The “Janus Face” of Platelets in Cancer


Abstract
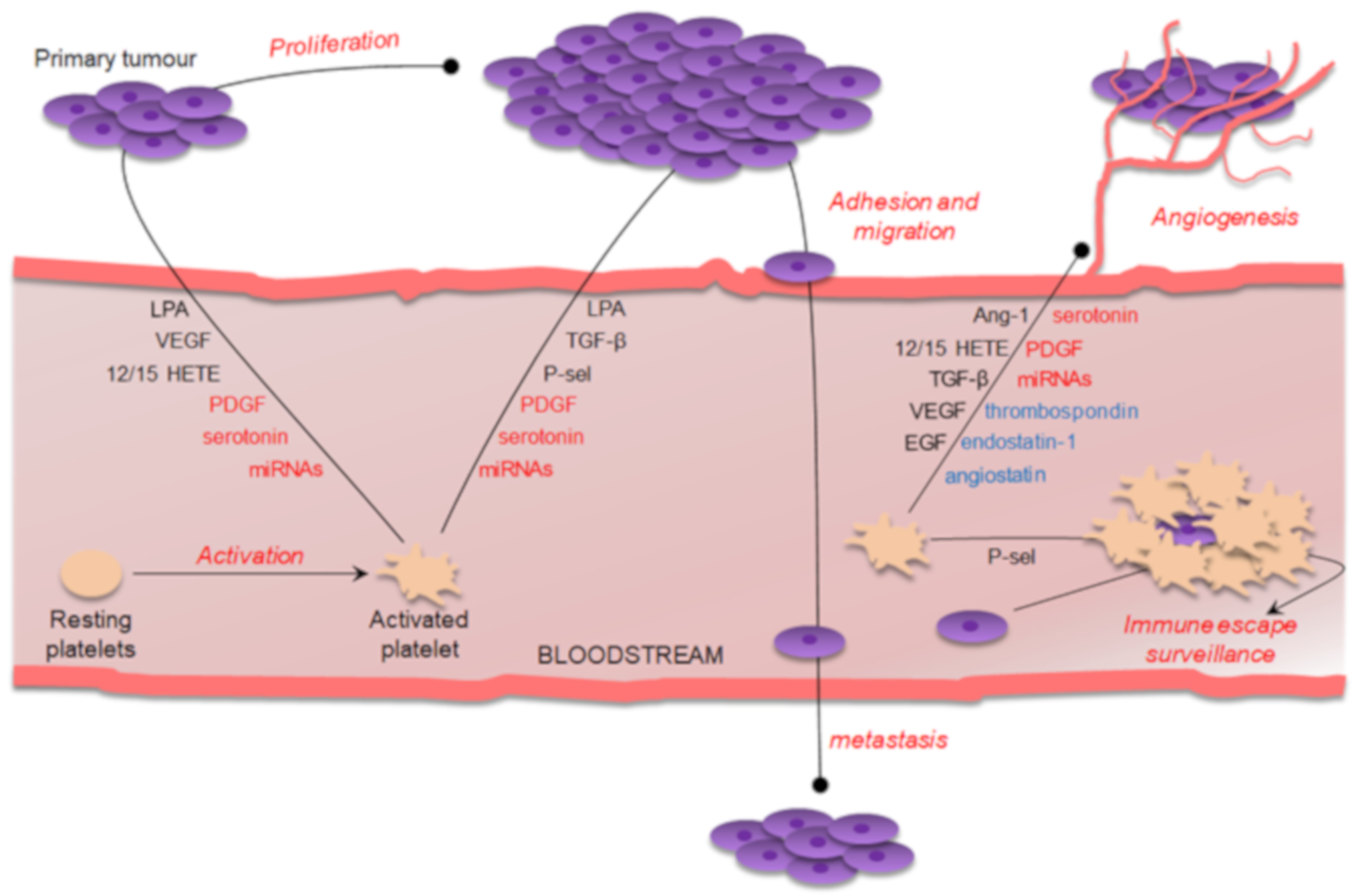
1. Introduction
2. Paraneoplastic Thrombocytosis and Thrombocytopenia in Cancer

{kind=link}
| Cancer | Study | Platelet Cut-Off | Main Findings | Ref. |
|---|---|---|---|---|
| Oesophageal |
| 450 × 109/L |
| [43] |
| 293 × 109/L |
| [33] | |
| 205 × 109/L |
| [34] | |
| 300 × 109/L |
| [41] | |
| 400 × 109/L |
| [39] | |
| 400 × 109/L |
| [40] | |
| Cervical |
| 400 × 109/L (seven studies) 300 × 109/L (six studies) 200 × 109/L (five studies) Not specified (one study) |
| [32] |
| Epithelial ovarian |
| 450 × 109/L |
| [28] |
| Lung |
| 300 × 109/L |
| [31] |
| 400 × 109/L (6 studies) 300 × 109/L (5 studies) 214.5 × 109/L (1 study) |
| [29] |
3. Platelet-Derived Bioactive Compounds
4. Platelet Microvesicle-Derived miRNAs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ang-1 | Angiopoetin-1 |
| COX | Cyclooxygenase |
| EGF | Epidermal growth factor |
| G-CSF | Granulocyte colony-stimulating factor |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GRO | Growth-regulated oncogene |
| HETE | Hydroxyeicosatraenoic acid |
| HGF | Hepatocyte growth factor |
| HOA | Homeobox |
| HR | Hazard ratio |
| JAK | Janus kinase |
| IBC | Inflammatory breast cancer |
| IL | Interleukin |
| LOX | Lipoxygenase |
| LPA | Lysophosphatidic acid |
| M-CSF | Macrophage colony-stimulating factor |
| miRNA | MicroRNA |
| MMP | Matrix metalloproteinase |
| MV | Microvesicle |
| PDGF | Platelet-derived growth factor |
| PUFA | Polyunsaturated fatty acid |
| SNP | Single nucleotide polymorphisms |
| STAT | Signal transducer and activator of transcription |
| TF | Tissue factor |
| TGF | Transforming growth factor |
| TPO | Thrombopoietin |
| VEGF | Vascular endothelial growth factor |
| VTE | Venous thromboembolism |
References
- Bizzozero, J. Ueber einen neuen Formbestandtheil des Blutes und dessen Rolle bei der Thrombose und der Blutgerinnung—Untersuchungen. Arch. Pathol. Anat. Physiol. Klin. Med. 1882, 90, 261–332. [Google Scholar] [CrossRef]
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Schulze, H.; Stegner, D. Imaging platelet biogenesis in vivo. Res. Pract. Thromb. Haemost. 2018, 2, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.H. Origin of Blood Platelets. Nature 1955, 176, 38. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.H. The histogenesis of the blood platelets. J. Morphol. 1910, 21, 263–278. [Google Scholar] [CrossRef]
- Vinholt, P.J. The role of platelets in bleeding in patients with thrombocytopenia and hematological disease. Clin. Chem. Lab. Med. 2019, 57, 1808–1817. [Google Scholar] [CrossRef]
- Mezger, M.; Nording, H.; Sauter, R.; Graf, T.; Heim, C.; von Bubnoff, N.; Ensminger, S.M.; Langer, H.F. Platelets and Immune Responses During Thromboinflammation. Front. Immunol. 2019, 10, 1731. [Google Scholar] [CrossRef]
- Assinger, A.; Schrottmaier, W.C.; Salzmann, M.; Rayes, J. Platelets in Sepsis: An Update on Experimental Models and Clinical Data. Front. Immunol. 2019, 10, 1687. [Google Scholar] [CrossRef]
- Mohammed, S.; Yu, J. Platelet-rich plasma injections: An emerging therapy for chronic discogenic low back pain. J. Spine Surg. 2018, 4, 115–122. [Google Scholar] [CrossRef]
- Nieswandt, B.; Kleinschnitz, C.; Stoll, G. Ischaemic stroke: A thrombo-inflammatory disease? J. Physiol. 2011, 589, 4115–4123. [Google Scholar] [CrossRef]
- Saluk-Bijak, J.; Dziedzic, A.; Bijak, M. Pro-Thrombotic Activity of Blood Platelets in Multiple Sclerosis. Cells 2019, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, K.; Bhattacharyya, M. Overview of Platelet Physiology: Its Hemostatic and Nonhemostatic Role in Disease Pathogenesis. Sci. World J. 2014, 2014, 781857. [Google Scholar] [CrossRef] [PubMed]
- Biino, G.; Santimone, I.; Minelli, C.; Sorice, R.; Frongia, B.; Traglia, M.; Ulivi, S.; Di Castelnuovo, A.; Gögele, M.; Nutile, T.; et al. Age- And Sex-Related Variations in Platelet Count in Italy: A Proposal of Reference Ranges Based on 40987 Subjects’ Data. PLoS ONE 2013, 8, e54289. [Google Scholar] [CrossRef] [PubMed]
- Eicher, J.D.; Lettre, G.; Johnson, A.D. The genetics of platelet count and volume in humans. Platelets 2018, 29, 125–130. [Google Scholar] [CrossRef]
- Catani, M.V.; Gasperi, V.; Evangelista, D.; Finazzi Agrò, A.; Avigliano, L.; Maccarrone, M. Anandamide extends platelets survival through CB1-dependent Akt signaling. Cell. Mol. Life Sci. 2010, 67, 601–610. [Google Scholar] [CrossRef]
- Grozovsky, R.; Giannini, S.; Hoffmeister, K.M. Novel mechanisms of platelet clearance and thrombopoietin regulation. Curr. Opin. Hematol. 2017, 22, 445–451. [Google Scholar] [CrossRef]
- Grozovsky, R.; Giannini, S.; Falet, H.; Hoffmeister, K.M. Novel mechanisms of platelet clearance and thrombopoietin regulation. Curr. Opin. Hematol. 2015, 22, 445–451. [Google Scholar] [CrossRef]
- Tefferi, A.; Thiele, J.; Orazi, A.; Kvasnicka, H.M.; Barbui, T.; Hanson, C.A.; Barosi, G.; Verstovsek, S.; Birgegard, G.; Mesa, R.; et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: Recommendations from an ad hoc international expert panel. Blood 2007, 110, 1092–1097. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Finazzi, G.; Vannucchi, A.M.; Tefferi, A. The 2016 revision of WHO classification of myeloproliferative neoplasms: Clinical and molecular advances. Blood Rev. 2016, 30, 453–459. [Google Scholar] [CrossRef]
- Song, J.; Hussaini, M.; Zhang, H.; Shao, H.; Qin, D.; Zhang, X.; Ma, Z.; Hussnain Naqvi, S.M.; Zhang, L.; Moscinski, L.C. Comparison of the Mutational Profiles of Primary Myelofibrosis, Polycythemia Vera, and Essential Thrombocytosis. Am. J. Clin. Pathol. 2017, 147, 444–452. [Google Scholar] [CrossRef]
- Chia, T.L.; Chesney, T.R.; Isa, D.; Mnatzakanian, G.; Colak, E.; Belmont, C.; Hirpara, D.; Veigas, P.V.; Acuna, S.A.; Rizoli, S.; et al. Thrombocytosis in splenic trauma: In-hospital course and association with venous thromboembolism. Injury 2017, 48, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Griesshammer, M.; Bangerter, M.; Sauer, T.; Wennauer, R.; Bergmann, L.; Heimpel, H. Aetiology and clinical significance of thrombocytosis: Analysis of 732 patients with an elevated platelet count. J. Intern. Med. 1999, 245, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Santhosh-Kumar, C.R.; Yohannan, M.D.; Higgy, K.E.; Al-Mashhadani, S.A. Thrombocytosis in adults: Analysis of 777 patients. J. Intern. Med. 1991, 229, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Francis, C.W.; Culakova, E.; Lyman, G.H. Risk factors for chemotherapy-associated venous thromboembolism in a prospective observational study. Cancer 2005, 104, 2822–2829. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Z.; Xu, R. Human cancer and platelet interaction, a potential therapeutic target. Int. J. Mol. Sci. 2018, 19, 1246. [Google Scholar] [CrossRef] [PubMed]
- Levin, J. Thrombocytosis Associated With Malignant Disease. Arch. Intern. Med. 1964, 114, 497. [Google Scholar] [CrossRef]
- Wojtukiewicz, M.Z.; Sierko, E.; Hempel, D.; Tucker, S.C.; Honn, K.V. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev. 2017, 36, 249–262. [Google Scholar] [CrossRef]
- Stone, R.L.; Nick, A.M.; McNeish, I.A.; Balkwill, F.; Han, H.D.; Bottsford-Miller, J.; Rupaimoole, R.; Armaiz-Pena, G.N.; Pecot, C.V.; Coward, J.; et al. Paraneoplastic Thrombocytosis in Ovarian Cancer. N. Engl. J. Med. 2012, 366, 610–618. [Google Scholar] [CrossRef]
- Zhang, X.; Ran, Y. Prognostic role of elevated platelet count in patients with lung cancer: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 5379–5387. [Google Scholar]
- Gu, D.; Szallasi, A. Thrombocytosis portends adverse prognosis in colorectal cancer: A meta-analysis of 5,619 patients in 16 individual studies. Anticancer Res. 2017, 37, 4717–4726. [Google Scholar]
- Ji, Y.; Sheng, L.; Du, X.; Qiu, G.; Su, D. Elevated platelet count is a strong predictor of poor prognosis in stage i non-small cell lung cancer patients. Platelets 2015, 26, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Yao, X.; Cen, D.; Zhi, Y.; Zhu, N.; Xu, L. Prognostic role of pretreatment thrombocytosis on survival in patients with cervical cancer: A systematic review and meta-analysis. World J. Surg. Oncol. 2019, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Oohira, G.; Okazumi, S.; Matsubara, H.; Nabeya, Y.; Hayashi, H.; Takeda, A.; Gunji, Y.; Ochiai, T. Thrombocytosis associated with poor prognosis in patients with esophageal carcinoma1 1No competing interests declared. J. Am. Coll. Surg. 2004, 198, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.F.; Huang, Y.; Lu, W.S.; Chen, Q.X. Preoperative platelet count in esophageal squamous cell carcinoma: Is it a prognostic factor? Langenbeck’s Arch. Surg. 2013, 398, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Lai, Y.; Myers, R.E.; Li, B.; Hyslop, T.; London, J.; Chatterjee, D.; Palazzo, J.P.; Burkart, A.L.; Zhang, K.; et al. Preoperative platelet count associates with survival and distant metastasis in surgically resected colorectal cancer patients. J. Gastrointest. Cancer 2013, 44, 293–304. [Google Scholar] [CrossRef]
- Lee, M.; Kim, S.W.; Nam, E.J.; Yim, G.W.; Kim, S.; Kim, Y.T. The impact of pretreatment thrombocytosis and persistent thrombocytosis after adjuvant chemotherapy in patients with advanced epithelial ovarian cancer. Gynecol. Oncol. 2011, 122, 238–241. [Google Scholar] [CrossRef]
- Harano, K.; Kogawa, T.; Wu, J.; Yuan, Y.; Cohen, E.N.; Lim, B.; Reuben, J.M.; Ueno, N.T. Thrombocytosis as a prognostic factor in inflammatory breast cancer. Breast Cancer Res. Treat. 2017, 166, 819–832. [Google Scholar] [CrossRef]
- Moschini, M.; Suardi, N.; Pellucchi, F.; Rocchini, L.; La Croce, G.; Capitanio, U.; Briganti, A.; Damiano, R.; Montorsi, F.; Colombo, R. Impact of preoperative thrombocytosis on pathological outcomes and survival in patients treated with radical cystectomy for bladder carcinoma. Anticancer Res. 2014, 34, 3225–3230. [Google Scholar]
- Dutta, S.; Crumley, A.B.C.; Fullarton, G.M.; Horgan, P.G.; McMillan, D.C. Comparison of the prognostic value of tumour and patient related factors in patients undergoing potentially curative resection of gastric cancer. Am. J. Surg. 2012, 204, 294–299. [Google Scholar] [CrossRef]
- Aminian, A.; Karimian, F.; Mirsharifi, R.; Alibakhshi, A.; Dashti, H.; Jahangiri, Y.; Safari, S.; Ghaderi, H.; Noaparast, M.; Hasani, S.; et al. Significance of platelet count in esophageal carcinomas. Saudi J. Gastroenterol. 2011, 17, 134. [Google Scholar] [CrossRef]
- Wang, J.; Liu, H.; Shao, N.; Tan, B.; Song, Q.; Jia, Y.; Cheng, Y. The clinical significance of preoperative plasma fibrinogen level and platelet count in resectable esophageal squamous cell carcinoma. World J. Surg. Oncol. 2015, 13, 157. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.B.; Gu, X.L.; Ma, X.Q.; Lv, T.F.; Wu, Y.; Xiao, Y.Y.; Yuan, D.M.; Li, Y.F.; Song, Y. Preoperative platelet count in predicting lymph node metastasis and prognosis in patients with non-small cell lung cancer. Neoplasma 2012, 60, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Agoston, A.T.; Srivastava, A.; Zheng, Y.; Bueno, R.; Odze, R.D.; Szallasi, Z. Paraneoplastic thrombocytosis is associated with increased mortality and increased rate of lymph node metastasis in oesophageal adenocarcinoma. Pathology 2017, 49, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Tranum, B.L.; Haut, A. Thrombocytosis: Platelet kinetics in neoplasia. J. Lab. Clin. Med. 1974, 84, 615–619. [Google Scholar]
- Kuter, D.J. The biology of thrombopoietin and thrombopoietin receptor agonists. Int. J. Hematol. 2013, 98, 10–23. [Google Scholar] [CrossRef]
- Tsukishiro, S.; Suzumori, N.; Nishikawa, H.; Arakawa, A.; Suzumori, K. Preoperative serum thrombopoietin levels are higher in patients with ovarian cancer than with benign cysts. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 140, 67–70. [Google Scholar] [CrossRef]
- Furuhashi, M.; Miyabe, Y.; Oda, H. A Case of Thrombopoietin-Producing Ovarian Carcinoma Confirmed by Immunohistochemistry. Gynecol. Oncol. 1999, 74, 278–281. [Google Scholar] [CrossRef]
- Weryńska, B.; Ramlau, R.; Podolak-Dawidziak, M.; Jankowska, R.; Prajs, I.; Usnarska-Zubkiewicz, L.; Kuliczkowski, K. Serum thrombopoietin levels in patients with reactive thrombocytosis due to lung cancer and in patients with essential thrombocythemia. Neoplasma 2003, 50, 447–451. [Google Scholar]
- Columbyova, L.; Loda, M.; Scadden, D.T. Thrombopoietin Receptor Expression in Human Cancer Cell Lines and Primary Tissues. Cancer Res. 1995, 55, 3509–3512. [Google Scholar]
- Besbes, S.; Shah, S.; Al-Dybiat, I.; Mirshahi, S.; Helfer, H.; Najah, H.; Fourgeaud, C.; Pocard, M.; Ghedira, I.; Soria, J.; et al. Thrombopoietin Secretion by Human Ovarian Cancer Cells. Int. J. Cell Biol. 2017, 2017, 1873834. [Google Scholar] [CrossRef]
- Hodge, D.R.; Hurt, E.M.; Farrar, W.L. The role of IL-6 and STAT3 in inflammation and cancer. Eur. J. Cancer 2005, 41, 2502–2512. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: Role in inflammatory thrombocytosis. Blood 2001, 98, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Wolber, E.-M.; Fandrey, J.; Frackowski, U.; Jelkmann, W. Hepatic Thrombopoietin mRNA Is Increased in Acute Inflammation. Thromb. Haemost. 2001, 86, 1421–1424. [Google Scholar] [PubMed]
- Wolber, E.M.; Jelkmann, W. Interleukin-6 increases thrombopoietin production in human hepatoma cells HepG2 and Hep3B. J. Interferon Cytokine Res. 2000, 20, 499–506. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Conze, D.; Weiss, L.; Regen, P.S.; Rincón, M.; Weaver, D.; Bhushan, A.; Johnson, P. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 2001, 61, 8851–8858. [Google Scholar]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef]
- Higashihara, M.; Sunaga, S.; Tange, T.; Oohashi, H.; Kurokawa, K. Increased secretion of lnterleukin-6 in malignant mesothelioma cells from a patient with marked thrombocytosis. Cancer 1992, 70, 2105–2108. [Google Scholar] [CrossRef]
- Shinriki, S.; Jono, H.; Ota, K.; Ueda, M.; Kudo, M.; Ota, T.; Oike, Y.; Endo, M.; Ibusuki, M.; Hiraki, A.; et al. Humanized anti-interleukin-6 receptor antibody suppresses tumor angiogenesis and in vivo growth of human oral squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 5426–5434. [Google Scholar] [CrossRef]
- Chen, F.; Teachey, D.T.; Pequignot, E.; Frey, N.; Porter, D.; Maude, S.L.; Grupp, S.A.; June, C.H.; Melenhorst, J.J.; Lacey, S.F. Measuring IL-6 and sIL-6R in serum from patients treated with tocilizumab and/or siltuximab following CAR T cell therapy. J. Immunol. Methods 2016, 434, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vainer, N.; Dehlendorff, C.; Johansen, J.S. Systematic literature review of IL-6 as a biomarker or treatment target in patients with gastric, bile duct, pancreatic and colorectal cancer. Oncotarget 2018, 9, 29820–29841. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Takahashi, T.; Nakamura, K.; Tsuyuoka, R.; Okuno, Y.; Enomoto, T.; Fukumoto, M.; Imura, H. Thrombocytosis in patients with tumors producing colony-stimulating factor. Blood 1992, 80, 2052–2059. [Google Scholar] [CrossRef] [PubMed]
- Assoian, R.K.; Komoriya, A.; Meyers, C.A.; Miller, D.M.; Sporn, M.B. Transforming growth factor-β in human platelets. Identification of a major storage site, purification, and characterization. J. Biol. Chem. 1983, 258, 7155–7160. [Google Scholar] [PubMed]
- Lian, S.; Zhai, X.; Wang, X.; Zhu, H.; Zhang, S.; Wang, W.; Wang, Z.; Huang, J. Elevated expression of growth-regulated oncogene-alpha in tumor and stromal cells predicts unfavorable prognosis in pancreatic cancer. Medicine 2016, 95, e4328. [Google Scholar] [CrossRef] [PubMed]
- Yung, M.M.H.; Tang, H.W.M.; Cai, P.C.H.; Leung, T.H.Y.; Ngu, S.F.; Chan, K.K.L.; Xu, D.; Yang, H.; Ngan, H.Y.S.; Chan, D.W. GRO-α and IL-8 enhance ovarian cancer metastatic potential via the CXCR2-mediated TAK1/NFκB signaling cascade. Theranostics 2018, 8, 1270–1285. [Google Scholar] [CrossRef]
- Phakathi, B.P.; Mannell, A.; Nietz, S. Early stage breast cancer with concomittant primary hyperparathyroidism and autoimmune thrombocytopenia: A case report. S. Afr. J. Surg. 2018, 56, 64–65. [Google Scholar] [CrossRef]
- Liebman, H.A. Thrombocytopenia in cancer patients. Thromb. Res. 2014, 133, S63–S69. [Google Scholar] [CrossRef]
- Khasraw, M.; Faraj, H.; Sheikha, A. Thrombocytopenia in solid tumors. Eur. J. Clin. Med. Oncol. 2010, 2, 89–92. [Google Scholar]
- Gaydos, L.A.; Freireich, E.J.; Mantel, N. The quantitative relation between platelet count and hemorrhage in patients with acute leukemia. N. Engl. J. Med. 1962, 266, 905–909. [Google Scholar] [CrossRef]
- Avvisati, G.; Tirindelli, M.C.; Annibali, O. Thrombocytopenia and hemorrhagic risk in cancer patients. Crit. Rev. Oncol. Hematol. 2003, 48, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Ghanavat, M.; Ebrahimi, M.; Rafieemehr, H.; Maniati, M.; Behzad, M.M.; Shahrabi, S. Thrombocytopenia in solid tumors: Prognostic significance. Oncol. Rev. 2019, 13, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Pooja, S.; Chaudhary, P.; Nayak, L.V.; Rajender, S.; Saini, K.S.; Deol, D.; Kumar, S.; Bid, H.K.; Konwar, R. Polymorphic variations in IL-1β, IL-6 and IL-10 genes, their circulating serum levels and breast cancer risk in Indian women. Cytokine 2012, 60, 122–128. [Google Scholar] [CrossRef]
- Rodríguez-Berriguete, G.; Sánchez-Espiridión, B.; Cansino, J.R.; Olmedilla, G.; Martínez-Onsurbe, P.; Sánchez-Chapado, M.; Paniagua, R.; Fraile, B.; Royuela, M. Clinical significance of both tumor and stromal expression of components of the IL-1 and TNF-α signaling pathways in prostate cancer. Cytokine 2013, 64, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Lin, B.; Ni, P.; Xu, H.; Huang, G. Interleukin-1B and interleukin-1 RN polymorphisms and gastric carcinoma risk: A meta-analysis. J. Gastroenterol. Hepatol. 2010, 25, 1604–1617. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.Y.; Yu, B.W.; Yang, Z.; Yang, S.S.; Bo, L.H.; Shan, X.Y.; Wang, H.J.; Zhu, Y.J.; Wu, X.S. Interleukin-1B 31 C>T polymorphism combined with Helicobacter pylori-modified gastric cancer susceptibility: Evidence from 37 studies. J. Cell. Mol. Med. 2016, 20, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Hefler, L.A.; Grimm, C.; Ackermann, S.; Malur, S.; Radjabi-Rahat, A.R.; Leodolter, S.; Beckmann, M.W.; Zeillinger, R.; Koelbl, H.; Tempfer, C.B. An interleukin-6 gene promoter polymorphism influences the biological phenotype of ovarian cancer. Cancer Res. 2003, 63, 3066–3068. [Google Scholar]
- Talar-Wojnarowska, R.; Gasiorowska, A.; Smolarz, B.; Romanowicz-Makowska, H.; Kulig, A.; Malecka-Panas, E. Clinical significance of interleukin-6 (Il-6) gene polymorphism and Il-6 serum level in pancreatic adenocarcinoma and chronic pancreatitis. Dig. Dis. Sci. 2009, 54, 683–689. [Google Scholar] [CrossRef]
- Takaku, M.; Grimm, S.A.; Wade, P.A. GATA3 in breast cancer: Tumor suppressor or oncogene? Gene Expr. 2015, 16, 163–168. [Google Scholar] [CrossRef]
- Li, Q.; Chen, C.; Ren, X.; Sun, W. DNA methylation profiling identifies the HOXA11 gene as an early diagnostic and prognostic molecular marker in human lung adenocarcinoma. Oncotarget 2017, 8, 33100–33109. [Google Scholar] [CrossRef][Green Version]
- Xia, B.; Shan, M.; Wang, J.; Zhong, Z.; Geng, J.; He, X.; Vu, T.; Zhang, D.; Pang, D. Homeobox A11 hypermethylation indicates unfavorable prognosis in breast cancer. Oncotarget 2017, 8, 9794–9805. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Ware, J. Pathophysiology 2: The Role of Platelets in Cancer Biology. In Cancer Treatment and Research; Springer: Cham, Switzerland, 2019; pp. 37–54. [Google Scholar]
- Kisucka, J.; Butterfield, C.E.; Duda, D.G.; Eichenberger, S.C.; Saffaripour, S.; Ware, J.; Ruggeri, Z.M.; Jain, R.K.; Folkman, J.; Wagner, D.D. Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc. Natl. Acad. Sci. USA 2006, 103, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Zuka, M.; Liu, J.; Russell, S.; Dent, J.; Guerrero, J.A.; Forsyth, J.; Maruszak, B.; Gartner, T.K.; Felding-Habermann, B.; et al. Platelet glycoprotein Ibα supports experimental lung metastasis. Proc. Natl. Acad. Sci. USA 2007, 104, 9024–9028. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Ho-Tin-Noé, B.; Schatzberg, D.; Yang, J.J.; Wagner, D.D. Increased Efficacy of Breast Cancer Chemotherapy in Thrombocytopenic Mice. Cancer Res. 2011, 71, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; Tai, L.H.; Falls, T.; De Souza, C.T.; Bell, J.C.; Carrier, M.; Atkins, H.; Boushey, R.; Auer, R.A. Surgical stress promotes the development of cancer metastases by a coagulation-dependent mechanism involving natural killer cells in a murine model. Ann. Surg. 2013, 258, 158–168. [Google Scholar] [CrossRef]
- Clar, K.L.; Hinterleitner, C.; Schneider, P.; Salih, H.R.; Maurer, S. Inhibition of NK reactivity against solid tumors by platelet-derived RANKL. Cancers 2019, 11, 277. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Farge, D.; Bounameaux, H.; Brenner, B.; Cajfinger, F.; Debourdeau, P.; Khorana, A.A.; Pabinger, I.; Solymoss, S.; Douketis, J.; Kakkar, A. International clinical practice guidelines including guidance for direct oral anticoagulants in the treatment and prophylaxis of venous thromboembolism in patients with cancer. Lancet Oncol. 2016, 17, e452–e466. [Google Scholar] [CrossRef]
- Zarà, M.; Canobbio, I.; Visconte, C.; Canino, J.; Torti, M.; Guidetti, G.F. Molecular mechanisms of platelet activation and aggregation induced by breast cancer cells. Cell. Signal. 2018, 48, 45–53. [Google Scholar] [CrossRef]
- Reddel, C.; Tan, C.; Chen, V. Thrombin Generation and Cancer: Contributors and Consequences. Cancers 2019, 11, 100. [Google Scholar] [CrossRef]
- Chang, J.; Jiang, L.; Wang, Y.; Yao, B.; Yang, S.; Zhang, B.; Zhang, M.-Z. 12/15 lipoxygenase regulation of colorectal tumorigenesis is determined by the relative tumor levels of its metabolite 12-HETE and 13-HODE in animal models. Oncotarget 2015, 6, 2879. [Google Scholar] [CrossRef] [PubMed]
- Duvernay, M.T.; Temple, K.J.; Maeng, J.G.; Blobaum, A.L.; Stauffer, S.R.; Lindsley, C.W.; Hamm, H.E. Contributions of Protease-Activated Receptors PAR1 and PAR4 to Thrombin-Induced GPIIbIIIa Activation in Human Platelets. Mol. Pharmacol. 2017, 91, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Dahlbäck, B. Pro- and anticoagulant properties of factor V in pathogenesis of thrombosis and bleeding disorders. Int. J. Lab. Hematol. 2016, 38, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Melvin, R.L.; Salsbury, F.R. Mechanistic insights into thrombin’s switch between “slow” and “fast” forms. Phys. Chem. Chem. Phys. 2017, 19, 24522–24533. [Google Scholar] [CrossRef] [PubMed]
- Abu Saadeh, F.; Langhe, R.; Galvin, D.M.; Toole, S.O.; O’Donnell, D.M.; Gleeson, N.; Norris, L.A. Procoagulant activity in gynaecological cancer patients; The effect of surgery and chemotherapy. Thromb. Res. 2016, 139, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Reitter, E.M.; Kaider, A.; Ay, C.; Quehenberger, P.; Marosi, C.; Zielinski, C.; Pabinger, I. Longitudinal analysis of hemostasis biomarkers in cancer patients during antitumor treatment. J. Thromb. Haemost. 2016, 14, 294–305. [Google Scholar] [CrossRef]
- Adams, G.N.; Rosenfeldt, L.; Frederick, M.; Miller, W.; Waltz, D.; Kombrinck, K.; McElhinney, K.E.; Flick, M.J.; Monia, B.P.; Revenko, A.S.; et al. Colon cancer growth and dissemination relies upon thrombin, Stromal PAR-1, and fibrinogen. Cancer Res. 2015, 75, 4235–4243. [Google Scholar] [CrossRef]
- Muqaku, B.; Eisinger, M.; Meier, S.M.; Tahir, A.; Pukrop, T.; Haferkamp, S.; Slany, A.; Reichle, A.; Gerner, C. Multi-omics analysis of serum samples demonstrates reprogramming of organ functions via systemic calcium mobilization and platelet activation in metastatic melanoma. Mol. Cell. Proteom. 2017, 16, 86–99. [Google Scholar] [CrossRef]
- McCarty, O.J.T.; Mousa, S.A.; Bray, P.F.; Konstantopoulos, K. Immobilized platelets support human colon carcinoma cell tethering, rolling, and firm adhesion under dynamic flow conditions. Blood 2000, 96, 1789–1797. [Google Scholar] [CrossRef]
- Li, S.S.; Ip, C.K.M.; Tang, M.Y.H.; Tang, M.K.S.; Tong, Y.; Zhang, J.; Hassan, A.A.; Mak, A.S.C.; Yung, S.; Chan, T.M.; et al. Sialyl Lewisx-P-selectin cascade mediates tumor–mesothelial adhesion in ascitic fluid shear flow. Nat. Commun. 2019, 10, 2406. [Google Scholar] [CrossRef]
- Del Conde, I.; Nabi, F.; Tonda, R.; Thiagarajan, P.; López, J.A.; Kleiman, N.S. Effect of P-selectin on phosphatidylserine exposure and surface-dependent thrombin generation on monocytes. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Apta, B.H.R.; Bonna, A.M.; Harper, M.T. Platelet P-selectin triggers rapid surface exposure of tissue factor in monocytes. Sci. Rep. 2019, 9, 13397. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Luan, Y.; Miao, X.; Sun, C.; Li, K.; Huang, Z.; Xu, D.; Zhang, M.; Kong, F.; Li, N. Platelet releasate promotes breast cancer growth and angiogenesis via VEGF-integrin cooperative signalling. Br. J. Cancer 2017, 117, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.G.; Wang, L.L.; Ma, D.C. Effects of vascular endothelial growth factors and their receptors on megakaryocytes and platelets and related diseases. Br. J. Haematol. 2018, 180, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Vega, S.; Kondo, A.; Suzuki, M.; Arai, H.; Jiapaer, S.; Sabit, H.; Nakada, M.; Ikeuchi, T.; Ishijima, M.; Arikawa-Hirasawa, E.; et al. Fibulin-7 is overexpressed in glioblastomas and modulates glioblastoma neovascularization through interaction with angiopoietin-1. Int. J. Cancer 2019, 145, 2157–2169. [Google Scholar] [CrossRef] [PubMed]
- Nissen, L.J.; Cao, R.; Hedlund, E.M.; Wang, Z.; Zhao, X.; Wetterskog, D.; Funa, K.; Bråkenhielm, E.; Cao, Y. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J. Clin. Investig. 2007, 117, 2766–2777. [Google Scholar] [CrossRef]
- Kumar, S.; Lu, B.; Davra, V.; Hornbeck, P.; Machida, K.; Birge, R.B. Crk tyrosine phosphorylation regulates PDGF-BB-inducible Src activation and breast tumorigenicity and metastasis. Mol. Cancer Res. 2018, 16, 173–183. [Google Scholar] [CrossRef]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, 0123–0138. [Google Scholar] [CrossRef]
- Hosaka, K.; Yang, Y.; Seki, T.; Nakamura, M.; Andersson, P.; Rouhi, P.; Yang, X.; Jensen, L.; Lim, S.; Feng, N.; et al. Tumour PDGF-BB expression levels determine dual effects of anti-PDGF drugs on vascular remodelling and metastasis. Nat. Commun. 2013, 4, 2129. [Google Scholar] [CrossRef]
- Battinelli, E.M.; Markens, B.A.; Italiano, J.E. Release of angiogenesis regulatory proteins from platelet alpha granules: Modulation of physiologic and pathologic angiogenesis. Blood 2011, 118, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Italiano, J.E.; Richardson, J.L.; Patel-Hett, S.; Battinelli, E.; Zaslavsky, A.; Short, S.; Ryeom, S.; Folkman, J.; Klement, G.L. Angiogenesis is regulated by a novel mechanism: Pro- and antiangiogenic proteins are organized into separate platelet α granules and differentially released. Blood 2008, 111, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Dymicka-Piekarska, V.; Matowicka-Karna, J.; Osada, J.; Kemona, H.; Butkiewicz, A.M. Changes in platelet CD 62P expression and soluble P-selectin concentration in surgically treated colorectal carcinoma. Adv. Med. Sci. 2006, 51, 304–308. [Google Scholar] [PubMed]
- Riedl, J.; Kaider, A.; Marosi, C.; Prager, G.W.; Eichelberger, B.; Assinger, A.; Pabinger, I.; Panzer, S.; Ay, C. Decreased platelet reactivity in patients with cancer is associated with high risk of venous thromboembolism and poor prognosis. Thromb. Haemost. 2017, 117, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Riedl, J.; Kaider, A.; Marosi, C.; Prager, G.; Eichelberger, B.; Koder, S.; Panzer, S.; Pabinger, I.; Ay, C. PO-63—Exhausted platelets in cancer patients with high risk of venous thromboembolism and poor prognosis. Thromb. Res. 2016, 140, S199–S200. [Google Scholar] [CrossRef]
- McCarty, M.F.; Somcio, R.J.; Stoeltzing, O.; Wey, J.; Fan, F.; Liu, W.; Bucana, C.; Ellis, L.M. Overexpression of PDGF-BB decreases colorectal and pancreatic cancer growth by increasing tumor pericyte content. J. Clin. Investig. 2007, 117, 2114–2122. [Google Scholar] [CrossRef]
- Üçüncü, M.; Serilmez, M.; Sarı, M.; Bademler, S.; Karabulut, S. The Diagnostic Significance of PDGF, EphA7, CCR5, and CCL5 Levels in Colorectal Cancer. Biomolecules 2019, 9, 464. [Google Scholar] [CrossRef]
- Aryal, B.; Shimizu, T.; Kadono, J.; Furoi, A.; Komokata, T.; Kitazono, I.; Koriyama, C.; Yamakuchi, M.; Hashiguchi, T.; Imoto, Y. Post-resection exhaustion of intra-platelet serotonin: Also an indicator of early hepatocellular carcinoma recurrence? J. Cancer 2017, 8, 3984–3991. [Google Scholar] [CrossRef]
- Holmes, C.E.; Levis, J.E.; Schneider, D.J.; Bambace, N.M.; Sharma, D.; Lal, I.; Wood, M.E.; Muss, H.B. Platelet phenotype changes associated with breast cancer and its treatment. Platelets 2016, 27, 703–711. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Salgado, R.; Weytjens, R.; Colpaert, C.; Benoy, I.; Huget, P.; Van Dam, P.; Prové, A.; Lemmens, J.; Vermeulen, P. Plasma fibrin D-dimer levels correlate with tumour volume, progression rate and survival in patients with metastatic breast cancer. Br. J. Cancer 2002, 86, 389–395. [Google Scholar] [CrossRef]
- Sallinen, H.; Heikura, T.; Koponen, J.; Kosma, V.M.; Heinonen, S.; Ylä-Herttuala, S.; Anttila, M. Serum angiopoietin-2 and soluble VEGFR-2 levels predict malignancy of ovarian neoplasm and poor prognosis in epithelial ovarian cancer. BMC Cancer 2014, 14, 696. [Google Scholar] [CrossRef] [PubMed]
- Faried, A.; Mobarak, L.; El Gohary, K.K.; El-deeb, H.H.; El-feky, S.; Ahmed, A.; Zaki, N.A.; Alkhalegy, A.A. Serum levels of Arginase Isoenzyme Activity, Alpha- Fetoprotein-L3 and Endostatin as Biomarkers for Hepatocellular Carcinoma in Egyptian Patients. Donn. J. Biomed. Res. 2016, 3, 1–5. [Google Scholar]
- Wang, Z.H.; Zhu, Z.T.; Xiao, X.Y.; Sun, J. Correlation of serum levels of endostatin with tumor stage in gastric cancer: A systematic review and meta-analysis. BioMed Res. Int. 2015, 2015, 623939. [Google Scholar] [CrossRef] [PubMed]
- Cymbaluk-Ploska, A.; Chudecka-Glaz, A.; Pius-Sadowska, E.; Machalilski, B.; Menkiszak, J. Thrombospondin-I concentrations behavior in plasma of patients with ovarian cancer. Cancer Biomark. 2017, 20, 31–39. [Google Scholar] [CrossRef]
- Rouanne, M.; Adam, J.; Goubar, A.; Robin, A.; Ohana, C.; Louvet, E.; Cormier, J.; Mercier, O.; Dorfmüller, P.; Fattal, S.; et al. Osteopontin and thrombospondin-1 play opposite roles in promoting tumor aggressiveness of primary resected non-small cell lung cancer. BMC Cancer 2016, 16, 483. [Google Scholar] [CrossRef]
- Wiśniewski, T.; Zyromska, A.; Makarewicz, R.; Zekanowska, E. Osteopontin and angiogenic factors as new biomarkers of prostate cancer. Urol. J. 2019, 16, 134–140. [Google Scholar]
- Drenberg, C.D.; Saunders, B.O.; Wilbanks, G.D.; Chen, R.; Nicosia, R.F.; Kruk, P.A.; Nicosia, S.V. Urinary angiostatin levels are elevated in patients with epithelial ovarian cancer. Gynecol. Oncol. 2010, 117, 117–124. [Google Scholar] [CrossRef]
- Nocito, A.; Dahm, F.; Jochum, W.; Jae, H.J.; Georgiev, P.; Bader, M.; Graf, R.; Clavien, P.A. Serotonin regulates macrophage-mediated angiogenesis in a mouse model of colon cancer allografts. Cancer Res. 2008, 68, 5152–5158. [Google Scholar] [CrossRef]
- Kelly, C.M.; Juurlink, D.N.; Gomes, T.; Duong-Hua, M.; Pritchard, K.I.; Austin, P.C.; Paszat, L.F. Selective serotonin reuptake inhibitors and breast cancer mortality in women receiving tamoxifen: A population based cohort study. BMJ 2010, 340, 355. [Google Scholar] [CrossRef]
- Alpini, G.; Invernizzi, P.; Gaudio, E.; Venter, J.; Kopriva, S.; Bernuzzi, F.; Onori, P.; Franchitto, A.; Coufal, M.; Frampton, G.; et al. Serotonin metabolism is dysregulated in cholangiocarcinoma, which has implications for tumor growth. Cancer Res. 2008, 68, 9184–9193. [Google Scholar] [CrossRef]
- Svejda, B.; Kidd, M.; Timberlake, A.; Harry, K.; Kazberouk, A.; Schimmack, S.; Lawrence, B.; Pfragner, R.; Modlin, I.M. Serotonin and the 5-HT7 receptor: The link between hepatocytes, IGF-1 and small intestinal neuroendocrine tumors. Cancer Sci. 2013, 104, 844–855. [Google Scholar] [CrossRef]
- Müller, K.; Gilbertz, K.P.; Meineke, V. Serotonin and ionizing radiation synergistically affect proliferation and adhesion molecule expression of malignant melanoma cells. J. Dermatol. Sci. 2012, 68, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Lübbe, A.S.; Huhnt, W. Microvessel diameters of human colon adenocarcinoma during acute treatment with serotonin. Int. J. Microcirc. Exp. 1994, 14, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Hughes, D.J.; Larkin, A.M.; Meiller, J.; Henry, M.; Meleady, P.; Lynch, V.; Pardini, B.; Naccarati, A.; Levy, M.; et al. Elevated levels of 14-3-3 proteins, serotonin, gamma enolase and pyruvate kinase identified in clinical samples from patients diagnosed with colorectal cancer. Clin. Chim. Acta 2015, 441, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Razik, A.; Elhelaly, R.; Elzehery, R.; El-Diasty, A.; Abed, S.; Elhammady, D.; Tawfik, A. Could serotonin be a potential marker for hepatocellular carcinoma? A prospective single-center observational study. Eur. J. Gastroenterol. Hepatol. 2016, 28, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, N.; Haeberle, L.; Schrott, K.M.; Wullich, B.; Krause, F.S. Serotonin used as prognostic marker of urological tumors. World J. Urol. 2008, 26, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, P.M.; Cattaneo, M.; Teresa Canciani, M.; Maniezzo, M.; Vaglini, M.; Cascinelli, N. Early presence of activated (‘exhausted’) platelets in malignant tumors (breast adenocarcinoma and malignant melanoma). Eur. J. Cancer Clin. Oncol. 1989, 25, 1413–1417. [Google Scholar] [CrossRef]
- Crescente, M.; Menke, L.; Chan, M.V.; Armstrong, P.C.; Warner, T.D. Eicosanoids in platelets and the effect of their modulation by aspirin in the cardiovascular system (and beyond). Br. J. Pharmacol. 2019, 176, 988–999. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, W.; Giroux, C.; Cai, Y.; Ekambaram, P.; Dilly, A.K.; Hsu, A.; Zhou, S.; Maddipati, K.R.; Liu, J.; et al. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J. Biol. Chem. 2011, 286, 33832–33840. [Google Scholar] [CrossRef]
- Porro, B.; Songia, P.; Squellerio, I.; Tremoli, E.; Cavalca, V. Analysis, physiological and clinical significance of 12-HETE: A neglected platelet-derived 12-lipoxygenase product. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 964, 26–40. [Google Scholar] [CrossRef]
- Rauzi, F.; Kirkby, N.S.; Edin, M.L.; Whiteford, J.; Zeldin, D.C.; Mitchell, J.A.; Warner, T.D. Aspirin inhibits the production of proangiogenic 15(S)-HETE by platelet cyclooxygenase-1. FASEB J. 2016, 30, 4256–4266. [Google Scholar] [CrossRef] [PubMed]
- Kanikarla-Marie, P.; Kopetz, S.; Hawk, E.T.; Millward, S.W.; Sood, A.K.; Gresele, P.; Overman, M.; Honn, K.; Menter, D.G. Bioactive lipid metabolism in platelet “first responder” and cancer biology. Cancer Metastasis Rev. 2018, 37, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, K.K.F.; Ho, J.M.W.; Chan, F.C.H.; Sung, J.J.Y. Long-term use of low-dose aspirin for cancer prevention: A 10-year population cohort study in Hong Kong. Int. J. Cancer 2019, 145, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Chen, C.C.; Chen, W.M.; Lu, K.Y.; Shen, T.L.; Jou, Y.C.; Shen, C.H.; Ohbayashi, N.; Kanaho, Y.; Huang, Y.L.; et al. LPA1/3 signaling mediates tumor lymphangiogenesis through promoting CRT expression in prostate cancer. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2018, 1863, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Zhang, W.C.; Zhang, J.L.; Zheng, C.J.; Zhu, H.; Yu, H.M.; Fan, L.M. Plasma levels of lysophosphatidic acid in ovarian cancer versus controls: A meta-analysis. Lipids Health Dis. 2015, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Shoaib, M.; Kuschner, C.; Nishikimi, M.; Becker, L.B.; Lee, A.T.; Kim, J. Challenges and inconsistencies in using lysophosphatidic acid as a biomarker for ovarian cancer. Cancers 2019, 11, 520. [Google Scholar] [CrossRef]
- Mezouar, S.; Mege, D.; Darbousset, R.; Farge, D.; Debourdeau, P.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Involvement of platelet-derived microparticles in tumor progression and thrombosis. Semin. Oncol. 2014, 41, 346–358. [Google Scholar] [CrossRef]
- Goubran, H.A.; Kotb, R.R.; Stakiw, J.; Emara, M.E.; Burnouf, T. Regulation of Tumor Growth and Metastasis: The Role of Tumor Microenvironment. Cancer Growth Metastasis 2014, 7, CGM-S11285. [Google Scholar] [CrossRef]
- Goubran, H.A.; Stakiw, J.; Radosevic, M.; Burnouf, T. Platelet-cancer interactions. Semin. Thromb. Hemost. 2014, 40, 296–305. [Google Scholar]
- Rak, J. Microparticles in cancer. Semin. Thromb. Hemost. 2010, 36, 888–906. [Google Scholar] [CrossRef]
- Lazar, S.; Goldfinger, L.E. Platelet Microparticles and miRNA Transfer in Cancer Progression: Many Targets, Modes of Action, and Effects Across Cancer Stages. Front. Cardiovasc. Med. 2018, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Janowska-Wieczorek, A.; Wysoczynski, M.; Kijowski, J.; Marquez-Curtis, L.; Machalinski, B.; Ratajczak, J.; Ratajczak, M.Z. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int. J. Cancer 2005, 113, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Prokopi, M.; Pula, G.; Mayr, U.; Devue, C.; Gallagher, J.; Xiao, Q.; Boulanger, C.M.; Westwood, N.; Urbich, C.; Willeit, J.; et al. Proteomic analysis reveals presence of platelet microparticles in endothelial progenitor cell cultures. Blood 2009, 114, 723–732. [Google Scholar] [CrossRef]
- Zarà, M.; Guidetti, G.; Boselli, D.; Villa, C.; Canobbio, I.; Seppi, C.; Visconte, C.; Canino, J.; Torti, M. Release of Prometastatic Platelet-Derived Microparticles Induced by Breast Cancer Cells: A Novel Positive Feedback Mechanism for Metastasis. TH Open 2017, 1, e155–e163. [Google Scholar] [CrossRef] [PubMed]
- Gasperi, V.; Vangapandu, C.; Savini, I.; Ventimiglia, G.; Adorno, G.; Catani, M.V. Polyunsaturated fatty acids modulate the delivery of platelet microvesicle-derived microRNAs into human breast cancer cell lines. J. Nutr. Biochem. 2019, 74, 108242. [Google Scholar] [CrossRef] [PubMed]
- Provost, P. The clinical significance of platelet microparticle-associated microRNAs. Clin. Chem. Lab. Med. 2017, 55, 657–666. [Google Scholar] [CrossRef]
- Liang, H.; Yan, X.; Pan, Y.; Wang, Y.; Wang, N.; Li, L.; Liu, Y.; Chen, X.; Zhang, C.Y.; Gu, H.; et al. MicroRNA-223 delivered by platelet-derived microvesicles promotes lung cancer cell invasion via targeting tumor suppressor EPB41L3. Mol. Cancer 2015, 14, 58. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, X.; Cao, F.; Wang, Y.; Shen, Y.; Yang, C.; Uzan, G.; Peng, B.; Zhang, D. Inhibiting inducible miR-223 further reduces viable cells in human cancer cell lines MCF-7 and PC3 treated by celastrol. BMC Cancer 2015, 15, 873. [Google Scholar] [CrossRef]
- Pinatel, E.M.; Orso, F.; Penna, E.; Cimino, D.; Elia, A.R.; Circosta, P.; Dentelli, P.; Brizzi, M.F.; Provero, P.; Taverna, D. miR-223 is a coordinator of breast cancer progression as revealed by bioinformatics predictions. PLoS ONE 2014, 9, e84859. [Google Scholar] [CrossRef]
- Sun, X.; Li, Y.; Zheng, M.; Zuo, W.; Zheng, W. MicroRNA-223 increases the sensitivity of triple-negative breast cancer stem cells to TRAIL-Induced apoptosis by targeting HAX-1. PLoS ONE 2016, 11, e0162754. [Google Scholar] [CrossRef]
- Shi, L.; Fisslthaler, B.; Zippel, N.; Frömel, T.; Hu, J.; Elgheznawy, A.; Heide, H.; Popp, R.; Fleming, I. MicroRNA-223 antagonizes angiogenesis by targeting β1 integrin and preventing growth factor signaling in endothelial cells. Circ. Res. 2013, 113, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Jiang, L.; Lin, Y.; Wu, X.; Wang, K.; He, Q.; Wang, X.; Li, W. Platelet microparticle-mediated transfer of miR-939 to epithelial ovarian cancer cells promotes epithelial to mesenchymal transition. Oncotarget 2017, 8, 97464–97475. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Chen, Y.; Song, H.; Xu, Y.; Wang, R.; Chen, L. Mir-24-3p downregulation contributes to VP16-DDP resistance in small-cell lung cancer by targeting ATG4A. Oncotarget 2015, 6, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.V.; Wurtzel, J.G.T.; Mao, G.F.; Rao, A.K.; Kolpakov, M.A.; Sabri, A.; Hoffman, N.E.; Rajan, S.; Tomar, D.; Madesh, M.; et al. Platelet microparticles infiltrating solid tumors transfer miRNAs that suppress tumor growth. Blood 2017, 130, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Liu, C.G.; Addario, A.; Peschle, C.; Scambia, G.; Ferlini, C. Role of microRNAs in drug-resistant ovarian cancer cells. Gynecol. Oncol. 2008, 111, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wu, H.; Liu, X.; Evans, B.R.; Medina, D.J.; Liu, C.G.; Yang, J.M. Role of MicroRNA miR-27a and miR-451 in the regulation of MDR1/P-glycoprotein expression in human cancer cells. Biochem. Pharmacol. 2008, 76, 582–588. [Google Scholar] [CrossRef]
- Anene, C.; Graham, A.M.; Boyne, J.; Roberts, W. Platelet microparticle delivered microRNA-Let-7a promotes the angiogenic switch. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 2633–2643. [Google Scholar] [CrossRef]
- Miao, X.; Rahman, M.F.U.; Jiang, L.; Min, Y.; Tan, S.; Xie, H.; Lee, L.; Wang, M.; Malmström, R.E.; Lui, W.O.; et al. Thrombin-reduced miR-27b attenuates platelet angiogenic activities in vitro via enhancing platelet synthesis of anti-angiogenic thrombospondin-1. J. Thromb. Haemost. 2018, 16, 791–801. [Google Scholar] [CrossRef]
- Wang, C.Z.; Yuan, P.; Li, Y. miR-126 regulated breast cancer cell invasion by targeting ADAM9. Int. J. Clin. Exp. Pathol. 2015, 8, 6547–6553. [Google Scholar] [PubMed]
- Liu, B.; Peng, X.C.; Zheng, X.L.; Wang, J.; Qin, Y.W. MiR-126 restoration down-regulate VEGF and inhibit the growth of lung cancer cell lines in vitro and in vivo. Lung Cancer 2009, 66, 169–175. [Google Scholar] [CrossRef]
- Penson, R.T.; Oliva, E.; Skates, S.J.; Glyptis, T.; Fuller, A.F.; Goodman, A.; Seiden, M.V. Expression of multidrug resistance-1 protein inversely correlates with paclitaxel response and survival in ovarian cancer patients: A study in serial samples. Gynecol. Oncol. 2004, 93, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.J.; Yoon, H.; Kim, S.W.; Kim, H.; Kim, Y.T.; Kim, J.H.; Kim, J.W.; Kim, S. MicroRNA expression profiles in serous ovarian carcinoma. Clin. Cancer Res. 2008, 14, 2690–2695. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, R.; Achinger-Kawecka, J.; Winter, S.; Fritz, P.; Lo, W.Y.; Schroth, W.; Brauch, H. Increased expression of miR-126 and miR-10a predict prolonged relapse-free time of primary oestrogen receptor-positive breast cancer following tamoxifen treatment. Eur. J. Cancer 2013, 49, 3598–3608. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Bruno, A.; Contursi, A.; Grande, R.; Patrignani, P. Platelets and extracellular vesicles in cancer: Diagnostic and therapeutic implications. Cancer Metastasis Rev. 2018, 37, 455–467. [Google Scholar] [CrossRef]
- Kim, H.K.; Song, K.S.; Park, Y.S.; Kang, Y.H.; Lee, Y.J.; Lee, K.R.; Kim, H.K.; Ryu, K.W.; Bae, J.M.; Kim, S. Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: Possible role of a metastasis predictor. Eur. J. Cancer 2003, 39, 184–191. [Google Scholar] [CrossRef]
- Wang, C.C.; Tseng, C.C.; Chang, H.C.; Huang, K.T.; Fang, W.F.; Chen, Y.M.; Yang, C.T.; Hsiao, C.C.; Lin, M.C.; Ho, C.K.; et al. Circulating microparticles are prognostic biomarkers in advanced non-small cell lung cancer patients. Oncotarget 2017, 8, 75952–75967. [Google Scholar] [CrossRef]

| Molecule | Main Findings | Role in Cancer | Ref. |
|---|---|---|---|
| P-selectin | ↑ tumor cell extravasation by promoting cancer cell interaction with platelets and endothelium ↑ platelet activation by increasing thrombin generation ↑ monocyte TF exposure | NEGATIVE | [98,100,101,102,103] |
| TF | Clotting cascade activation ↑ cancer cell survival, ↑ angiogenesis, ↑ tumor growth, ↑ metastasis | [100] | |
| VEGF | ↑cancer growth and angiogenesis ↑ MK maturation | [101] | |
| EGF | ↑ mesenchymal and epithelial cell proliferation ↑ pro-angiogenic effect of other cytokines | [102] | |
| Ang-1 | ↑ vessel development and maturation | [103] | |
| PDGF-BB | ↑ cancer cell proliferation, survival and invasion ↑ tumor stroma changes ↑ blood vessel maturation | [104,105] | |
| ↓ tumor cell growth and dissemination ↓ metastasis | POSITIVE | [106] | |
| Endostatin, TSP-1, angiostatin | ↓ angiogenesis | [107,108] |
| miRNA | Experimental Settings | Main Findings | Targets | Ref. |
|---|---|---|---|---|
| miR-223 | Platelet MV delivey to lung A549 cancer cells | ↑ cell invasion | EPB41L3 | [158] |
| Transfection of breast MCF-7 and prostate PC-3 cancer cells | ↓ vitality, ↑ effects of the anti-tumor celastrol | NF-κB | [159] | |
| Transfection of breast MDA-MB-231 and MCF-7 cancer cells Incubation of MDA-MB-231 cells with CM derived from stable transduced MEFs or HEK293 cells | ↓ migration, ↑ anoikis cell death, ↑ sensitivity to chemotherapy | STAT5A | [160] | |
| Transfection of breast MCF-7, SKBR3, MDA-MB-231 and MDA-MB-435 cancer cells | ↑ sensitivity to TRAIL-induced apoptosis | HAX-1 | [161] | |
| Transient transfection of primary endothelial cells | ↓ formation of new blood vessels | endothelial β1 integrin | [162] | |
| miR-939 | Platelet MV delivey to ovarian SKOV3 cancer cells | ↑ epithelial to mesenchymal transition | E-cadherin and vimentin | [163] |
| miR-24-3p | Transfection of small-cell lung H446 cancer cells | resistance to etoposide plus cisplatin therapy | ATG4A | [164] |
| miR-24 | Platelet MV delivey to Lewis lung and colon MC-38 carcinoma cells | ↓ tumor growth, ↑ apoptosis | mt-Nd2 and Snora75 | [165] |
| miR-130a | miRNA microarray in drug-resistant ovarian A2780 carcinoma cells Transfection of cervix HeLa carcinoma cells | drug resistance | M-CSF | [166] |
| miR-27a, miR-451 | Transfection of MDR ovarian A2780 and cervical KB-V1 carcinoma cells | MDR1 | [167] | |
| miR-let-7a, miR-27b | Platelet MV delivey to primary endothelial cells | ↑ endothelial tube formation | thrombospondin-1 | [168,169] |
| miR-126 | Transfection of breast MDAMB231 and MCF7 cancer cells | ↓ cancer progression | ADAM9 | [170] |
| Transfection of breast BT-549 cancer cells Platelet MV delivey to breast BT-549, MDA-MB-468, BT-20 and MCF-7 cancer cells | cell cycle arrest, ↓ migration, ↑ sensitivity to cisplatin | ND | [156] | |
| Transfection of lung A549, Y-90 and SPC-A1 carcinoma cells | ↑ proliferation | VEGF | [171] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catani, M.V.; Savini, I.; Tullio, V.; Gasperi, V. The “Janus Face” of Platelets in Cancer. Int. J. Mol. Sci. 2020, 21, 788. https://doi.org/10.3390/ijms21030788
Catani MV, Savini I, Tullio V, Gasperi V. The “Janus Face” of Platelets in Cancer. International Journal of Molecular Sciences. 2020; 21(3):788. https://doi.org/10.3390/ijms21030788
Chicago/Turabian StyleCatani, Maria Valeria, Isabella Savini, Valentina Tullio, and Valeria Gasperi. 2020. "The “Janus Face” of Platelets in Cancer" International Journal of Molecular Sciences 21, no. 3: 788. https://doi.org/10.3390/ijms21030788
APA StyleCatani, M. V., Savini, I., Tullio, V., & Gasperi, V. (2020). The “Janus Face” of Platelets in Cancer. International Journal of Molecular Sciences, 21(3), 788. https://doi.org/10.3390/ijms21030788