The Roles of FoxO Transcription Factors in Regulation of Bone Cells Function

 , , ,
, , ,
Abstract
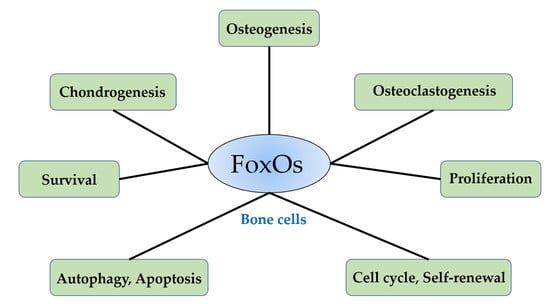
1. Introduction
2. The FoxO Family
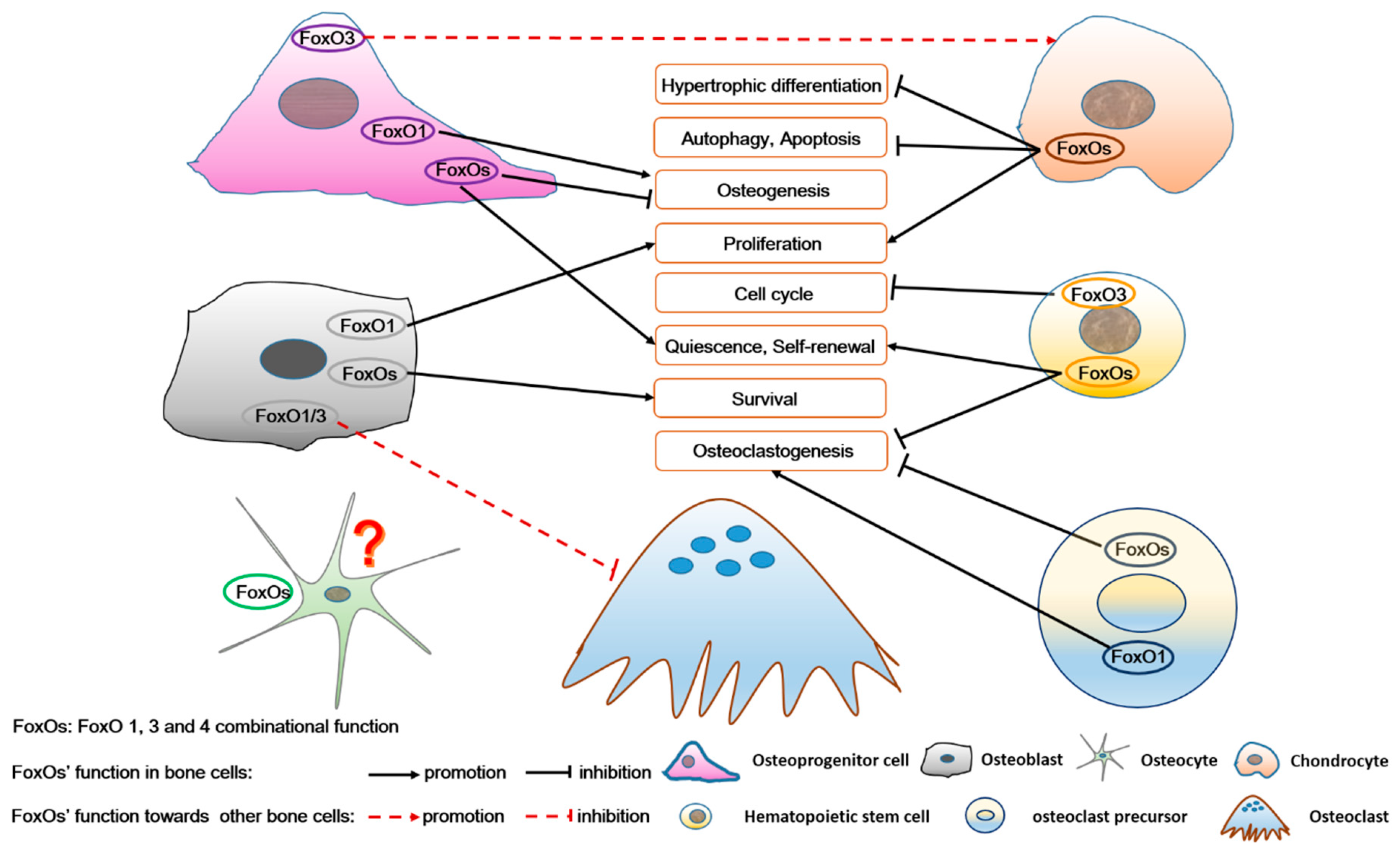
3. FoxOs and Bone Cells
3.1. Bidirectional Regulation of FoxOs in Osteoprogenitor Cells
3.1.1. FoxOs Promote Osteogenesis
3.1.2. FoxOs Repress Osteogenesis
3.2. FoxOs Improve Osteoblasts Function
3.3. FoxOs Positively Regulate Hematopoietic Stem Cell Activities
3.4. FoxOs Regulate Osteoclastogenesis
3.4.1. FoxOs Activate Osteoclastogenesis
3.4.2. FoxOs Suppress Osteoclastogenesis
3.4.3. FoxOs, Expressed in Osteoblasts, Indirectly Repress Osteoclastogenesis
3.5. FoxOs Facilitate Chondrogenesis
4. Conclusion and Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FoxOs | Forkhead box class O family member proteins |
| LPS | Lipopolysaccharide |
| TNF-α | Tumor necrosis factor α |
| MSCs | Mesenchymal stem cells |
| Runx2 | Runt-related transcription factor 2 |
| ALP | Alkaline phosphatase |
| OCN | Osteocalcin |
| CFU | Colony forming unit |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| PPREs | PPAR-response elements |
| PCNA | Proliferating cell nuclear antigen |
| ROS | Reactive oxygen species |
| FABP4 | Fatty acid-binding protein 4 |
| 4-HNE | 4-hydroxynonenal |
| ATF4 | Activating transcription factor 4 |
| BFR | Bone formation rate |
| HSC | Hematopoietic stem cell |
| Gpx-1 | Glutathione peroxidase 1 |
| M-CSF | Macrophage-colony stimulating factor |
| RANK | Receptor activator of NF-κ B |
| RANKL | Receptor activator of NF-κ B ligand |
| BMMCs | Bone marrow mononuclear cells |
| NFATc1 | Nuclear factor of activated T cells 1 |
| Akt | Protein kinase B |
| SIRT-1 | Sirtuin 1 |
| TRAP | Tartrate-resistant acid phosphatase |
| MAPKs | Mitogen-activated protein kinases |
| NF-κB | Nuclear factor kappa-B |
| HO-1 | Hemeoxygenase-1 |
| Mst 1 | Mammalian Sterile 20-like kinase 1 |
| OPG | Osteoprotegerin |
| AP-1 | Activator protein 1 |
| PI3K | phosphoinositide 3-kinase |
| OA | Osteoarthritis |
| PTP1B | Protein tyrosine phosphatase 1B |
| CTKO | Chondrocyte triple knock-out |
| PTEN | Phosphatase and tensin homolog deleted on chromosome ten |
References
- Carter, M.E.; Brunet, A. FOXO transcription factors. Curr. Biol. 2007, 17, R113–R114. [Google Scholar] [CrossRef] [PubMed]
- Avila-Flores, A.; Arranz-Nicolas, J.; Merida, I. Transcriptional Activity of FOXO Transcription Factors Measured by Luciferase Assays. In FOXO Transcription Factors; Springer: Berlin, Germany, 2019; pp. 91–102. [Google Scholar]
- Genin, E.C.; Caron, N.; Vandenbosch, R.; Nguyen, L.; Malgrange, B. Concise review: Forkhead pathway in the control of adult neurogenesis. Stem Cells 2014, 32, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.S.; Binderup, T.; Jensen, K.T.; Therkelsen, I.; Borup, R.; Nilsson, E.; Multhaupt, H.; Bouchard, C.; Quistorff, B.; Kjær, A. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J. 2011, 30, 4554–4570. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, G.; Kaestner, K.H. SnapShot: Forkhead transcription factors I. Cell 2007, 130, 1160. [Google Scholar] [CrossRef]
- Calnan, D.; Brunet, A. The foxo code. Oncogene 2008, 27, 2276. [Google Scholar] [CrossRef]
- Henderson, S.T.; Johnson, T.E. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 2001, 11, 1975–1980. [Google Scholar] [CrossRef]
- Webb, A.E.; Brunet, A. FOXO transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83. [Google Scholar] [CrossRef]
- Kim, H.-N.; Iyer, S.; Ring, R.; Almeida, M. The role of FoxOs in bone health and disease. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 127, pp. 149–163. [Google Scholar]
- Bullock, M. FOXO factors and breast cancer: Outfoxing endocrine resistance. Endocr. Relat. Cancer 2016, 23, R113–R130. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Y.; Graves, D.T. FOXO transcription factors: Their clinical significance and regulation. BioMed Res. Int. 2014, 2014, 925350. [Google Scholar] [CrossRef]
- De Brachène, A.C.; Demoulin, J.-B. FOXO transcription factors in cancer development and therapy. Cell. Mol. Life Sci. 2016, 73, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Tsitsipatis, D.; Klotz, L.-O.; Steinbrenner, H. Multifaceted functions of the forkhead box transcription factors FoxO1 and FoxO3 in skin. Biochim. Biophys. Acta. Gen. Subj. 2017, 1861, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; da Fontoura, C.S.; Moreno, M.; Holton, N.E.; Sweat, M.; Sweat, Y.; Lee, M.K.; Arbon, J.; Bidlack, F.B.; Thedens, D.R. FoxO6 regulates Hippo signaling and growth of the craniofacial complex. PLoS Genet. 2018, 14, e1007675. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Gong, Y.; Xu, L.; Zhou, M.; Li, J.; Song, J. Bidirectional regulation of osteogenic differentiation by the FOXO subfamily of Forkhead transcription factors in mammalian MSCs. Cell Prolif. 2019, 52, e12540. [Google Scholar] [CrossRef]
- Fu, Z.; Tindall, D. FOXOs, cancer and regulation of apoptosis. Oncogene 2008, 27, 2312. [Google Scholar] [CrossRef]
- Iyer, S.; Ambrogini, E.; Bartell, S.M.; Han, L.; Roberson, P.K.; de Cabo, R.; Jilka, R.L.; Weinstein, R.S.; O’Brien, C.A.; Manolagas, S.C. FOXOs attenuate bone formation by suppressing Wnt signaling. J. Clin. Investig. 2013, 123, 3409–3419. [Google Scholar] [CrossRef]
- Brent, M.M.; Anand, R.; Marmorstein, R. Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure 2008, 16, 1407–1416. [Google Scholar] [CrossRef]
- Murtaza, G.; Khan, A.K.; Rashid, R.; Muneer, S.; Hasan, S.M.F.; Chen, J. FOXO transcriptional factors and long-term living. Oxid. Med. Cell. Longev. 2017, 2017, 3494289. [Google Scholar] [CrossRef]
- Almeida, M. Unraveling the role of FoxOs in bone—insights from mouse models. Bone 2011, 49, 319–327. [Google Scholar] [CrossRef]
- Teixeira, C.C.; Liu, Y.; Thant, L.M.; Pang, J.; Palmer, G.; Alikhani, M. Foxo1, a novel regulator of osteoblast differentiation and skeletogenesis. J. Biol. Chem. 2010, 285, 31055–31065. [Google Scholar] [CrossRef]
- Komori, T. Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res. 2010, 339, 189. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, A.; Burgering, B.M. Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 440. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, M.F.; Flowers, S.; Bhattacharya, R.; Faibish, D.; Behl, Y.; Kotton, D.N.; Gerstenfeld, L.; Moran, E.; Graves, D.T. FOXO1 modulates osteoblast differentiation. Bone 2011, 48, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Ambrogini, E.; Almeida, M.; Martin-Millan, M.; Paik, J.-H.; DePinho, R.A.; Han, L.; Goellner, J.; Weinstein, R.S.; Jilka, R.L.; O’Brien, C.A. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 2010, 11, 136–146. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017, 13, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Armoni, M.; Harel, C.; Karni, S.; Chen, H.; Bar-Yoseph, F.; Ver, M.R.; Quon, M.J.; Karnieli, E. FOXO1 represses peroxisome proliferator-activated receptor-gamma1 and -gamma2 gene promoters in primary adipocytes. A novel paradigm to increase insulin sensitivity. J. Biol. Chem. 2006, 281, 19881–19891. [Google Scholar] [CrossRef]
- Fan, W.; Imamura, T.; Sonoda, N.; Sears, D.D.; Patsouris, D.; Kim, J.J.; Olefsky, J.M. FOXO1 transrepresses peroxisome proliferator-activated receptor γ transactivation, coordinating an insulin-induced feed-forward response in adipocytes. J. Biol. Chem. 2009, 284, 12188–12197. [Google Scholar] [CrossRef]
- Muller, F.L.; Lustgarten, M.S.; Jang, Y.; Richardson, A.; Van Remmen, H. Trends in oxidative aging theories. Free Radic. Biol. Med. 2007, 43, 477–503. [Google Scholar] [CrossRef]
- Almeida, M.; Han, L.; Martin-Millan, M.; O’Brien, C.A.; Manolagas, S.C. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting β-catenin from T cell factor-to forkhead box O-mediated transcription. J. Biol. Chem. 2007, 282, 27298–27305. [Google Scholar] [CrossRef]
- Almeida, M.; Han, L.; Martin-Millan, M.; Plotkin, L.I.; Stewart, S.A.; Roberson, P.K.; Kousteni, S.; O’Brien, C.A.; Bellido, T.; Parfitt, A.M. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007, 282, 27285–27297. [Google Scholar] [CrossRef]
- Bennett, C.N.; Longo, K.A.; Wright, W.S.; Suva, L.J.; Lane, T.F.; Hankenson, K.D.; MacDougald, O.A. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. 2005, 102, 3324–3329. [Google Scholar] [CrossRef] [PubMed]
- Babij, P.; Zhao, W.; Small, C.; Kharode, Y.; Yaworsky, P.J.; Bouxsein, M.L.; Reddy, P.S.; Bodine, P.V.; Robinson, J.A.; Bhat, B. High bone mass in mice expressing a mutant LRP5 gene. J. Bone Miner. Res. 2003, 18, 960–974. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Porter, N.A.; Brash, A.R. Routes to 4-hydroxynonenal: Fundamental issues in the mechanisms of lipid peroxidation. J. Biol. Chem. 2008, 283, 15539–15543. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Ambrogini, E.; Han, L.; Manolagas, S.C.; Jilka, R.L. Increased lipid oxidation causes oxidative stress, increased peroxisome proliferator-activated receptor-γ expression, and diminished pro-osteogenic Wnt signaling in the skeleton. J. Biol. Chem. 2009, 284, 27438–27448. [Google Scholar] [CrossRef]
- Kang, S.; Bennett, C.N.; Gerin, I.; Rapp, L.A.; Hankenson, K.D.; MacDougald, O.A. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein α and peroxisome proliferator-activated receptor γ. J. Biol. Chem. 2007, 282, 14515–14524. [Google Scholar] [CrossRef]
- Okamura, M.; Kudo, H.; Wakabayashi, K.-i.; Tanaka, T.; Nonaka, A.; Uchida, A.; Tsutsumi, S.; Sakakibara, I.; Naito, M.; Osborne, T.F. COUP-TFII acts downstream of Wnt/β-catenin signal to silence PPARγ gene expression and repress adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 5819–5824. [Google Scholar] [CrossRef]
- Sharma, C.; Pradeep, A.; Wong, L.; Rana, A.; Rana, B. Peroxisome proliferator-activated receptor γ activation can regulate β-catenin levels via a proteasome-mediated and adenomatous polyposis coli-independent pathway. J. Biol. Chem. 2004, 279, 35583–35594. [Google Scholar] [CrossRef]
- Atashi, F.; Modarressi, A.; Pepper, M.S. The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: A review. Stem Cells Dev. 2015, 24, 1150–1163. [Google Scholar] [CrossRef]
- Rached, M.-T.; Kode, A.; Xu, L.; Yoshikawa, Y.; Paik, J.-H.; DePinho, R.A.; Kousteni, S. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 2010, 11, 147–160. [Google Scholar] [CrossRef]
- Clemens, T.L.; Karsenty, G. The osteoblast: An insulin target cell controlling glucose homeostasis. J. Bone Miner. Res. 2011, 26, 677–680. [Google Scholar] [CrossRef]
- Yang, S.; Xu, H.; Yu, S.; Cao, H.; Fan, J.; Ge, C.; Fransceschi, R.T.; Dong, H.H.; Xiao, G. Foxo1 mediates insulin-like growth factor 1 (IGF1)/insulin regulation of osteocalcin expression by antagonizing Runx2 in osteoblasts. J. Biol. Chem. 2011, 286, 19149–19158. [Google Scholar] [CrossRef] [PubMed]
- Rached, M.-T.; Kode, A.; Silva, B.C.; Jung, D.Y.; Gray, S.; Ong, H.; Paik, J.-H.; DePinho, R.A.; Kim, J.K.; Karsenty, G. FoxO1 expression in osteoblasts regulates glucose homeostasis through regulation of osteocalcin in mice. J. Clin. Investig. 2010, 120, 357–368. [Google Scholar] [CrossRef]
- Behl, Y.; Siqueira, M.; Ortiz, J.; Li, J.; Desta, T.; Faibish, D.; Graves, D.T. Activation of the acquired immune response reduces coupled bone formation in response to a periodontal pathogen. J. Immunol. 2008, 181, 8711–8718. [Google Scholar] [CrossRef]
- Bigarella, C.L.; Li, J.; Rimmelé, P.; Liang, R.; Sobol, R.W.; Ghaffari, S. FOXO3 transcription factor is essential for protecting hematopoietic stem and progenitor cells from oxidative DNA damage. J. Biol. Chem. 2017, 292, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, S.; Zhang, X.; Luciano, J.P.; Mungamuri, S.K.; Marinkovic, D.; Vercherat, C.; Sarkar, A.; Grisotto, M.; Taneja, R.; Ghaffari, S. Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress-mediated homeostasis of hematopoietic stem cells. J. Biol. Chem. 2008, 283, 25692–25705. [Google Scholar] [CrossRef] [PubMed]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef]
- Buck, D.W.; Dumanian, G.A. Bone biology and physiology: Part I. The fundamentals. Plast. Reconstr. Surg. 2012, 129, 1314–1320. [Google Scholar] [CrossRef]
- Boyce, B. Advances in the regulation of osteoclasts and osteoclast functions. J. Dent. Res. 2013, 92, 860–867. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, G.; Jeon, H.H.; Elazizi, M.; La, L.B.; Hameedaldeen, A.; Xiao, E.; Tian, C.; Alsadun, S.; Choi, Y. FOXO1 mediates RANKL-induced osteoclast formation and activity. J. Immunol. 2015, 194, 2878–2887. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Takayanagi, H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol. Metab. 2012, 23, 582–590. [Google Scholar] [CrossRef]
- Sugatani, T.; Hruska, K.A. Akt1/Akt2 and mammalian target of rapamycin/Bim play critical roles in osteoclast differentiation and survival, respectively, whereas Akt is dispensable for cell survival in isolated osteoclast precursors. J. Biol. Chem. 2005, 280, 3583–3589. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, N.; Kugimiya, F.; Oshima, Y.; Ohba, S.; Ikeda, T.; Saito, T.; Shinoda, Y.; Kawasaki, Y.; Ogata, N.; Hoshi, K. Akt1 in osteoblasts and osteoclasts controls bone remodeling. PLoS ONE 2007, 2, e1058. [Google Scholar] [CrossRef]
- Bartell, S.M.; Kim, H.-N.; Ambrogini, E.; Han, L.; Iyer, S.; Ucer, S.S.; Rabinovitch, P.; Jilka, R.L.; Weinstein, R.S.; Zhao, H. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H 2 O 2 accumulation. Nat. Commun. 2014, 5, 3773. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.K.; Choi, Y.G.; Baik, J.Y.; Han, S.Y.; Jeong, D.-w.; Bae, Y.S.; Kim, N.; Lee, S.Y. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 2005, 106, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Guan, H.; Xie, L.; Mi, B.; Fang, Z.; Li, J.; Li, F. FOXO1 inhibits osteoclastogenesis partially by antagnozing MYC. Sci. Rep. 2015, 5, 16835. [Google Scholar] [CrossRef]
- Ferber, E.C.; Peck, B.; Delpuech, O.; Bell, G.P.; East, P.; Schulze, A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012, 19, 968. [Google Scholar] [CrossRef]
- Peck, B.; Ferber, E.C.; Schulze, A. Antagonism between FOXO and MYC regulates cellular powerhouse. Front. Oncol. 2013, 3, 96. [Google Scholar] [CrossRef]
- Ke, K.; Safder, M.; Sul, O.-J.; Kim, W.-K.; Suh, J.-H.; Joe, Y.; Chung, H.-T.; Choi, H.-S. Hemeoxygenase-1 maintains bone mass via attenuating a redox imbalance in osteoclast. Mol. Cell. Endocrinol. 2015, 409, 11–20. [Google Scholar] [CrossRef]
- Kim, H.-N.; Han, L.; Iyer, S.; de Cabo, R.; Zhao, H.; O’Brien, C.A.; Manolagas, S.C.; Almeida, M. Sirtuin1 suppresses osteoclastogenesis by deacetylating FoxOs. Mol. Endocrinol. 2015, 29, 1498–1509. [Google Scholar] [CrossRef]
- Reszka, A.A.; Halasy-Nagy, J.M.; Masarachia, P.J.; Rodan, G.A. Bisphosphonates act directly on the osteoclast to induce caspase cleavage of mst1 kinase during apoptosis A link between inhibition of the mevalonate pathway and regulation of an apoptosis-promoting kinase. J. Biol. Chem. 1999, 274, 34967–34973. [Google Scholar] [CrossRef]
- Jang, S.-W.; Yang, S.-J.; Srinivasan, S.; Ye, K. Akt phosphorylates MstI and prevents its proteolytic activation, blocking FOXO3 phosphorylation and nuclear translocation. J. Biol. Chem. 2007, 282, 30836–30844. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337. [Google Scholar] [CrossRef]
- Domazetovic, V.; Marcucci, G.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Oxidative stress in bone remodeling: Role of antioxidants. Clin. Cases Miner. Bone Metab. 2017, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Hasegawa, A.; Saito, M.; Asahara, H.; Iwamoto, Y.; Lotz, M. Dysregulated FOXO transcription factors in articular cartilage in aging and osteoarthritis. Osteoarthr. Cartil. 2014, 22, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; Verlinden, L.; Maes, C.; Beullens, I.; Gysemans, C.; Paik, J.-H.; DePinho, R.A.; Bouillon, R.; Carmeliet, G.; Verstuyf, A. Forkhead box O transcription factors in chondrocytes regulate endochondral bone formation. J. Steroid Biochem. Mol. Biol. 2016, 164, 337–343. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Alvarez-Garcia, O.; Mokuda, S.; Nagira, K.; Olmer, M.; Gamini, R.; Miyata, K.; Akasaki, Y.; Su, A.I.; Asahara, H. FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci. Transl. Med. 2018, 10, eaan0746. [Google Scholar] [CrossRef]
- Djouad, F.; Bony, C.; Canovas, F.; Fromigue, O.; Reme, T.; Jorgensen, C.; Noel, D. Transcriptomic analysis identifies Foxo3A as a novel transcription factor regulating mesenchymal stem cell chrondrogenic differentiation. Cloning Stem Cells 2009, 11, 407–416. [Google Scholar] [CrossRef]
- Akasaki, Y.; Alvarez-Garcia, O.; Saito, M.; Caramés, B.; Iwamoto, Y.; Lotz, M.K. FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis Rheumatol. 2014, 66, 3349–3358. [Google Scholar] [CrossRef]
- Fujita, N.; Matsushita, T.; Ishida, K.; Kubo, S.; Matsumoto, T.; Takayama, K.; Kurosaka, M.; Kuroda, R. Potential involvement of SIRT1 in the pathogenesis of osteoarthritis through the modulation of chondrocyte gene expressions. J. Orthop. Res. 2011, 29, 511–515. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. SIRT1: Regulation of longevity via autophagy. Cell. Signal. 2009, 21, 1356–1360. [Google Scholar] [CrossRef]
- Gagarina, V.; Gabay, O.; Dvir-Ginzberg, M.; Lee, E.J.; Brady, J.K.; Quon, M.J.; Hall, D.J. SirT1 enhances survival of human osteoarthritic chondrocytes by repressing protein tyrosine phosphatase 1B and activating the insulin-like growth factor receptor pathway. Arthritis Rheum. 2010, 62, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.; Tang, B.L. Sirtuins’ modulation of autophagy. J. Cell. Physiol. 2013, 228, 2262–2270. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Miyamoto, T.; Fujita, N.; Kubota, Y.; Ito, K.; Takubo, K.; Miyamoto, K.; Ninomiya, K.; Suzuki, T.; Iwasaki, R. Reactive oxygen species induce chondrocyte hypertrophy in endochondral ossification. J. Exp. Med. 2007, 204, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Ford-Hutchinson, A.F.; Ali, Z.; Lines, S.E.; Hallgrímsson, B.; Boyd, S.K.; Jirik, F.R. Inactivation of Pten in osteo-chondroprogenitor cells leads to epiphyseal growth plate abnormalities and skeletal overgrowth. J. Bone Miner. Res. 2007, 22, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Rokutanda, S.; Fujita, T.; Kanatani, N.; Yoshida, C.A.; Komori, H.; Liu, W.; Mizuno, A.; Komori, T. Akt regulates skeletal development through GSK3, mTOR, and FoxOs. Dev. Biol. 2009, 328, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Kayal, R.A.; Siqueira, M.; Alblowi, J.; McLean, J.; Krothapalli, N.; Faibish, D.; Einhorn, T.A.; Gerstenfeld, L.C.; Graves, D.T. TNF-α mediates diabetes-enhanced chondrocyte apoptosis during fracture healing and stimulates chondrocyte apoptosis Through FOXO1. J. Bone Miner. Res. 2010, 25, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
- Alblowi, J.; Kayal, R.A.; Siqueria, M.; McKenzie, E.; Krothapalli, N.; McLean, J.; Conn, J.; Nikolajczyk, B.; Einhorn, T.A.; Gerstenfeld, L. High levels of tumor necrosis factor-α contribute to accelerated loss of cartilage in diabetic fracture healing. Am. J. Pathol. 2009, 175, 1574–1585. [Google Scholar] [CrossRef]
- Alblowi, J.; Tian, C.; Siqueira, M.F.; Kayal, R.A.; McKenzie, E.; Behl, Y.; Gerstenfeld, L.; Einhorn, T.A.; Graves, D.T. Chemokine expression is upregulated in chondrocytes in diabetic fracture healing. Bone 2013, 53, 294–300. [Google Scholar] [CrossRef]
- Manolagas, S.C. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010, 31, 266–300. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FoxOs | Effects on Bone | Functions in Osteoprogenitor Cell | Mechanisms | Cell/Mice Models | References |
|---|---|---|---|---|---|
| FoxO1 | skeletogenesis (+); craniofacial development (+); craniofacial area (+) | osteogenesis differentiation (+); calcification culture (+) | FoxO1 interacts with Runx2 promoter (+); Runx2, ALP and OCN expression (+) | C3H10T1/2 cells with FoxO1 overexpression or silencing; mice with downregulated FoxO1 expression in developing embryos in vivo/embryonic tibiae ex vivo | [22] |
| osteoblast differentiation (+); osteoblast proliferation (-) | depletion/overexpression of FoxO1 in MC3T3-E1 cells | [25] | |||
| FoxO1, 3, and 4 | oxidative stress (-); skeletal homeostasis (+) | CFU-osteoblasts (+); | conditional deletion of FoxO1, 3, and 4 in 3-month-old mice | [21] | |
| osteogenic differentiation (+) | Runx2, Osterix, and ALP expression (+) | [26] | |||
| Adipogenesis (-) | PPARγ (-) | global deletion of FoxO1, 3, and 4 in mice | [21] | ||
| bone mass (-); osteoblast numbers (-) | osteoprogenitor cells proliferation (-) | Wnt/β-catenin signaling (-); cyclin D1 expression (-) | mice lacking FoxO1, 3, and 4 in bipotential progenitors of osteoblast and adipocytes (expressing Osterix1) | [18] |
| FoxOs | Effects on Bone | Functions in Osteoblast | Mechanisms | Cell/Mice Models | References |
|---|---|---|---|---|---|
| FoxO1, 3, and 4 | bone mass (+); bone formation rate (BFR) (+) | osteoblast number (+); osteoblast apoptosis (-); oxidative stress (-) | osteoblast number (+) through osteoblastogenesis (+); osteoblast apoptosis (-) through a cell-autonomous mechanism that enhances oxidative stress | conditional deletion of FoxO1, 3, and 4 in 3-month-old mice | [26] |
| FoxO1 | BFR (+); bone volume (+) | osteoblast numbers (+); oxidative stress (-) | ROS activates the p53 signaling cascade, inducing cell cycle arrest and limiting osteoblast proliferation. | FoxOs deletion mice in bone | |
| FoxO3 | vertebral bone mass (+); BFR (+) | osteoblast number (+), osteoblast apoptosis (-), oxidative stress (-) | ROS (-); phosphorylation of p66 Shc (-) | mice overexpressing FoxO3 under the control of the osteocalcin promoter | |
| FoxO1 | bone mass (+), BFR (+) bone volume (+) | osteoblast proliferation (+), oxidative stress (-) | FoxO1 interacts with ATF4 and promotes amino acid import to favor the protein synthesis, such as glutathione. FoxO1 reduces ROS, activating a p53 signaling cascade, then promoting cell cycle. | FoxO1 deletion in mice from collagen1a1 expressing cells | [41] |
| FoxOs | Effects on Bone | Functions in Hematopoietic Stem Cell | Mechanisms | Cell/Mice Models | References |
|---|---|---|---|---|---|
| FoxO3 | oxidative DNA damage in HSC and progenitor cell (-) | ROS (-); the base excision repair pathway (+) | mice model with FoxO3−/− in HSC | [47] | |
| FoxO3 | HSC quiescence (+); HSC G2/M transition (-) | ROS-independent modulations of ATM and p16INK4a and ROS-mediated activation of p53/p21CIP1/WAF1/Sdi1 tumor suppressor pathways (-) | [46] | ||
| FoxO3 | bone mass (+) | osteoclast number (-) | mice overexpressing FoxO3 in monocyte/macrophage lineage cells | [21] | |
| FoxO1, 3, and 4 | HSC quiescence (+); HSC compartment survival (+) | partly by impairing detoxification of ROS, FoxOs decreases HSC apoptosis and HSC-specific entry into the S/G2/M and G1 phases of the cell cycle | conditional deletion of FoxO1, 3, and 4 in the adult mice hematopoietic system | [48] |
| FoxOs | Effects on Bone | Functions in Osteoclast and Progenitor Cell | Mechanisms | Cell/Mice Models | References |
|---|---|---|---|---|---|
| FoxO1, 3, and 4 | bone resorption (-) | osteoclast progenitor proliferation (-); osteoclast lifespan (-) | FoxOs upregulate the H2O2-inactivating enzyme catalase and attenuates H2O2 accumulation | FoxO1,3,4f/f; LysM-Cre C57BL/6 mice; transgenic C57BL/6 mice: mitochondria-targeted catalase in osteoclasts | [55] |
| FoxO1, 3, and 4 | bone mass (+); BFR (+) | osteoclast progenitor numbers (-); osteoclast apoptosis (-) | FoxOs increase the expression of the osteoclast-specific markers like the calcitonin receptor, TRAP, and cathepsin K. | conditional deletion of FoxO1, 3, and 4 in 3-month-old mice | [26] |
| FoxO3 | vertebral bone mass (+); BFR (+) | osteoclast progenitor numbers (-); osteoclast numbers (-) | mice overexpressing FoxO3 under the control of the osteocalcin promoter | ||
| FoxO1 | bone resorption (-); osteoclast surface (-) | FoxO1 deletion in mice from collagen1a1 expressing cells | [41] | ||
| FoxO1 | osteoclast differentiation (-); osteoclast activity (-) | MAPKs, NF-κB and AP-1 (-); MYC activity (-); ROS (-); | bone marrow mononuclear cells; RAW264.7 cells | [57] | |
| FoxO1 | osteoclastogenesis of calvarial bone (+) | osteoclastogenesis (+); osteoclast activity (+); osteoclast precursor migration (+) | FoxO1 activates osteoclast formation by mediating the effect of RANKL on NFATc1 and several downstream effectors; FoxO1 deletion or knockdown reduces M-CSF induced RANK expression and migration of osteoclast precursors. | LyzM.Cre+FoxO1 L/L mice; BMMs or RAW264.7 cells transfected with FoxO1 siRNA | [51] |
| FoxOs | Effects on Bone | Functions in Chondrocyte | Mechanisms | Cell/Mice Models | References |
|---|---|---|---|---|---|
| FoxO1, FoxO3 | chondrocyte viability (+); chondrocyte apoptosis (-) | FoxO1 and FoxO3 up-regulated antioxidant proteins and autophagy-related proteins, but decreased expression of ADAMTS-4 and chemerin. | human articular chondrocyte transfected into siFoxO1 and siFoxO3 | [70] | |
| FoxO1, 3, and 4 | hypertrophic zone of the growth plate (-); overall body and tail length at eight weeks of age (-); hyperkyphosis (-) | expression of genes involved in redox homeostasis (+) | FoxO1,3a,4f/f; Collagen2-Cre mice; | [67] | |
| FoxO1, 3, and 4 | total body and tail length at 1 month of age (-); height of the proliferative zone of proximal tibial growth plate at P7 and 1 month (-); articular cartilage thicker at 1 or 2 months of age (-); OA-like changes developed in cartilage, synovium, and subchondral bone between 4 and 6 months of age (-) | chondrocyte proliferation (-); cell density (+); | autophagy and antioxidant defense genes (+); Prg4 expression (+) | Col2Cre-FoxO1, 3, and 4 triple knockout mice (Col2Cre-TKO); Col2Cre-FoxO1 knockout mice | [68] |
| FoxO3 or 4 | no cartilage abnormalities until 18 months of age | Col2Cre-FoxO3 or 4 single knockout mice | |||
| FoxO1, 3, and 4 | spontaneous cartilage degradation and OA severity in a surgical model or treadmill running of skeletally mature mice (-) | cell density (+); | Prg4 expression (+) | deletion of FoxO1/3/4 in mature mice using Aggrecan-CreERT2 | |
| FoxO3 | cell apoptosis (+); chondrogenic differentiation (-) | expression level of markers specific for mature (aggrecan, collagen II) and hypertrophic (collagen X) chondrocytes (-) | multipotent mesenchymal stromal cells (MSCs) | [69] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; Su, P.; Yin, C.; Lin, X.; Wang, X.; Gao, Y.; Patil, S.; War, A.R.; Qadir, A.; Tian, Y.; et al. The Roles of FoxO Transcription Factors in Regulation of Bone Cells Function. Int. J. Mol. Sci. 2020, 21, 692. https://doi.org/10.3390/ijms21030692
Ma X, Su P, Yin C, Lin X, Wang X, Gao Y, Patil S, War AR, Qadir A, Tian Y, et al. The Roles of FoxO Transcription Factors in Regulation of Bone Cells Function. International Journal of Molecular Sciences. 2020; 21(3):692. https://doi.org/10.3390/ijms21030692
Chicago/Turabian StyleMa, Xiaoli, Peihong Su, Chong Yin, Xiao Lin, Xue Wang, Yongguang Gao, Suryaji Patil, Abdul Rouf War, Abdul Qadir, Ye Tian, and et al. 2020. "The Roles of FoxO Transcription Factors in Regulation of Bone Cells Function" International Journal of Molecular Sciences 21, no. 3: 692. https://doi.org/10.3390/ijms21030692
APA StyleMa, X., Su, P., Yin, C., Lin, X., Wang, X., Gao, Y., Patil, S., War, A. R., Qadir, A., Tian, Y., & Qian, A. (2020). The Roles of FoxO Transcription Factors in Regulation of Bone Cells Function. International Journal of Molecular Sciences, 21(3), 692. https://doi.org/10.3390/ijms21030692