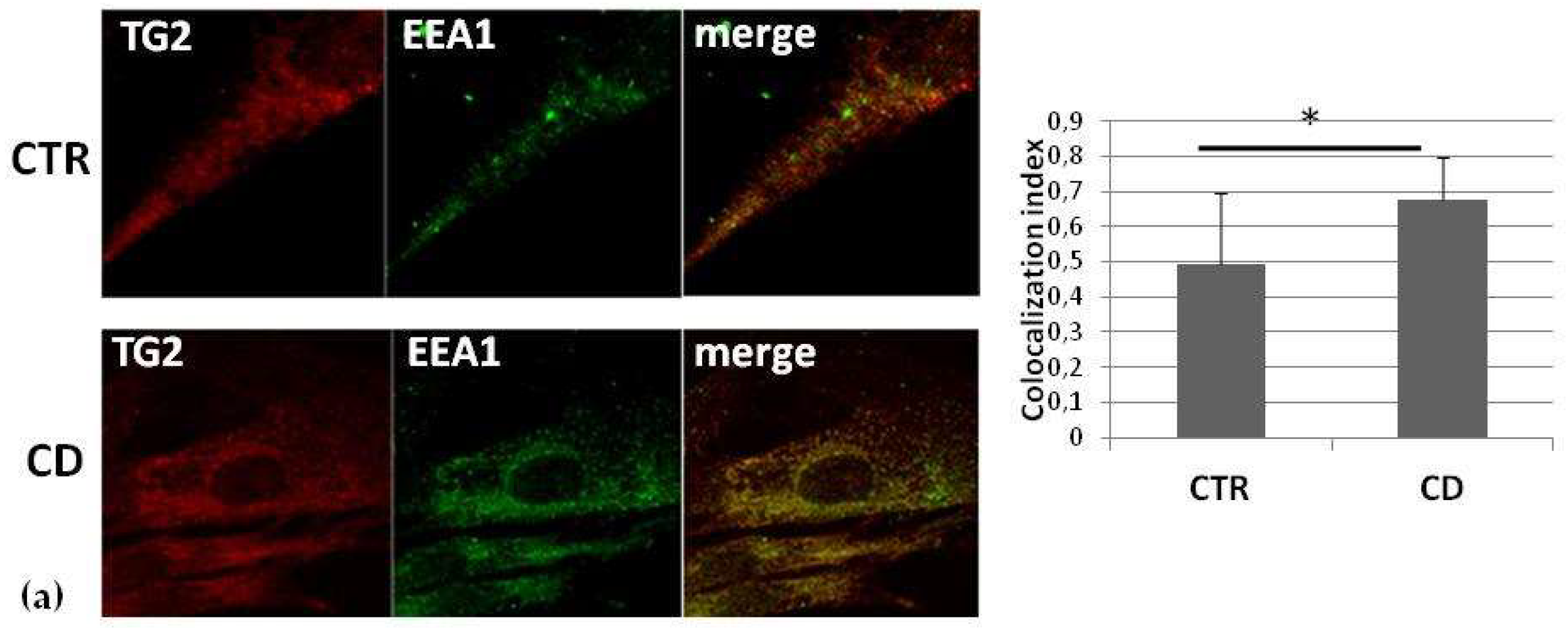
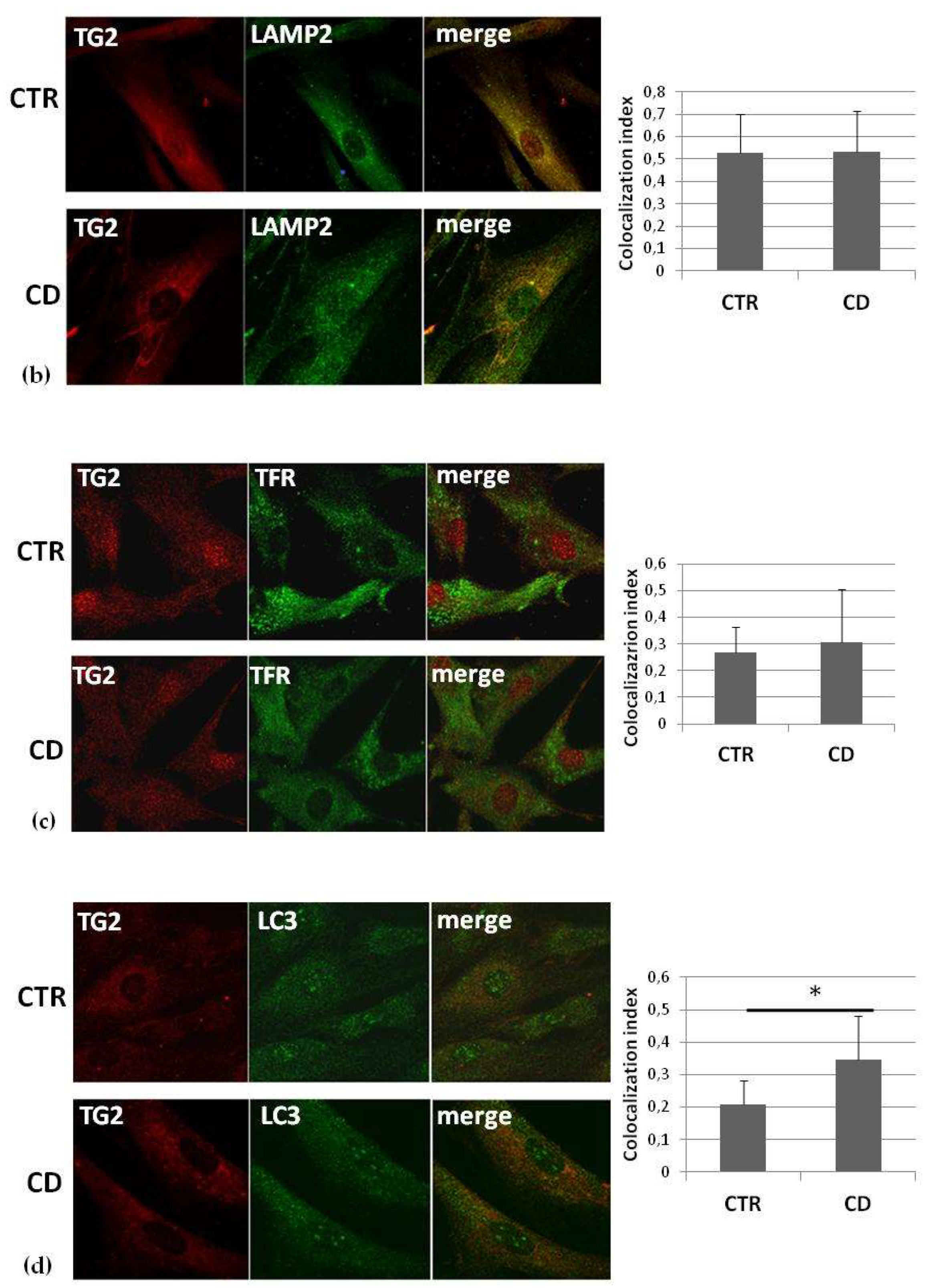
2.3. Intracellular Colocalization of TG2 with Vesicular Markers
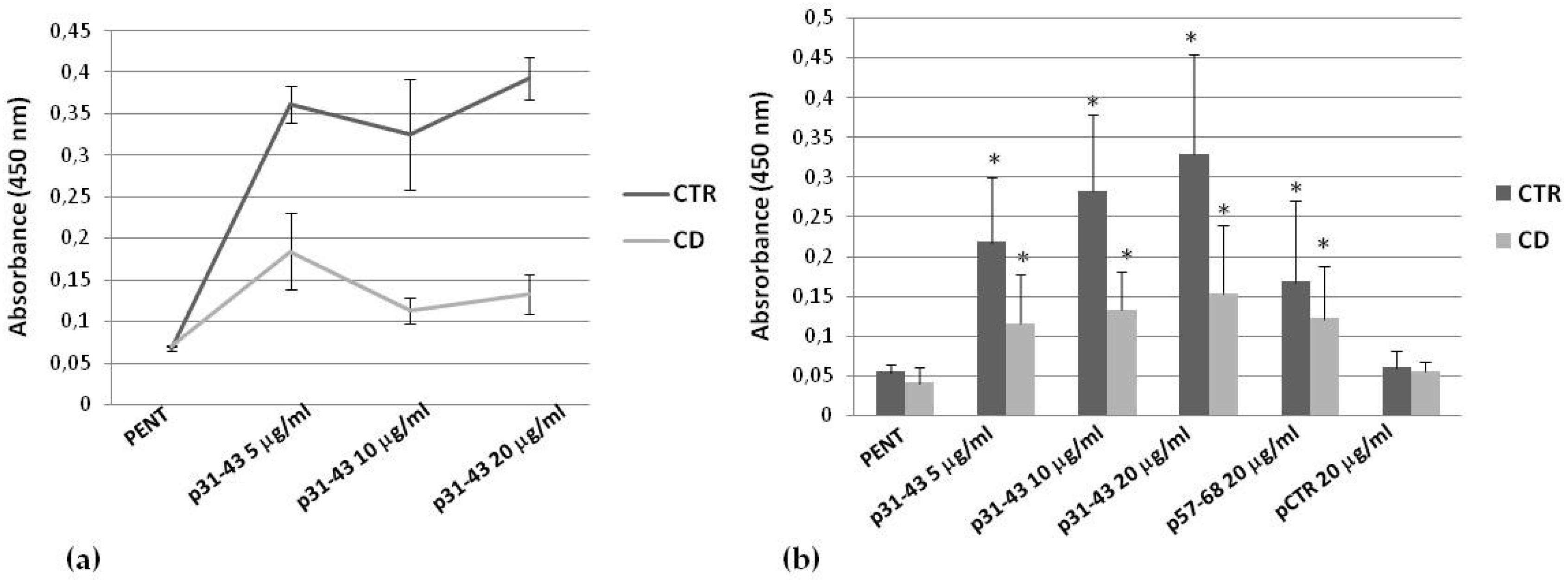
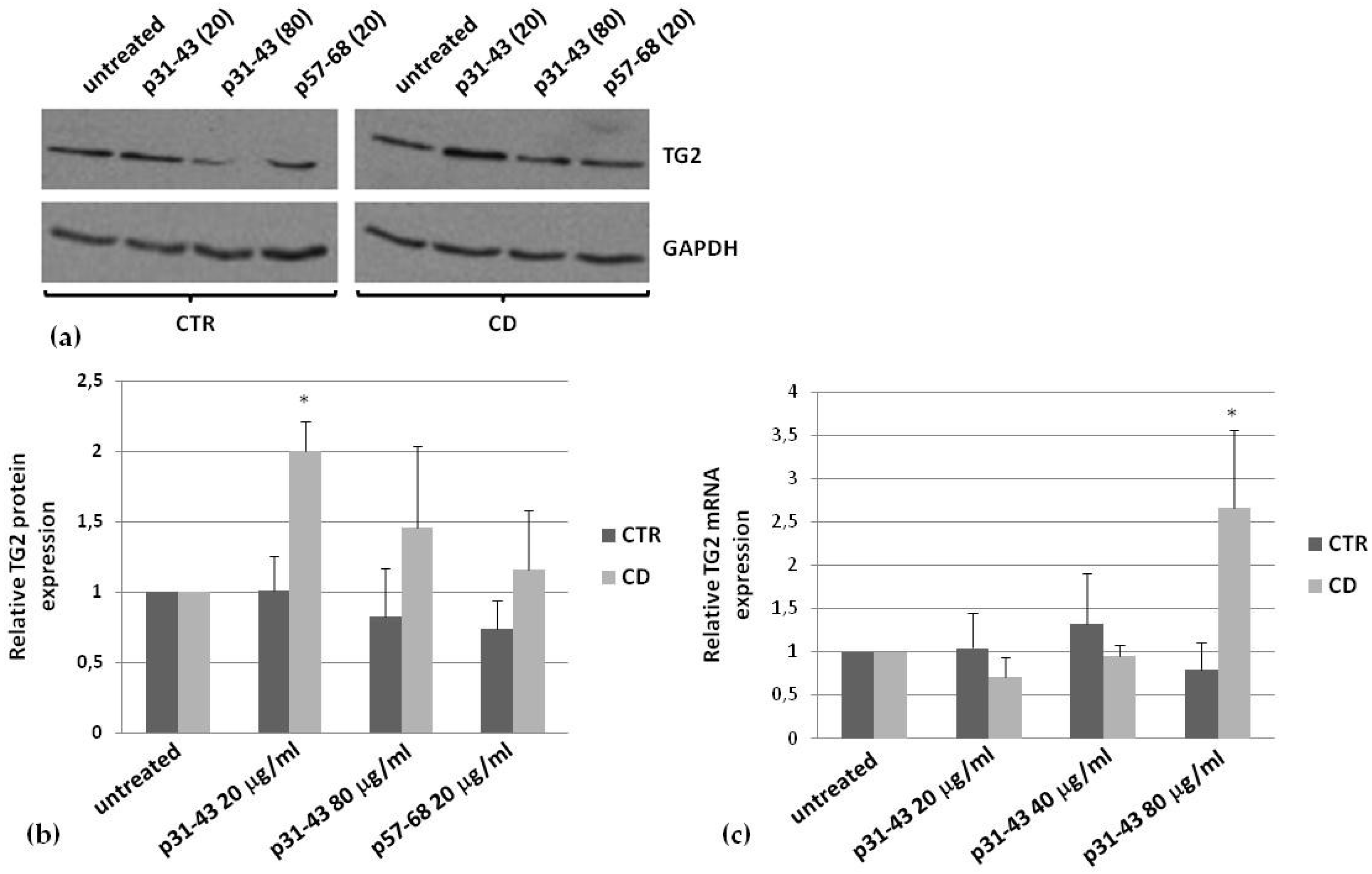
2.4. Effects of p31–43 on TG2 Activity and Expression
3. Discussion
Conclusions
4. Materials and Methods
4.1. Primary Fibroblasts Culture
4.2. Peptides
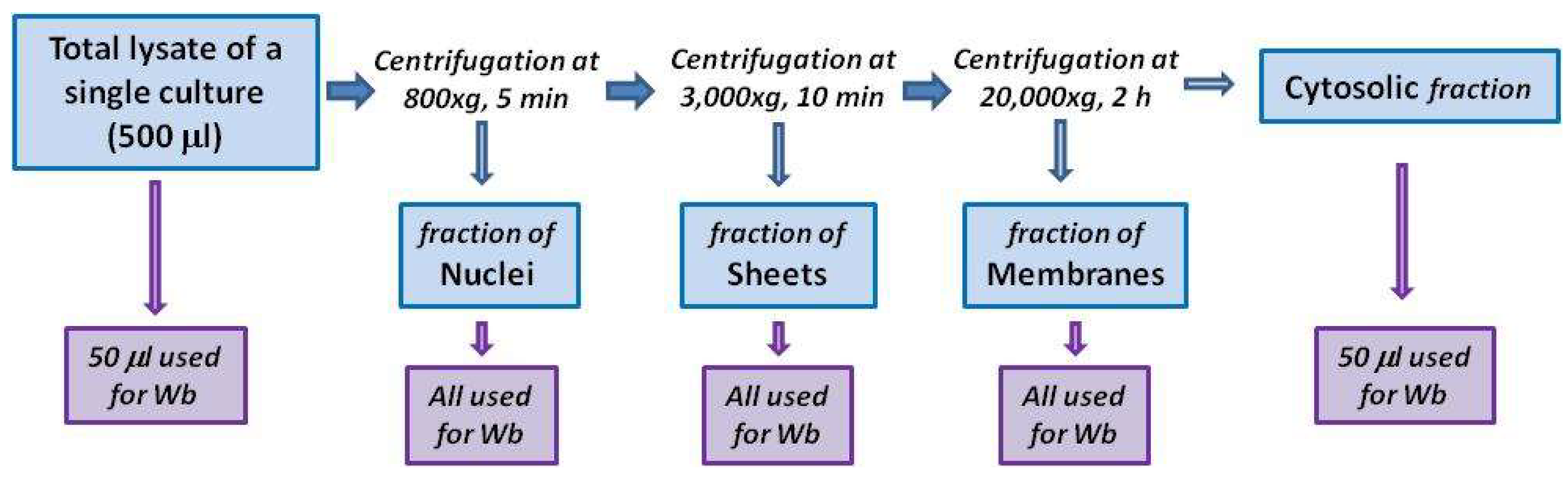
4.3. Subcellular Fractionation
4.4. Detection of Membrane-Surface TG2
4.5. Confocal Microscopy
4.6. In Situ TG2 Activity
4.7. Western Blot
4.8. Real-Time PCR
4.9. Statistics
4.10. Ethic Statement
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TG2 | type 2 transglutaminase |
| CD | celiac disease |
| HLA | human leucocytes antigen |
| p31–43 | α-gliadin peptide 31–43 |
| p57–68 | α-gliadin peptide 57–68 |
| p229–246 | A-gliadin peptide 229–246 |
| LAMP2 | lysosome-associated membrane protein 2 |
| EEA1 | early endosome antigen 1 |
| LC3 | microtubule-associated protein1A/1B-light chain 3 |
| PBS | phosphate-buffered saline |
References
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell. Biol. 2003, 4, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417. [Google Scholar] [CrossRef] [PubMed]
- Nurminskaya, M.V.; Belkin, A.M. Cellular functions of tissue transglutaminase. Int. Rev. Cell. Mol. Biol. 2012, 294, 1–97. [Google Scholar] [CrossRef] [PubMed]
- Esposito, C.; Caputo, I. Mammalian transglutaminases. Identification of substrates as a key to physiological function and physiopathological relevance. FEBS J. 2005, 272, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Kanchan, K.; Fuxreiter, M.; Fésüs, L. Physiological, pathological, and structural implications of non-enzymatic protein-protein interactions of the multifunctional human transglutaminase 2. Cell. Mol. Life Sci. 2015, 72, 3009–3035. [Google Scholar] [CrossRef] [PubMed]
- Ientile, R.; Caccamo, D.; Griffin, M. Tissue transglutaminase and the stress response. Amino Acids 2007, 33, 385–394. [Google Scholar] [CrossRef]
- Kumar, S.; Mehta, K. Tissue transglutaminase, inflammation, and cancer: How intimate is the relationship? Amino Acids 2013, 44, 81–88. [Google Scholar] [CrossRef]
- Ruan, Q.; Johnson, G.V. Transglutaminase 2 in neurodegenerative disorders. Front. Biosci. 2007, 12, 891–904. [Google Scholar] [CrossRef][Green Version]
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 2009, 89, 991–1023. [Google Scholar] [CrossRef]
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [CrossRef]
- Sollid, L.M. Coeliac disease: Dissecting a complex inflammatory disorder. Nat. Rev. Immunol. 2002, 2, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Stamnaes, J.; Sollid, L.M. Celiac disease: Autoimmunity in response to food antigen. Semin. Immunol. 2015, 27, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Vader, L.W.; de Ru, A.; van der Wal, Y.; Kooy, Y.M.; Benckhuijsen, W.; Mearin, M.L.; Drijfhout, J.W.; van Veelen, P.; Koning, F. Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. J. Exp. Med. 2002, 195, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Sollid, L.M.; Jabri, B. Celiac disease and transglutaminase 2: A model for posttranslational modification of antigens and HLA association in the pathogenesis of auto-immune disorders. Curr. Opin. Immunol. 2011, 23, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Sollid, L.M.; Molberg, Ø.; McAdam, S.; Lundin, K.E.A. Auto-antibodies in celiac disease: Tissue transglutaminase–guilt by association? Gut 1997, 41, 851–852. [Google Scholar] [CrossRef] [PubMed]
- Martucciello, S.; Lavric, M.; Toth, B.; Korponay-Szabo, I.; Nadalutti, C.; Myrsky, E.; Rauhavirta, T.; Esposito, C.; Sulic, A.M.; Sblattero, D.; et al. RhoB is associated with the antiangiogenic effects of celiac patient transglutaminase 2-targeted autoantibodies. J. Mol. Med. (Berl.) 2012, 90, 817–826. [Google Scholar] [CrossRef]
- Kalliokoski, S.; Sulic, A.M.; Korponay-Szabó, I.R.; Szondy, Z.; Frias, R.; Perez, M.A.; Martucciello, S.; Roivainen, A.; Pelliniemi, L.J.; Esposito, C.; et al. Celiac disease-specific TG2-targeted auto-antibodies inhibit angiogenesis ex vivo and in vivo in mice by interfering with endothelial cell dynamics. PLoS ONE 2013, 8, e65887. [Google Scholar] [CrossRef]
- Paolella, G.; Caputo, I.; Marabotti, A.; Lepretti, M.; Salzano, A.M.; Scaloni, A.; Vitale, M.; Zambrano, N.; Sblattero, D.; Esposito, C. Celiac anti-type 2 transglutaminase antibodies induce phosphoproteome modification in intestinal epithelial Caco-2 cells. PLoS ONE 2013, 8, e84403. [Google Scholar] [CrossRef]
- Caputo, I.; Lepretti, M.; Secondo, A.; Martucciello, S.; Paolella, G.; Sblattero, D.; Barone, M.V.; Esposito, C. Anti-tissue transglutaminase antibodies activate intracellular tissue transglutaminase by modulating cytosolic Ca2+ homeostasis. Amino Acids 2013, 44, 251–260. [Google Scholar] [CrossRef]
- Kalliokoski, S.; Piqueras, V.O.; Frías, R.; Sulic, A.M.; Määttä, J.A.; Kähkönen, N.; Viiri, K.; Huhtala, H.; Pasternack, A.; Laurila, K.; et al. Transglutaminase 2-specific coeliac disease auto-antibodies induce morphological changes and signs of inflammation in the small-bowel mucosa of mice. Amino Acids 2017, 49, 529–540. [Google Scholar] [CrossRef]
- Martucciello, S.; Paolella, G.; Esposito, C.; Lepretti, M.; Caputo, I. Anti-type 2 transglutaminase antibodies as modulators of type 2 transglutaminase functions: A possible pathological role in celiac disease. Cell. Mol. Life Sci. 2018, 75, 4107–4124. [Google Scholar] [CrossRef] [PubMed]
- Barone, M.V.; Troncone, R.; Auricchio, S. Gliadin peptides as tiggers of the proliferative and stress/innate Immune response of the celiac small sntestinal mucosa. Int. J. Mol. Sci. 2014, 5, 20518–20537. [Google Scholar] [CrossRef] [PubMed]
- Caputo, I.; Barone, M.V.; Lepretti, M.; Martucciello, S.; Nista, I.; Troncone, R.; Auricchio, S.; Sblattero, D.; Esposito, C. Celiac anti-tissue transglutaminase antibodies interfere with the uptake of alpha gliadin peptide 31-43 but not of peptide 57-68 by epithelial cells. Biochim. Biophys. Acta 2010, 1802, 717–727. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Paolella, G.; Lepretti, M.; Barone, M.V.; Nanayakkara, M.; Di Zenzo, M.; Sblattero, D.; Auricchio, S.; Esposito, C.; Caputo, I. Celiac anti-type 2 transglutaminase antibodies induce differential effects in fibroblasts from celiac disease patients and from healthy subjects. Amino Acids 2017, 49, 541–550. [Google Scholar] [CrossRef]
- Skovbjerg, H.; Anthonsen, D.; Knudsen, E.; Sjöström, H. Deamidation of gliadin peptides in lamina propria: Implications for celiac disease. Dig. Dis. Sci. 2008, 53, 2917–2924. [Google Scholar] [CrossRef]
- Vilasi, S.; Sirangelo, I.; Irace, G.; Caputo, I.; Barone, M.V.; Esposito, C.; Ragone, R. Interaction of ‘toxic’ and ‘immunogenic’ A-gliadin peptides with a membrane-mimetic environment. J. Mol. Recognit. 2010, 23, 322–328. [Google Scholar] [CrossRef]
- Paolella, G.; Lepretti, M.; Martucciello, S.; Nanayakkara, M.; Auricchio, S.; Esposito, C.; Barone, M.V.; Caputo, I. The toxic alpha-gliadin peptide 31-43 enters cells without a surface membrane receptor. Cell. Biol. Int. 2018, 42, 112–120. [Google Scholar] [CrossRef]
- Nanayakkara, M.; Kosova, R.; Lania, G.; Sarno, M.; Gaito, A.; Galatola, M.; Greco, L.; Cuomo, M.; Troncone, R.; Auricchio, S.; et al. A celiac cellular phenotype, with altered LPP sub-cellular distribution, is inducible in controls by the toxic gliadin peptide P31-43. PLoS ONE 2013, 8, e79763. [Google Scholar] [CrossRef]
- Nanayakkara, M.; Lania, G.; Maglio, M.; Kosova, R.; Sarno, M.; Gaito, A.; Discepolo, V.; Troncone, R.; Auricchio, S.; Auricchio, R.; et al. Enterocyte proliferation and signaling are constitutively altered in celiac disease. PLoS ONE 2013, 8, e76006. [Google Scholar] [CrossRef]
- Lania, G.; Nanayakkara, M.; Maglio, M.; Auricchio, R.; Porpora, M.; Conte, M.; De Matteis, M.M.; Rizzo, R.; Luini, A.; Discepolo, V.; et al. Constitutive alterations in vesicular trafficking increase the sensitivity of cells from celiac disease patients to gliadin. Commun. Biol. 2019, 2, 190. [Google Scholar] [CrossRef]
- Zemskov, E.A.; Mikhailenko, I.; Hsia, R.C.; Zaritskaya, L.; Belkin, A.M. Unconventional Secretion of Tissue Transglutaminase Involves Phospholipid-Dependent Delivery into Recycling Endosomes. PLoS ONE 2001, 6, e19414. [Google Scholar] [CrossRef] [PubMed]
- D’Eletto, M.; Farrace, M.G.; Rossin, F.; Strappazzon, F.; Giacomo, G.D.; Cecconi, F.; Melino, G.; Sepe, S.; Moreno, S.; Fimia, G.M.; et al. Type 2 transglutaminase is involved in the autophagy-dependent clearance of ubiquitinated proteins. Cell Death Differ. 2012, 19, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- D’Eletto, M.; Farrace, M.G.; Falasca, L.; Reali, V.; Oliverio, S.; Melino, G.; Griffin, M.; Fimia, G.M.; Piacentini, M. Transglutaminase 2 is involved in autophagosome maturation. Autophagy 2009, 5, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Verhaar, R.; Drukarch, B.; Bol, J.G.; Jongenelen, C.A.; Wilhelmus, M.M. Tissue transglutaminase cross-links beclin 1 and regulates autophagy in MPP+-treated human SH-SY5Y cells. Neurochem. Int. 2013, 62, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, L.; Villella, V.R.; Piacentini, M.; Raia, V.; Kroemer, G. Defective proteostasis in celiac disease as a new therapeutic target. Cell Death Dis. 2019, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Manai, F.; Azzalin, A.; Gabriele, F.; Martinelli, C.; Morandi, M.; Biggiogera, M.; Bozzola, M.; Comincini, S. The in vitro effects of enzymatic digested gliadin on the functionality of the autophagy process. Int. J. Mol. Sci. 2018, 19, 635. [Google Scholar] [CrossRef]
- Caputo, I.; Secondo, A.; Lepretti, M.; Paolella, G.; Auricchio, S.; Barone, M.V.; Esposito, C. Gliadin peptides induce tissue transglutaminase activation and ER-stress through Ca2+ mobilization in Caco-2 cells. PLoS ONE 2012, 7, e45209. [Google Scholar] [CrossRef]
- Spector, D.L.; Goldman, R.D.; Leinwand, L.A. Cells: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2000. [Google Scholar]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal violet assay for determining viability of cultured cells. Cold Spring Harb. Protoc. 2016. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Investigated Features | Findings in CD Versus Control Fibroblasts |
|---|---|
| Association to the membrane | More evident |
| Association to the cell surface | More evident |
| Colocalization with EEA1 | More evident |
| Colocalization with LAMP2 | Similar |
| Colocalization with the transferrin-receptor | Similar |
| Colocalization with LC3 | More evident |
| Intracellular activity induced by p31–43 | Less evident |
| Expression induced by p31–43 | More evident |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolella, G.; Nanayakkara, M.; Sposito, S.; Lepretti, M.; Auricchio, S.; Esposito, C.; Barone, M.V.; Martucciello, S.; Caputo, I. Constitutive Differential Features of Type 2 Transglutaminase in Cells Derived from Celiac Patients and from Healthy Subjects. Int. J. Mol. Sci. 2020, 21, 1231. https://doi.org/10.3390/ijms21041231
Paolella G, Nanayakkara M, Sposito S, Lepretti M, Auricchio S, Esposito C, Barone MV, Martucciello S, Caputo I. Constitutive Differential Features of Type 2 Transglutaminase in Cells Derived from Celiac Patients and from Healthy Subjects. International Journal of Molecular Sciences. 2020; 21(4):1231. https://doi.org/10.3390/ijms21041231
Chicago/Turabian StylePaolella, Gaetana, Merlin Nanayakkara, Silvia Sposito, Marilena Lepretti, Salvatore Auricchio, Carla Esposito, Maria Vittoria Barone, Stefania Martucciello, and Ivana Caputo. 2020. "Constitutive Differential Features of Type 2 Transglutaminase in Cells Derived from Celiac Patients and from Healthy Subjects" International Journal of Molecular Sciences 21, no. 4: 1231. https://doi.org/10.3390/ijms21041231
APA StylePaolella, G., Nanayakkara, M., Sposito, S., Lepretti, M., Auricchio, S., Esposito, C., Barone, M. V., Martucciello, S., & Caputo, I. (2020). Constitutive Differential Features of Type 2 Transglutaminase in Cells Derived from Celiac Patients and from Healthy Subjects. International Journal of Molecular Sciences, 21(4), 1231. https://doi.org/10.3390/ijms21041231