RasGRP2 Structure, Function and Genetic Variants in Platelet Pathophysiology


Abstract
1. RasGRPs General Description
Cell Signal Transduction Is a Finely Regulated Process That Relies on Several Control Hubs
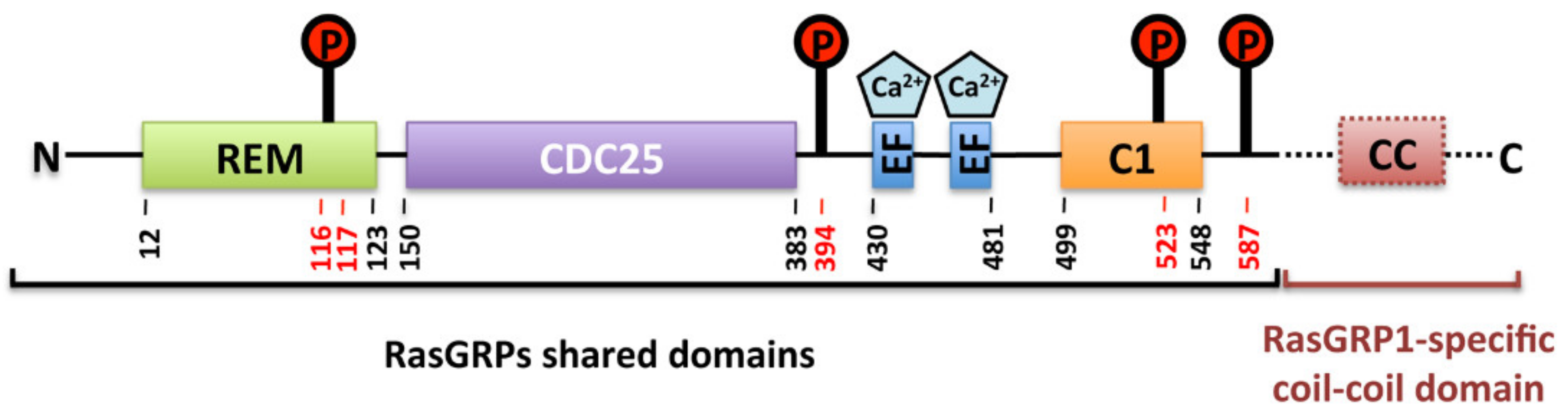
2. RasGRPs Structure and Domain Organization
3. RasGRP2
4. RasGRP2 Functions in Platelets
5. RasGRP2 Functions Outside Platelets
6. RasGRP2 Activity Regulation
6.1. Calcium
6.2. Phosphorylation
6.3. C1 Domain
7. RasGRP2 Variants and RasGRP2-Related Bleeding Disorders
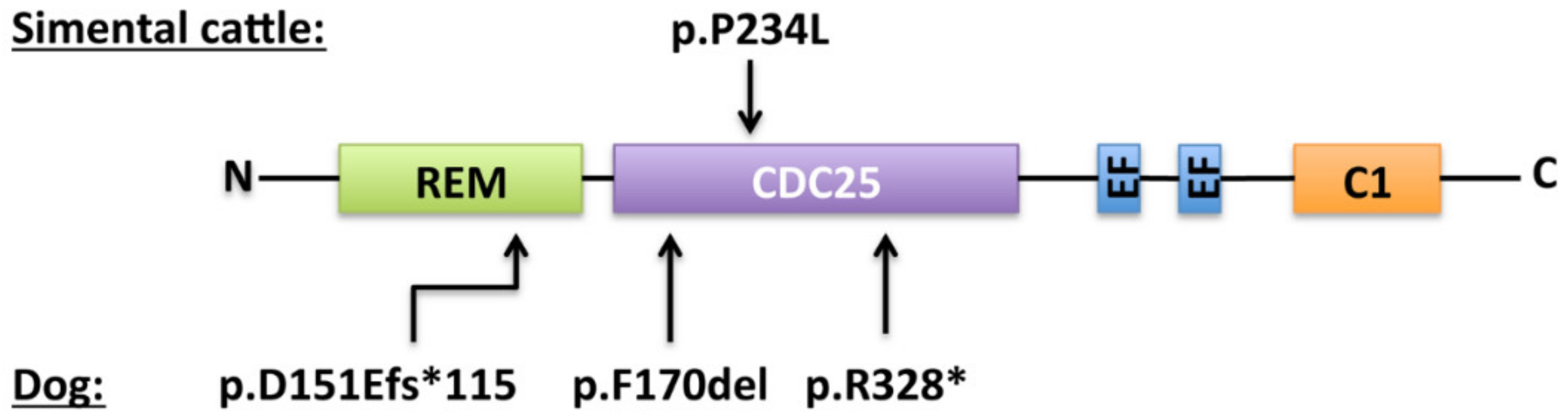
7.1. In Animals
7.2. In humans
7.3. Diagnosis
7.4. RASGRP2 Gene Variations
7.5. Patient Management
Author Contributions
Funding
Conflicts of Interest
References
- Rojas, A.M.; Fuentes, G.; Rausell, A.; Valencia, A. The Ras protein superfamily: Evolutionary tree and role of conserved amino acids. J. Cell Biol. 2012, 196, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Schweins, T.; Geyer, M.; Scheffzek, K.; Warshel, A.; Kalbitzer, H.R.; Wittinghofer, A. Substrate-assisted catalysis as a mechanism for GTP hydrolysis of p21ras and other GTP-binding proteins. Nat. Genet. 1995, 2, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Shimizu, T.; Toma-Fukai, S. Structural Insights into the Regulation Mechanism of Small GTPases by GEFs. Molecules 2019, 24, 3308. [Google Scholar] [CrossRef] [PubMed]
- Boriack-Sjodin, P.A.; Margarit, S.M.; Bar-Sagi, D.; Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. [Google Scholar] [CrossRef]
- Rehmann, H.; Das, J.; Knipscheer, P.; Wittinghofer, A.; Bos, J.L. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature 2006, 439, 625–628. [Google Scholar] [CrossRef]
- Kawasaki, H.; Springett, G.M.; Toki, S.; Canales, J.J.; Harlan, P.; Blumenstiel, J.P.; Chen, E.J.; Bany, I.A.; Mochizuki, N.; Ashbacher, A. A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc. Natl. Acad. Sci. USA 1998, 95, 13278–13283. [Google Scholar] [CrossRef]
- Ebinu, J.O. RasGRP, a Ras Guanyl Nucleotide- Releasing Protein with Calcium- and Diacylglycerol-Binding Motifs. Science 1998, 280, 1082–1086. [Google Scholar] [CrossRef]
- Dower, N.A.; Stang, S.L.; Bottorff, D.A.; Ebinu, J.O.; Dickie, P.; Ostergaard, H.L.; Stone, J.C. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat. Immunol. 2000, 1, 317–321. [Google Scholar] [CrossRef]
- Lee, S.H.; Yun, S.; Lee, J.; Kim, M.J.; Piao, Z.-H.; Jeong, M.; Chung, J.W.; Kim, T.-D.; Yoon, S.R.; Greenberg, P.D.; et al. RasGRP1 is required for human NK cell function. J. Immunol. Baltim. Md 1950 2009, 183, 7931–7938. [Google Scholar]
- Crittenden, J.R.; Bergmeier, W.; Zhang, Y.; Piffath, C.L.; Liang, Y.; Wagner, D.D.; Housman, D.E.; Graybiel, A.M. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat. Med. 2004, 10, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Cifuni, S.M.; Wagner, D.D.; Bergmeier, W. CalDAG-GEFI and protein kinase C represent alternative pathways leading to activation of integrin alphaIIbbeta3 in platelets. Blood 2008, 112, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Carbo, C.; Duerschmied, D.; Goerge, T.; Hattori, H.; Sakai, J.; Cifuni, S.M.; White, G.C.; Chrzanowska-Wodnicka, M.; Luo, H.R.; Wagner, D.D. Integrin-independent role of CalDAG-GEFI in neutrophil chemotaxis. J. Leukoc. Boil. 2010, 88, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Ghandour, H.; Cullere, X.; Alvarez, A.; Luscinskas, F.W.; Mayadas, T.N. Essential role for Rap1 GTPase and its guanine exchange factor CalDAG-GEFI in LFA-1 but not VLA-4 integrin–mediated human T-cell adhesion. Blood 2007, 110, 3682–3690. [Google Scholar] [CrossRef]
- Nakamura, H.; Shimamura, S.; Yasuda, S.; Kono, M.; Kono, M.; Fujieda, Y.; Kato, M.; Oku, K.; Bohgaki, T.; Shimizu, T.; et al. Ectopic RASGRP2 (CalDAG-GEFI) expression in rheumatoid synovium contributes to the development of destructive arthritis. Ann. Rheum. Dis. 2018, 77, 1765–1772. [Google Scholar] [CrossRef]
- Nagamine, K.; Matsuda, A.; Hori, T. Identification of the gene regulatory region in human rasgrp2 gene in vascular endothelial cells. Boil. Pharm. Bull. 2010, 33, 1138–1142. [Google Scholar] [CrossRef]
- Yamashita, S.; Mochizuki, N.; Ohba, Y.; Tobiume, M.; Okada, Y.; Sawa, H.; Nagashima, K.; Matsuda, M. CalDAG-GEFIII activation of Ras, R-ras, and Rap1. J. Biol. Chem. 2000, 275, 25488–25493. [Google Scholar] [CrossRef]
- Teixeira, C.; Stang, S.L.; Zheng, Y.; Beswick, N.S.; Stone, J.C. Integration of DAG signaling systems mediated by PKC-dependent phosphorylation of RasGRP3. Blood 2003, 102, 1414–1420. [Google Scholar] [CrossRef]
- Roberts, D.M.; Anderson, A.L.; Hidaka, M.; Swetenburg, R.L.; Patterson, C.; Stanford, W.L.; Bautch, V.L. A Vascular Gene Trap Screen Defines RasGRP3 as an Angiogenesis-Regulated Gene Required for the Endothelial Response to Phorbol Esters. Mol. Cell. Biol. 2004, 24, 10515–10528. [Google Scholar] [CrossRef]
- Botelho, R.J.; Harrison, R.E.; Stone, J.C.; Hancock, J.F.; Philips, M.R.; Jongstra-Bilen, J.; Mason, D.; Plumb, J.; Gold, M.R.; Grinstein, S. Localized diacylglycerol-dependent stimulation of Ras and Rap1 during phagocytosis. J. Biol. Chem. 2009, 284, 28522–28532. [Google Scholar] [CrossRef]
- Yang, Y.; Li, L.; Wong, G.W.; Krilis, S.A.; Madhusudhan, M.S.; Sali, A.; Stevens, R.L. RasGRP4, a new mast cell-restricted Ras guanine nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Identification of defective variants of this signaling protein in asthma, mastocytosis, and mast cell leukemia patients and demonstration of the importance of RasGRP4 in mast cell development and function. J. Biol. Chem. 2002, 277, 25756–25774. [Google Scholar] [PubMed]
- Suire, S.; Lécureuil, C.; Anderson, K.E.; Damoulakis, G.; Niewczas, I.; Davidson, K.; Guillou, H.; Pan, D.; Clark, J.; Hawkins, P.T.; et al. GPCR activation of Ras and PI3Kc in neutrophils depends on PLCb2/b3 and the RasGEF RasGRP4. EMBO J. 2012, 31, 3118–3129. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Fuller, D.M.; Zhang, W. The role of Ras guanine nucleotide releasing protein 4 in Fc epsilonRI-mediated signaling, mast cell function, and T cell development. J. Biol. Chem. 2012, 287, 8135–8143. [Google Scholar] [CrossRef] [PubMed]
- Ksionda, O.; Limnander, A.; Roose, J.P. RasGRP Ras guanine nucleotide exchange factors in cancer. Front. Boil. 2013, 8, 508–532. [Google Scholar] [CrossRef] [PubMed]
- Lewit-Bentley, A.; Réty, S. EF-hand calcium-binding proteins. Curr. Opin. Struct. Boil. 2000, 10, 637–643. [Google Scholar] [CrossRef]
- Johnson, J.E.; Goulding, R.E.; Ding, Z.; Partovi, A.; Anthony, K.V.; Beaulieu, N.; Tazmini, G.; Cornell, R.B.; Kay, R.J. Differential membrane binding and diacylglycerol recognition by C1 domains of RasGRPs. Biochem. J. 2007, 406, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Murphy, R.; Kerrigan, S.W.; Bertoni, A.; Stuhlmann, H.; Nakano, T.; Leavitt, A.D.; Shattil, S.J. Megakaryocytes derived from embryonic stem cells implicate CalDAG-GEFI in integrin signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 12819–12824. [Google Scholar] [CrossRef]
- Zang, C.; Luyten, A.; Chen, J.; Liu, X.S.; Shivdasani, R.A. NF-E2, FLI1 and RUNX1 collaborate at areas of dynamic chromatin to activate transcription in mature mouse megakaryocytes. Sci. Rep. 2016, 6, 30255. [Google Scholar] [CrossRef]
- Shiraga, M.; Ritchie, A.; Aidoudi, S.; Baron, V.; Wilcox, D.; White, G.; Ybarrondo, B.; Murphy, G.; Leavitt, A.; Shattil, S. Primary megakaryocytes reveal a role for transcription factor NF-E2 in integrin alpha IIb beta 3 signaling. J. Cell Biol. 1999, 147, 1419–1430. [Google Scholar] [CrossRef]
- Choi, J.; Baldwin, T.M.; Wong, M.; Bolden, J.E.; Fairfax, K.A.; Lucas, E.C.; Cole, R.; Biben, C.; Morgan, C.; Ramsay, K.A.; et al. Haemopedia RNA-seq: A database of gene expression during haematopoiesis in mice and humans. Nucleic Acids Res. 2019, 47, D780–D785. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2014, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Jaśkiewicz, A.; Pająk, B.; Orzechowski, A. The Many Faces of Rap1 GTPase. Int. J. Mol. Sci. 2018, 19, 2848. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [PubMed]
- Stefanini, L.; Lee, R.H.; Paul, D.S.; O’Shaughnessy, E.C.; Ghalloussi, D.; Jones, D.I.; Boulaftali, Y.; Poe, K.O.; Piatt, R.; Kechele, D.O.; et al. Functional redundancy between RAP1 isoforms in murine platelet production and function. Blood 2018, 132, 1951–1962. [Google Scholar] [CrossRef]
- Schultess, J.; Danielewski, O.; Smolenski, A.P. Rap1GAP2 is a new GTPase-activating protein of Rap1 expressed in human platelets. Blood 2005, 105, 3185–3192. [Google Scholar] [CrossRef] [PubMed]
- Lorenowicz, M.J.; Van Gils, J.; De Boer, M.; Hordijk, P.L.; Fernandez-Borja, M. Epac1-Rap1 signaling regulates monocyte adhesion and chemotaxis. J. Leukoc. Boil. 2006, 80, 1542–1552. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodeling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Bird, G.S.; Aziz, O.; Lievremont, J.-P.; Wedel, B.J.; Trebak, M.; Vazquez, G.; Putney, J.W. Mechanisms of phospholipase C-regulated calcium entry. Curr. Mol. Med. 2004, 4, 291–301. [Google Scholar] [CrossRef]
- Quinton, T.M.; Kim, S.; Dangelmaier, C.; Dorsam, R.T.; Jin, J.; Daniel, J.L.; Kunapuli, S.P. Protein kinase C- and calcium-regulated pathways independently synergize with Gi pathways in agonist-induced fibrinogen receptor activation. Biochem. J. 2002, 368, 535–543. [Google Scholar] [CrossRef]
- Quinton, T.M.; Ozdener, F.; Dangelmaier, C.; Daniel, J.L.; Kunapuli, S.P. Glycoprotein VI–mediated platelet fibrinogen receptor activation occurs through calcium-sensitive and PKC-sensitive pathways without a requirement for secreted ADP. Blood 2002, 99, 3228–3234. [Google Scholar] [CrossRef]
- Stolla, M.; Stefanini, L.; Roden, R.C.; Chavez, M.; Hirsch, J.; Greene, T.; Ouellette, T.D.; Maloney, S.F.; Diamond, S.L.; Poncz, M.; et al. The kinetics of αIIbβ3 activation determines the size and stability of thrombi in mice: Implications for antiplatelet therapy. Blood 2011, 117, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Franke, B.; van Triest, M.; de Bruijn, K.M.; van Willigen, G.; Nieuwenhuis, H.K.; Negrier, C.; Akkerman, J.W.; Bos, J.L. Sequential regulation of the small GTPase Rap1 in human platelets. Mol. Cell. Biol. 2000, 20, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Woulfe, D.; Jiang, H.; Mortensen, R.; Yang, J.; Brass, L.F. Activation of Rap1B by G(i) family members in platelets. J. Biol. Chem. 2002, 277, 23382–23390. [Google Scholar] [CrossRef] [PubMed]
- Lova, P.; Paganini, S.; Hirsch, E.; Barberis, L.; Wymann, M.; Sinigaglia, F.; Balduini, C.; Torti, M. A selective role for phosphatidylinositol 3,4,5-trisphosphate in the Gi-dependent activation of platelet Rap1B. J. Biol. Chem. 2003, 278, 131–138. [Google Scholar] [CrossRef]
- Lova, P.; Paganini, S.; Sinigaglia, F.; Balduini, C.; Torti, M. A Gi-dependent pathway is required for activation of the small GTPase Rap1B in human platelets. J. Biol. Chem. 2002, 277, 12009–12015. [Google Scholar] [CrossRef]
- Rowley, J.W.; Oler, A.J.; Tolley, N.D.; Hunter, B.N.; Low, E.N.; Nix, D.A.; Yost, C.C.; Zimmerman, G.A.; Weyrich, A.S. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood 2011, 118, e101–e111. [Google Scholar] [CrossRef]
- Simon, L.M.; Edelstein, L.C.; Nagalla, S.; Woodley, A.B.; Chen, E.S.; Kong, X.; Ma, L.; Fortina, P.; Kunapuli, S.; Holinstat, M.; et al. Human platelet microRNA-mRNA networks associated with age and gender revealed by integrated plateletomics. Blood 2014, 123, e37–e45. [Google Scholar] [CrossRef]
- Stefanini, L.; Paul, D.S.; Robledo, R.F.; Chan, E.R.; Getz, T.M.; Campbell, R.A.; Kechele, D.O.; Casari, C.; Piatt, R.; Caron, K.M.; et al. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J. Clin. Investig. 2015, 125, 1419–1432. [Google Scholar] [CrossRef]
- Stefanini, L.; Bergmeier, W. Negative regulators of platelet activation and adhesion. J. Thromb. Haemost. 2017, 16, 220–230. [Google Scholar] [CrossRef]
- Brass, L.F.; Diamond, S.L.; Stalker, T.J. Platelets and hemostasis: A new perspective on an old subject. Blood Adv. 2016, 1, 5–9. [Google Scholar] [CrossRef]
- Grover, S.P.; Bergmeier, W.; Mackman, N. Recent highlights of ATVB: Platelet signaling pathways and new inhibitors. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e28–e35. [Google Scholar] [CrossRef] [PubMed]
- Lagarrigue, F.; Kim, C.; Ginsberg, M.H. The Rap1-RIAM-talin axis of integrin activation and blood cell function. Blood 2016, 128, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Piatt, R.; Paul, D.S.; Lee, R.H.; McKenzie, S.E.; Parise, L.V.; Cowley, D.O.; Cooley, B.C.; Bergmeier, W. Mice Expressing Low Levels of CalDAG-GEFI Exhibit Markedly Impaired Platelet Activation With Minor Impact on Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, L.; Roden, R.C.; Bergmeier, W. CalDAG-GEFI is at the nexus of calcium-dependent platelet activation. Blood 2009, 114, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Wodnicka, M.; Smyth, S.S.; Schoenwaelder, S.M.; Fischer, T.H.; White, G.C., II. Rap1b is required for normal platelet function and hemostasis in mice. J. Clin. Investig. 2005, 115, 680. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, L.; Boulaftali, Y.; Ouellette, T.D.; Holinstat, M.; Désiré, L.; Leblond, B.; Andre, P.; Conley, P.B.; Bergmeier, W. Rap1-Rac1 circuits potentiate platelet activation. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 434–441. [Google Scholar] [CrossRef]
- Bernardi, B.; Guidetti, G.F.; Campus, F.; Crittenden, J.R.; Graybiel, A.M.; Balduini, C.; Torti, M. The small GTPase Rap1b regulates the cross talk between platelet integrin alpha2beta1 and integrin alphaIIbbeta3. Blood 2006, 107, 2728–2735. [Google Scholar] [CrossRef]
- Canault, M.; Ghalloussi, D.; Grosdidier, C.; Guinier, M.; Perret, C.; Chelghoum, N.; Germain, M.; Raslova, H.; Peiretti, F.; Morange, P.E.; et al. Human CalDAG-GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. J. Exp. Med. 2014, 211, 1349–1362. [Google Scholar] [CrossRef]
- Lozano, M.L.; Cook, A.; Bastida, J.M.; Paul, D.S.; Iruin, G.; Cid, A.R.; Adan-Pedroso, R.; Ramón González-Porras, J.; Hernández-Rivas, J.M.; Fletcher, S.J.; et al. Novel mutations in RASGRP2, which encodes CalDAG-GEFI, abrogate Rap1 activation, causing platelet dysfunction. Blood 2016, 128, 1282–1289. [Google Scholar]
- Sevivas, T.; Bastida, J.M.; Paul, D.S.; Caparros, E.; Palma-Barqueros, V.; Coucelo, M.; Marques, D.; Ferrer-Marín, F.; González-Porras, J.R.; Vicente, V.; et al. Identification of two novel mutations in RASGRP2 affecting platelet CalDAG-GEFI expression and function in patients with bleeding diathesis. Platelets 2018, 29, 192–195. [Google Scholar]
- Ahmad, F.; Boulaftali, Y.; Greene, T.K.; Ouellette, T.D.; Poncz, M.; Feske, S.; Bergmeier, W. Relative contributions of stromal interaction molecule 1 and CalDAG-GEFI to calcium-dependent platelet activation and thrombosis. J. Thromb. Haemost. JTH 2011, 9, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Boulaftali, Y.; Owens, A.P.; Beale, A.; Piatt, R.; Casari, C.; Lee, R.H.; Conley, P.B.; Paul, D.S.; Mackman, N.; Bergmeier, W. CalDAG-GEFI Deficiency Reduces Atherosclerotic Lesion Development in Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Bergmeier, W.; Goerge, T.; Wang, H.-W.; Crittenden, J.R.; Baldwin, A.C.W.; Cifuni, S.M.; Housman, D.E.; Graybiel, A.M.; Wagner, D.D. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J. Clin. Investig. 2007, 117, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Stadtmann, A.; Brinkhaus, L.; Mueller, H.; Rossaint, J.; Bolomini-Vittori, M.; Bergmeier, W.; Van Aken, H.; Wagner, D.D.; Laudanna, C.; Ley, K.; et al. Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur. J. Immunol. 2011, 41, 2074–2085. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, K.; Shimonaka, M.; Kinashi, T. Rap1-mediated lymphocyte function-associated antigen-1 activation by the T cell antigen receptor is dependent on phospholipase C-gamma1. J. Biol. Chem. 2004, 279, 11875–11881. [Google Scholar] [CrossRef] [PubMed]
- Caloca, M.-J.; Zugaza, J.L.; Vicente-Manzanares, M.; Bustelo, X.R.; Sanchez-Madrid, F. F-actin-dependent Translocation of the Rap1 GDP/GTP Exchange Factor RasGRP2. J. Boil. Chem. 2004, 279, 20435–20446. [Google Scholar] [CrossRef]
- Dupuy, A.J.; Morgan, K.; von Lintig, F.C.; Shen, H.; Acar, H.; Hasz, D.E.; Jenkins, N.A.; Copeland, N.G.; Boss, G.R.; Largaespada, D.A. Activation of the Rap1 guanine nucleotide exchange gene, CalDAG-GEF I, in BXH-2 murine myeloid leukemia. J. Biol. Chem. 2001, 276, 11804–11811. [Google Scholar] [CrossRef]
- Riches, J.C.; O’Donovan, C.J.; Kingdon, S.J.; McClanahan, F.; Clear, A.J.; Neuberg, D.S.; Werner, L.; Croce, C.M.; Ramsay, A.G.; Rassenti, L.Z.; et al. Trisomy 12 chronic lymphocytic leukemia cells exhibit upregulation of integrin signaling that is modulated by NOTCH1 mutations. Blood 2014, 123, 4101–4110. [Google Scholar]
- Mele, S.; Devereux, S.; Pepper, A.G.; Infante, E.; Ridley, A.J. Calcium-RasGRP2-Rap1 signaling mediates CD38-induced migration of chronic lymphocytic leukemia cells. Blood Adv. 2018, 2, 1551–1561. [Google Scholar] [CrossRef]
- Nagamine, K.; Matsuda, A.; Asashima, M.; Hori, T. XRASGRP2 expression during early development of Xenopus embryos. Biochem. Biophys. Res. Commun. 2008, 372, 886–891. [Google Scholar] [CrossRef]
- Sato, T.; Takino, J.-I.; Nagamine, K.; Nishio, K.; Hori, T. RASGRP2 Suppresses Apoptosis via Inhibition of ROS Production in Vascular Endothelial Cells. Sci. World J. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Takino, J.; Sato, T.; Nagamine, K.; Hori, T. The inhibition of Bax activation-induced apoptosis by RasGRP2 via R-Ras-PI3K-Akt signaling pathway in the endothelial cells. Sci. Rep. 2019, 9, 16717. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Deng, Y. Disrupted striatal neuron inputs and outputs in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 250–280. [Google Scholar] [CrossRef] [PubMed]
- Crittenden, J.R.; Dunn, D.E.; Merali, F.I.; Woodman, B.; Yim, M.; Borkowska, A.E.; Frosch, M.P.; Bates, G.P.; Housman, D.E.; Lo, D.C.; et al. CalDAG-GEFI down-regulation in the striatum as a neuroprotective change in Huntington’s disease. Hum. Mol. Genet. 2010, 19, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Crittenden, J.R.; Cantuti-Castelvetri, I.; Saka, E.; Keller-McGandy, C.E.; Hernandez, L.F.; Kett, L.R.; Young, A.B.; Standaert, D.G.; Graybiel, A.M. Dysregulation of CalDAG-GEFI and CalDAG-GEFII predicts the severity of motor side-effects induced by anti-parkinsonian therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 2892–2896. [Google Scholar] [CrossRef]
- Nagai, T.; Nakamuta, S.; Kuroda, K.; Nakauchi, S.; Nishioka, T.; Takano, T.; Zhang, X.; Tsuboi, D.; Funahashi, Y.; Nakano, T.; et al. Phosphoproteomics of the Dopamine Pathway Enables Discovery of Rap1 Activation as a Reward Signal In Vivo. Neuron 2016, 89, 550–565. [Google Scholar] [CrossRef]
- Iwig, J.S.; Vercoulen, Y.; Das, R.; Barros, T.; Limnander, A.; Che, Y.; Pelton, J.G.; Wemmer, D.E.; Roose, J.P.; Kuriyan, J. Structural analysis of autoinhibition in the Ras-specific exchange factor RasGRP1. eLife 2013, 2, e00813. [Google Scholar] [CrossRef]
- Siess, W. Molecular mechanisms of platelet activation. Physiol. Rev. 1989, 69, 58–178. [Google Scholar] [CrossRef]
- Cook, A.A.; Deng, W.; Ren, J.; Li, R.; Sondek, J.; Bergmeier, W. Calcium-induced structural rearrangements release autoinhibition in the Rap-GEF CalDAG-GEFI. J. Boil. Chem. 2018, 293, 8521–8529. [Google Scholar] [CrossRef]
- Guidetti, G.F.; Manganaro, D.; Consonni, A.; Canobbio, I.; Balduini, C.; Torti, M. Phosphorylation of the guanine-nucleotide-exchange factor CalDAG-GEFI by protein kinase A regulates Ca(2+)-dependent activation of platelet Rap1b GTPase. Biochem. J. 2013, 453, 115–123. [Google Scholar] [CrossRef]
- Subramanian, H.; Zahedi, R.P.; Sickmann, A.; Walter, U.; Gambaryan, S. Phosphorylation of CalDAG-GEFI by protein kinase A prevents Rap1b activation. J. Thromb. Haemost. 2013, 11, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Beck, F.; Geiger, J.; Gambaryan, S.; Solari, F.A.; Dell’Aica, M.; Loroch, S.; Mattheij, N.J.; Mindukshev, I.; Pötz, O.; Jurk, K.; et al. Temporal quantitative phosphoproteomics of ADP stimulation reveals novel central nodes in platelet activation and inhibition. Blood 2017, 129, e1–e12. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Cook, A.A.; Bergmeier, W.; Sondek, J. A negative-feedback loop regulating ERK1/2 activation and mediated by RasGPR2 phosphorylation. Biochem. Biophys. Res. Commun. 2016, 474, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Bergmeier, W.; Canault, M.; Alessi, M.-C.; Paul, D.S.; Nurden, P.; Pillois, X.; Jy, W.; Ahn, Y.S.; Nurden, A.T. Phenotype analysis and clinical management in a large family with a novel truncating mutation in RASGRP2, the CalDAG-GEFI encoding gene. Res. Pract. Thromb. Haemost. 2017, 1, 128–133. [Google Scholar] [CrossRef]
- Sarker, M.; Goliaei, A.; Golesi, F.; Poggi, M.; Cook, A.; Khan, M.A.I.; Temple, B.R.; Stefanini, L.; Canault, M.; Bergmeier, W.; et al. Subcellular localization of Rap1 GTPase activator CalDAG-GEFI is orchestrated by interaction of its atypical C1 domain with membrane phosphoinositides. J. Thromb. Haemost. 2019. [Google Scholar] [CrossRef]
- Steficek, B.A.; Thomas, J.S.; McConnell, M.F.; Bell, T.G. A primary platelet disorder of consanguineous simmental cattle. Thromb. Res. 1993, 72, 145–153. [Google Scholar] [CrossRef]
- Aebi, M.; Wiedemar, N.; Zanolari, P.; Drögemüller, C. Inherited thrombopathia in Simmental cattle. Schweiz Arch Tierheilkd 2016, 158, 102–108. [Google Scholar] [CrossRef]
- Boudreaux, M.K.; Schmutz, S.M.; French, P.S. Calcium Diacylglycerol Guanine Nucleotide Exchange Factor I (CalDAG-GEFI) Gene Mutations in a Thrombopathic Simmental Calf. Veter- Pathol. 2007, 44, 932–935. [Google Scholar] [CrossRef]
- Boudreaux, M.K.; Catalfamo, J.L.; Klok, M. Calcium-diacylglycerol guanine nucleotide exchange factor I gene mutations associated with loss of function in canine platelets. Transl. Res. J. Lab. Clin. Med. 2007, 150, 81–92. [Google Scholar] [CrossRef][Green Version]
- Yun, J.W.; Lee, K.-O.; Jung, C.W.; Oh, S.-Y.; Kim, S.-H.; Choi, C.W.; Kim, H.-J. Hereditary platelet function disorder from RASGRP2 gene mutations encoding CalDAG-GEFI identified by whole-exome sequencing in a Korean woman with severe bleeding. Haematologica 2019, 104, e274–e276. [Google Scholar] [CrossRef]
- Westbury, S.K.; Canault, M.; Greene, D.; Bermejo, E.; Hanlon, K.; Lambert, M.P.; Millar, C.M.; Nurden, P.; Obaji, S.G.; Revel-Vilk, S.; et al. Expanded repertoire of RASGRP2 variants responsible for platelet dysfunction and severe bleeding. Blood 2017, 130, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Lyu, S.J.; Ren, W.R.; Zhu, H.L.; Liu, T. [The clinical characteristics and molecular pathogenesis of a variant Glanzmann’s thrombasthenia-like pedigree]. Zhonghua Xue Ye Xue Za Zhi 2018, 39, 807–811. [Google Scholar]
- Lunghi, B.; Lecchi, A.; Santacroce, R.; Scavone, M.; Paniccia, R.; Artoni, A.; Gachet, C.; Castaman, G.; Margaglione, M.; Bernardi, F.; et al. Severe bleeding and absent ADP-induced platelet aggregation associated with inherited combined CalDAG-GEFI and P2Y12 deficiencies. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed]
- Manukjan, G.; Wiegering, V.A.; Reindl, T.; Strauß, G.; Klopocki, E.; Schulze, H.; Andres, O. Novel variants in FERMT3 and RASGRP2-Genetic linkage in Glanzmann-like bleeding disorders. Pediatr. Blood Cancer 2019, 67, e28078. [Google Scholar] [CrossRef]
- Bermejo, E.; Alberto, M.F.; Paul, D.S.; Cook, A.A.; Nurden, P.; Luceros, A.S.; Nurden, A.T.; Bergmeier, W. Marked bleeding diathesis in patients with platelet dysfunction due to a novel mutation in RASGRP2, encoding CalDAG-GEFI (p.Gly305Asp). Platelets 2018, 29, 84–86. [Google Scholar] [CrossRef]
- Kato, H.; Nakazawa, Y.; Kurokawa, Y.; Kashiwagi, H.; Morikawa, Y.; Morita, D.; Banno, F.; Honda, S.; Kanakura, Y.; Tomiyama, Y. Human CalDAG-GEFI deficiency increases bleeding and delays αIIbβ3 activation. Blood 2016, 128, 2729–2733. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database J. Biol. Databases Curation 2018, 2018, 1. [Google Scholar]
- Kuijpers, T.W.; van Bruggen, R.; Kamerbeek, N.; Tool, A.T.J.; Hicsonmez, G.; Gurgey, A.; Karow, A.; Verhoeven, A.J.; Seeger, K.; Sanal, O.; et al. Natural history and early diagnosis of LAD-1/variant syndrome. Blood 2007, 109, 3529–3537. [Google Scholar] [CrossRef]
- Poon, M.-C.; Di Minno, G.; D’Oiron, R.; Zotz, R. New Insights into the Treatment of Glanzmann Thrombasthenia. Transfus. Med. Rev. 2016, 30, 92–99. [Google Scholar] [CrossRef]
- Canault, M.; Saultier, P.; Fauré, S.; Poggi, M.; Nurden, A.T.; Nurden, P.; Morange, P.E.; Alessi, M.-C.; Gris, J.-C. Peripartum bleeding management in a patient with CalDAG-GEFI deficiency. Haemoph. Off. J. World Fed. Hemoph. 2017, 23, e533–e535. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Variation | Variant Type | Protein Effect | Sex | Platelet Expression | Age at Diagnosis | Age at Presentation | Bleeding Symptoms | Ref | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E | SC | M | DE | S | GI | ||||||||
| c.742G>T | Missense | p.Gly248Trp | F | Yes | 55 | 1 year | [58] | ||||||
| H | Yes | 53 | 1 year | ||||||||||
| H | Yes | 49 | 1 year | ||||||||||
| c.337C>T | Stop codon | p.Arg113* | F | No | 55 | Lifelong | [59] | ||||||
| M | No | 46 | Childhood | ||||||||||
| c.1142C>T | Missense | p.Ser381Phe | M | No | 9 | lifelong | [59] | ||||||
| c.1142C>T c.659G>A | Missense Missense | p.Ser381Phe p.Arg220Glu | F | N/D | 41 | Early childhood | [90] | ||||||
| c.925A>T c.1081_1083delCTG | Stop codon Deletion | p.Lys309* p.Leu360del | F | No | 16 | Before 3 years | [89] | ||||||
| c.706C>T | Stop codon | p.Gln236* | M | No | 8 | 1 year | [60] | ||||||
| c.887G>A | Missense | p.Cys296Tyr | F | No | 4 | First year | [60] | ||||||
| c.914G>A | Missense | p.Gly305Asp | M | No | 9 | Early childhood | [89,91] | ||||||
| M | Yes (Residual) | 24 | 2 years | ||||||||||
| c.199delAA | Frameshift | p.Asn67Leufs*24 | F | No | 23 | 1 year | [91] | ||||||
| c.372-3C>G | Splice variant | p.(Pro125*) | F | N/D | 24 | 2 years | [91] | ||||||
| c.990C>G | Missense | p.Asn330Lys | F | N/D | 21 | 5 years | [91] | ||||||
| c.778G>T c.886T>C | Stop codon Missense | p.Glu260* p.Cys296Arg | F | No | 20 | 1 year | [91] | ||||||
| c.1482InsG | Frameshift | p.Arg494Alafs*54 | M | N/D | 60 | 5 years | [91] | ||||||
| c.1482InsG c. 542T>C | Frameshift Missense | p.Arg494Alafs*54 p.Phe181Ser | M | N/D | 13 | 1 year | [91] | ||||||
| c.1490delT | Frameshift | p.Phe497Serfs*22 | M | Yes (Lower MW) | 45 | 5 years | [84,91] | ||||||
| F | Yes (Lower MW) | 55 | During the first year | ||||||||||
| c.1490delT c.1033G>C | Frameshift Missense | p.Phe497Serfs*22 p.Ala345Pro | M | N/D | 57 | 5 years | [91] | ||||||
| c.1490delT c.866A>G | Frameshift Missense | p.Phe497Serfs*22 p.Tyr289Cys | M | N/D | 61 | 4 years | [91] | ||||||
| c.74-1G>C | Splice variant | p.(Asp25Ala*15) | M | N/D | 9 | 3 years | [92] | ||||||
| c.337delC | Deletion | p.Arg113Aspfs*6 | M | N/D | 9 | During the first year | [93] | ||||||
| c.742G>C | Missense | p.Gly248Arg | F | N/D | 15 | Early childhood | [94] | ||||||
| Newly identified variant | |||||||||||||
| c.1507G>T | Stop codon | p.Glu503* | M | N/D | 14 | Early childhood | |||||||
| % of patients | 96 | 84 | 73 | 40 | 40 | 16 | |||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canault, M.; Alessi, M.-C. RasGRP2 Structure, Function and Genetic Variants in Platelet Pathophysiology. Int. J. Mol. Sci. 2020, 21, 1075. https://doi.org/10.3390/ijms21031075
Canault M, Alessi M-C. RasGRP2 Structure, Function and Genetic Variants in Platelet Pathophysiology. International Journal of Molecular Sciences. 2020; 21(3):1075. https://doi.org/10.3390/ijms21031075
Chicago/Turabian StyleCanault, Matthias, and Marie-Christine Alessi. 2020. "RasGRP2 Structure, Function and Genetic Variants in Platelet Pathophysiology" International Journal of Molecular Sciences 21, no. 3: 1075. https://doi.org/10.3390/ijms21031075
APA StyleCanault, M., & Alessi, M.-C. (2020). RasGRP2 Structure, Function and Genetic Variants in Platelet Pathophysiology. International Journal of Molecular Sciences, 21(3), 1075. https://doi.org/10.3390/ijms21031075