1. Introduction
Myalgic encephalomyelitis (ME), also referred to as chronic fatigue syndrome (CFS), is a condition which is little understood, its hallmarks being chronic, unexplained fatigue and the debilitating overexertion “payback” termed post exertional malaise (PEM) experienced by patients [
1]. PEM can occur after even the simplest of daily physical tasks and may be accompanied by symptoms affecting a range of body systems. The lack of objective, timely, and accurate diagnostic criteria leaves patients for long periods without a clear diagnosis or an informed understanding of the condition [
1,
2]. It is paramount that fundamental molecular explanations for the underlying pathophysiology of ME/CFS and reliable biomarkers are pursued. These could lead to reliable, faster diagnosis and, in the longer term, rational, effective treatments.
Mitochondrial function is of interest to ME/CFS research as a bioenergetic explanation for the recurrent fatigue and the myriad links between the disorder’s characteristic immune inflammation and the mitochondria [
3]. Evidence for mitochondrial dysfunction in ME/CFS has been sought in the last decade, yet remains inconsistent. Reduced mitochondrial biogenesis but not normalized respiratory chain enzyme activities have been reported in the muscle of ME/CFS individuals [
4]. Muscle mitochondrial biogenesis is upregulated by exercise [
5], so this reduction is likely to be caused by the necessarily reduced exercise that ME/CFS patients can undertake. Mitochondrial respiratory function in ME/CFS neutrophils [
6,
7] and peripheral blood mononuclear cells (PBMCs) is reportedly reduced [
8] or unchanged [
9], yet the oxidative phosphorylation (OXPHOS) complexes appear normal [
10,
11], while the expression of genes encoding mitochondrial proteins in patient saliva, platelets, and lymphocytes is elevated [
12,
13,
14]. Most recently, mitochondrial respiration was found to be unchanged in resting and stimulated CD4
+ and CD8
+ T cells, with the sole exceptions of a small reduction in the proton leak in resting and ATP synthesis in stimulated CD8
+ cells [
15].
Differences between patient and control serum and urine metabolomes have been attributed to reduced provision of acetyl CoA to the TCA cycle caused by a defect in glycolysis [
16] or by a defect in pyruvate dehydrogenase (PDH) [
17]. While inconsistent, both proposals draw attention to a potential role in ME/CFS for impaired provision of reducing equivalents to mitochondrial OXPHOS by the TCA cycle. Despite their shared conviction that ME/CFS cells have fundamental problems in energy metabolism, the conflicting reports on the nature of these problems prompted us to reexamine the issue of mitochondrial function and its regulation in ME/CFS cells.
A key regulator of mitochondrial function is TORC1 (Target of Rapamycin Complex I) which regulates cell growth and energetics in a variety of cellular stress-sensing pathways [
18]. This pathway upregulates the expression of nuclear-encoded mitochondrial proteins [
19], among which are subunits of the OXPHOS complexes [
20]. Despite this connection, and the agency of TORC1 within a complex regulatory network which responds to intracellular stressors including energy supply, dysregulation of this signaling pathway has not yet been investigated in ME/CFS cells.
To clarify the roles of aberrant mitochondrial function and TORC1 signaling in ME/CFS, we have compared parameters of mitochondrial function in immortalized lymphocytes (termed lymphoblasts) from patient blood with those from healthy age- and gender-matched controls. We found that mitochondrial function in ME/CFS lymphoblasts is indeed abnormal, with an isolated Complex V deficiency accompanied by elevated capacity of Complexes I to IV, decreased membrane potential, upregulation of TORC1 activity and elevated expression of diverse mitochondrial proteins involved ATP-generating catabolic pathways. This pattern of changes in mitochondrial function in ME/CFS lymphoblasts is distinct from what we observed using the same approach to other neurological conditions, for example the mitochondrial hyperactivity we reported previously in Parkinson’s disease lymphoblasts [
21] and fragile X-associated tremor/ataxia syndrome lymphoblasts [
22]. It suggests a model involving a primary deficiency in Complex V function, combined with homeostatic, compensatory upregulation of TORC1 activity and mitochondrial protein expression.
3. Discussion
Previous steady state measurements and metabolic flux measurements of mitochondrial respiratory function in ME/CFS lymphocytes have suggested that in ME/CFS cells there is either a generalized reduction [
6,
7,
8] or little change [
9,
15] in mitochondrial activity and respiratory capacity. However, functionally normal OXPHOS Complex I to IV activity has also been reported in ME/CFS lymphocytes [
11,
41], while the expression of mitochondrial proteins is upregulated in patient saliva, platelets and lymphocytes [
12,
13,
23]. Elevated nonmitochondrial ATP production has also been reported in ME/CFS lymphocytes [
11].
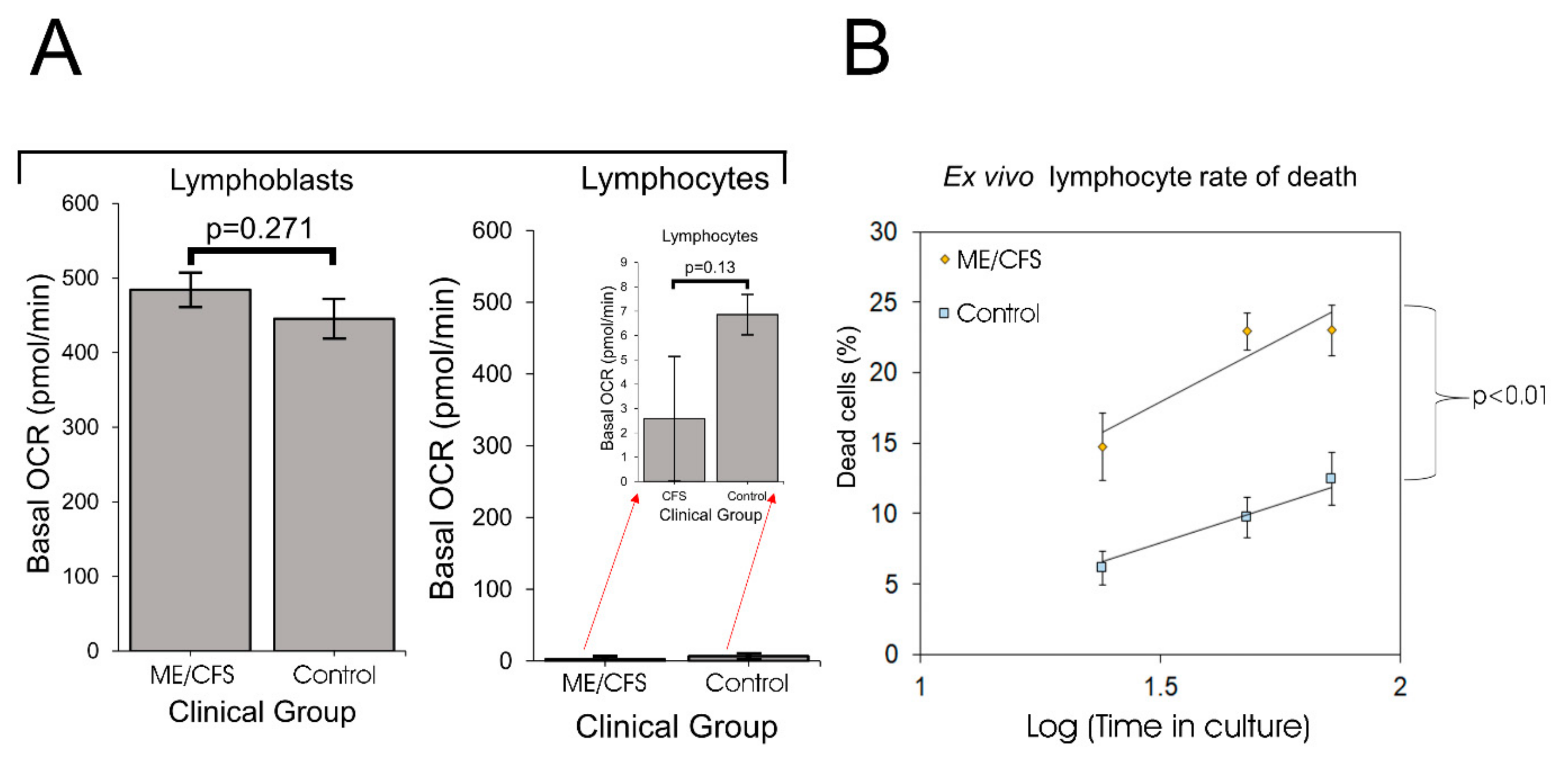
In this work we have resolved these inconsistencies by revisiting the issue of mitochondrial function and capacity in immortalized lymphocytes (lymphoblastoid cell lines or lymphoblasts). Although ex vivo lymphocyte populations are well suited for rapid characterization of relatively stable molecular features, such as the patterns of cell surface antigens they express, they may be less well suited to studying rapidly labile processes such as metabolism, mitochondrial function or intracellular signaling activities. As we showed here, unactivated ex vivo lymphocytes are metabolically quiescent and dying. A difference between patient and control groups in the depth of this quiescence and/or the extent of cell death may thus explain the previously reported reduction in mitochondrial activities in ME/CFS lymphocytes. One recent study examined mitochondrial respiratory function in resting and stimulated CD4
+ and CD8
+ T-cells from ME/CFS subjects and controls [
15]. The only significant differences found were small reductions in the ME/CFS CD8
+ lymphocytes in the proton leak in resting and ATP synthesis in stimulated cells. Surprisingly however, the overnight stimulation (with anti-CD3/anti-CD28 beads plus IL2) made almost no difference to mitochondrial respiratory function in these cells. This suggests that under the conditions used, the mitochondria in these cells were still quiescent after the overnight activation stimulus.
We found that not only were PBMCs quiescent, but the fraction of dead cells after 1–3 days incubation in culture medium was dramatically greater for ME/CFS lymphocytes than for control lymphocytes. It is likely that in some previous studies, the ME/CFS lymphocytes assayed for mitochondrial activity included a higher proportion of dead cells than did the controls. Tomas et al. (2017) found no significant difference in the viability of fresh and frozen PBMCs and no difference between ME/CFS and control samples [
8], a result that accords with our regression analysis of the death rates of PBMCs in culture (
Figure 1). However, these authors followed the common procedure in their Seahorse assays of incubating the cells overnight in situ in the assay plates before the respirometry measurements were done. Although they did not detect significant differences between frozen and fresh PBMCs in the Trypan Blue staining
before assay, they did observe consistently significant reductions in respiration of frozen compared to fresh cells, again
after overnight incubation in assay medium. Our results suggest that by this time there may have been a significant fraction of dead cells both in their control and ME/CFS samples and that this fraction may have been higher in the ME/CFS samples. The greater mortality rates for ME/CFS lymphocytes are not surprising given that pharmacological inhibition of mitochondrial respiration, including Complex V impairment, has long been known to result in apoptotic cell death in ex vivo lymphoid cells [
42].
By contrast, the lymphoblastoid cell lines (LCLs or lymphoblasts) used in this work are metabolically active lymphoid cells that may better represent activated lymphocytes, which drive inflammation in vivo [
43]. The use of cultured lymphoblasts in this way as models of not only metabolically active lymphoid but other cell types, both in health and disease, has been reviewed in detail previously [
44,
45]. Because the viral genome exists in multiple copies as a circular episome in the latent state in infected cells, it does not disrupt the genome, but affects gene expression patterns in favor of proliferation, as does B cell activation by other means. Lymphoblasts have thus been used both in genetic and genomic studies, including the well-known 1000 Genomes Project [
46]. They have also been used, as here, in functional studies of diverse, complex diseases, including autism, schizophrenia, Alzheimer’s, and Parkinson’s disease [
21,
45]. Whereas PBMCs, used ex vivo in some other ME/CFS studies, are a complex mixture of cell types, EBV-mediated immortalization and culture selects B cells from this population, because the EBV receptors are expressed in B cells. It has been reported previously that the cell type composition of PBMCs from ME/CFS patients is different from healthy controls [
47] and such differences could also contribute to observed functional differences in the mixed populations in ex vivo PBMCs. We chose not to clone our cell lines, but as a result of the selective nature of EBV infection, they can be expected to be more homogeneous in cell type than the PBMCs from which they were selected.
As EBV-infected B cells begin to proliferate, around 250 genes become hypomethylated [
48]. Many of these genes already exhibit low levels of methylation and high levels of expression in resting B cells, while many others exhibit hypomethylation and overexpression during B cell proliferation. They fall into 6 major Gene Ontology (GO) categories—immune response, homophilic cell adhesion, humoral immune response, B cell receptor signaling pathway, inflammatory response and chemotaxis genes. In the whole cell proteomes of lymphoblasts from both healthy and ME/CFS individuals, we observed expression of many B-cell specific proteins whose expression was not significantly different in ME/CFS and control cell lines (data not shown). The overall pattern of expression is consistent with lymphoblasts having had their normal B cell transcription and proliferation program activated by the virus [
48]. Importantly, this phenotype is stable through up to 180 cell doublings, unlike other in vitro methods of B cell activation (such as antigen stimulation) which induce similar changes in gene expression that increase and decline over only a few days and culminate in cell death.
Like primary cell cultures of other cell types such as myoblasts (which die after a few doublings) or fibroblasts (which progressively enter senescence between
ca. 30–60 doublings), lymphoblasts eventually die off after
ca. 160–180 doublings [
45]. Thereafter, the original lymphoblast population may be replaced by cells bearing mutational changes in the genome that support ongoing proliferation. At this point the cells may no longer be representative of activated forms of the B lymphocytes from which they were derived. In the work reported here, none of the lymphoblast cultures were allowed to proceed through more than a handful of cell doublings before storage or use in experiments.
During their more limited life spans, primary cell lines like myoblasts and fibroblasts undergo a progressive process called replicative senescence as part of which their relevant phenotypic and molecular features may change [
49]. Fibroblast and myoblast gene expression programs are also tightly regulated by contact inhibition so that their phenotypes can be dramatically affected by their density in culture [
50]. Furthermore, their life span and phenotypes may be affected by the age of the donor from whose tissues they were derived. These various sources of phenotypic differences can be disentangled but this requires care, appropriate controls and may need larger samples or more experiments to account for the additional variables. Provided long periods of culture approaching their replicative life span limits are avoided, lymphoblasts do not present these problems. They are not contact-inhibited and their metabolic phenotypes are stable in culture and storage, depending only, as we have shown here for ME/CFS and previously for Parkinson’s disease [
21], on the disease state, not the gender or age of the person from whom they were isolated. Despite these advantages, it will be important in future work to determine whether the mitochondrial and cell signaling anomalies in ME/CFS lymphoblasts are also observed in other cell types.
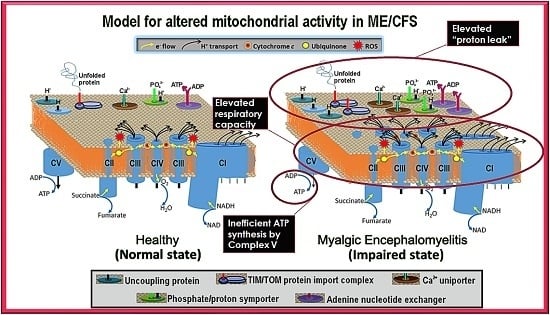
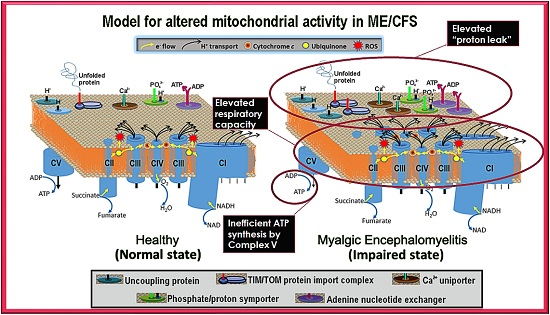
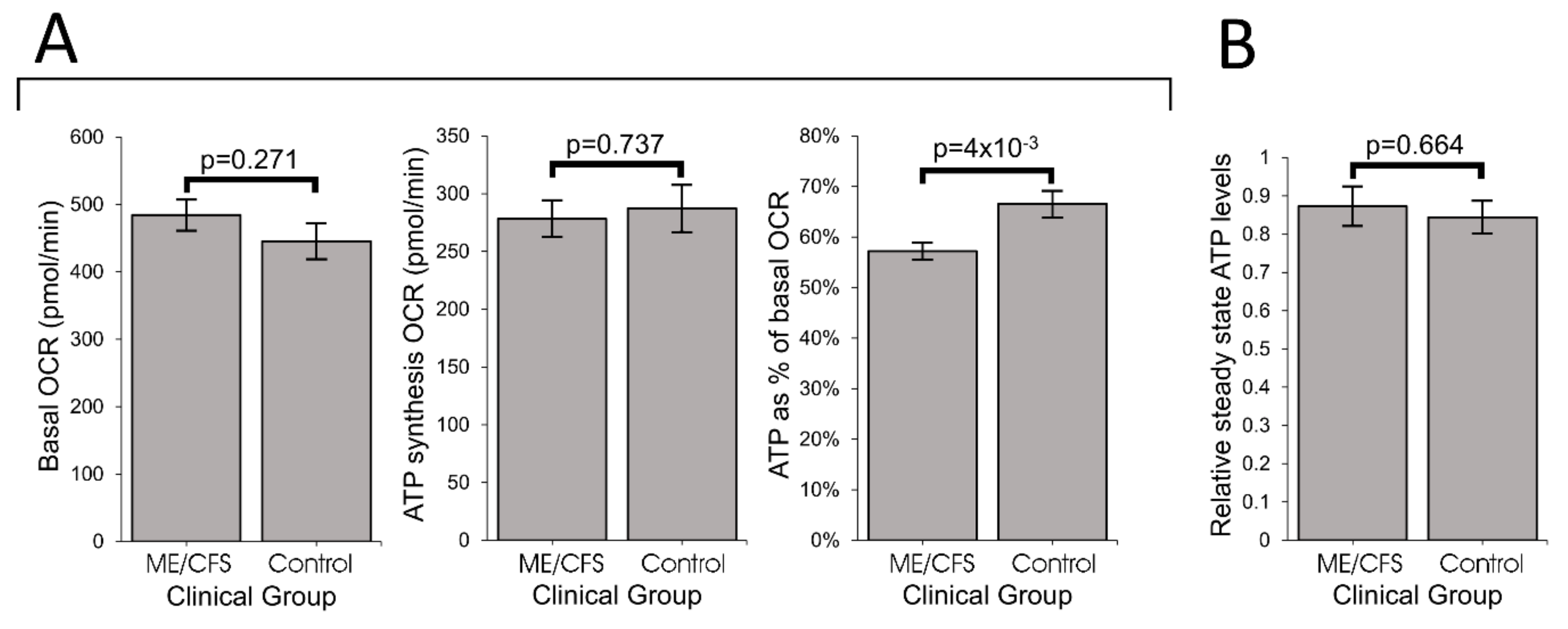
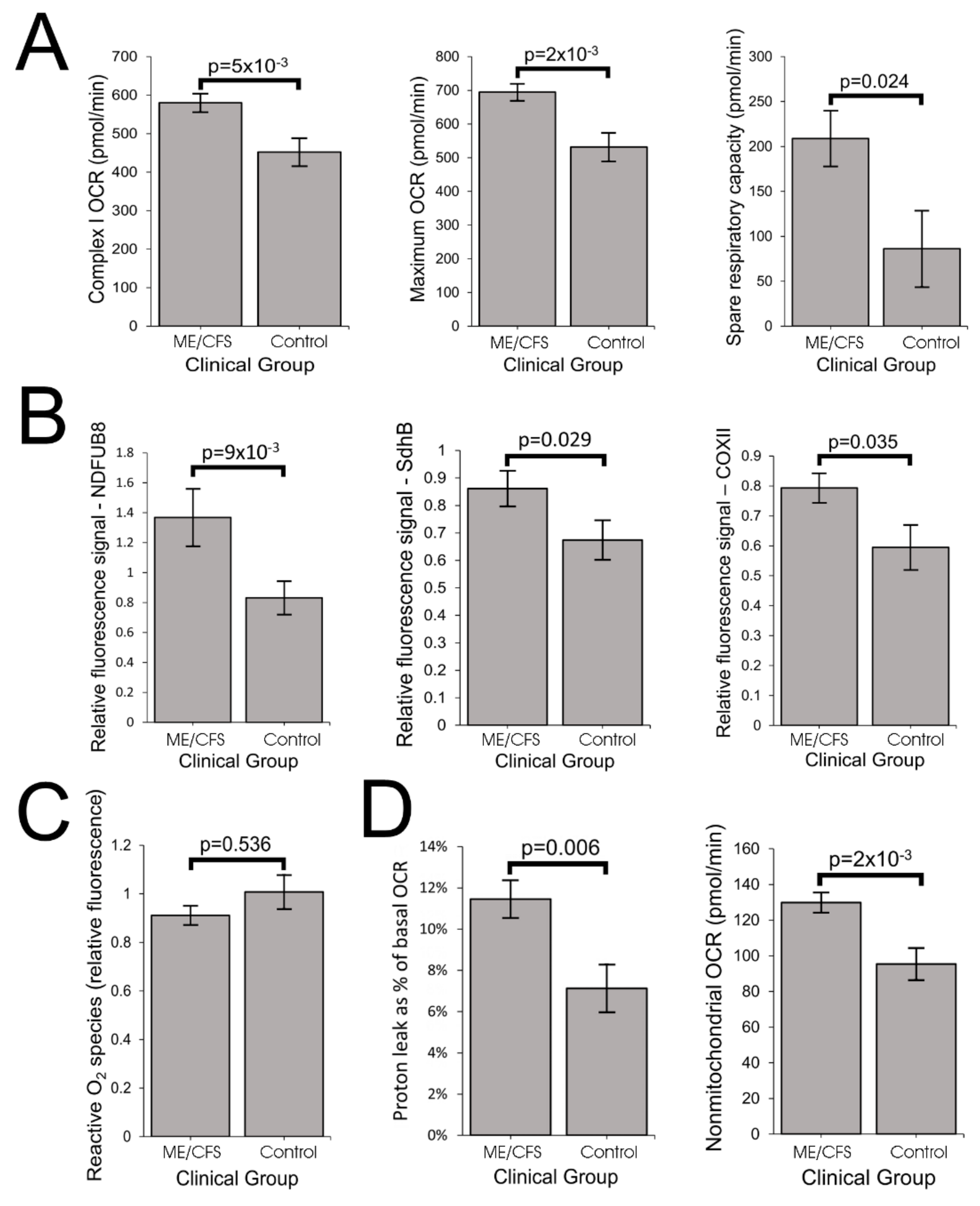
Our results showed that ME/CFS lymphoblasts exhibit an isolated Complex V inefficiency that is accompanied by upregulation of mitochondrial protein expression, including mitochondrial respiratory complexes and enzymes involved in the TCA cycle, fatty acid uptake and β-oxidation. These findings confirm that these ME/CFS cells do indeed exhibit a mitochondrial deficiency in ATP generation, but reveal that, in lymphoblasts at least, this specifically involves Complex V rather than a generalized reduction in all mitochondrial functions. This profile of mitochondrial dysfunction in intact ME/CFS lymphoblasts is distinct from the mitochondrial hyperactivity we previously found in Parkinson’s disease lymphoblasts [
21], so cannot be a simple reflection of neuroinflammatory processes believed to occur in both diseases.
What might cause such a mitochondrial Complex V inefficiency? Three possibilities are a mutation affecting one of the Complex V subunits or assembly proteins, an elevation of the relative use of the proton motive force for other purposes (“proton leak”) making less available for ATP synthesis or a dysregulation of Complex V itself. A mutational defect in Complex V seems unlikely in view of the failure of previous investigations to uncover any single nucleotide polymorphisms in Complex V genes that associate with the disease state [
51,
52]. The second possibility, that Complex V is inefficient in these ME/CFS cells because of the elevated use of the proton motive force by other processes, is suggested by the elevated proton leak we measured in ME/CFS lymphoblasts. However, the ME/CFS mitochondria have excess unused respiratory capacity and respiratory complex levels. These would indicate that the electron transport capacity in ME /CFS lymphoblasts is more than sufficient to allow Complex V to operate at normal efficiency. Dysregulatory inhibition of Complex V is a third possibility. It is known that mitochondrial ATP synthase activity can be regulated by a variety of proteins, small molecules and signaling pathways, some of them by acting through Complex V’s own inhibitory subunit AIF1 [
53,
54]. These possible causes for Complex V inefficiency in ME/CFS lymphoblast mitochondria should be investigated in future work.
It is possible that the dysregulation of Complex V and mitochondrial function in ME/CFS lymphoblasts arises because they respond differently to EBV infection than do control lymphoblasts. We showed that the EBV genome copy number did not differ between the participant groups and, in any case, had no effect on any of our assays. Nevertheless, it is possible that in ME/CFS lymphoblasts, EBV reactivates more readily to enter the lytic cycle and this in turn affects mitochondrial function. This is an attractive idea that, if true, would raise the possibility that EBV may also reactivate more readily in infected ME/CFS B-cells in vivo. Despite the appeal of this hypothesis, the profile of mitochondrial changes we observed is not consistent with the reported effects of EBV on mitochondria in the lytic cycle. These include
decreases in ROS production, mitochondrial membrane potential, expression of mitochondrial proteins, and mitochondrial biogenesis [
55,
56]. Although we did observe decreased mitochondrial membrane potential in ME/CFS lymphoblasts, we found no change in ROS levels, no change in mitochondrial mass or genome copy number and an elevation not a reduction in expression of mitochondrial proteins. We are unaware of any evidence that EBV causes an isolated inefficiency in Complex V coupled with an elevated proton leak and an increase in maximum OCR and Complex I activity. Instead of EBV reactivation causing mitochondrial dysfunction in ME/CFS lymphoblasts, it is also possible that the reverse occurs—namely that mitochondrial dysfunction causes the virus to be more readily reactivated. This possibility is suggested by the fact that EBV is reactivated by cellular stress and could potentially contribute to the post-exertional malaise that characterizes ME/CFS. The interactions between EBV and mitochondrial function in B-cells and lymphoblasts from ME/CFS patients are clearly worth pursuing in future work.
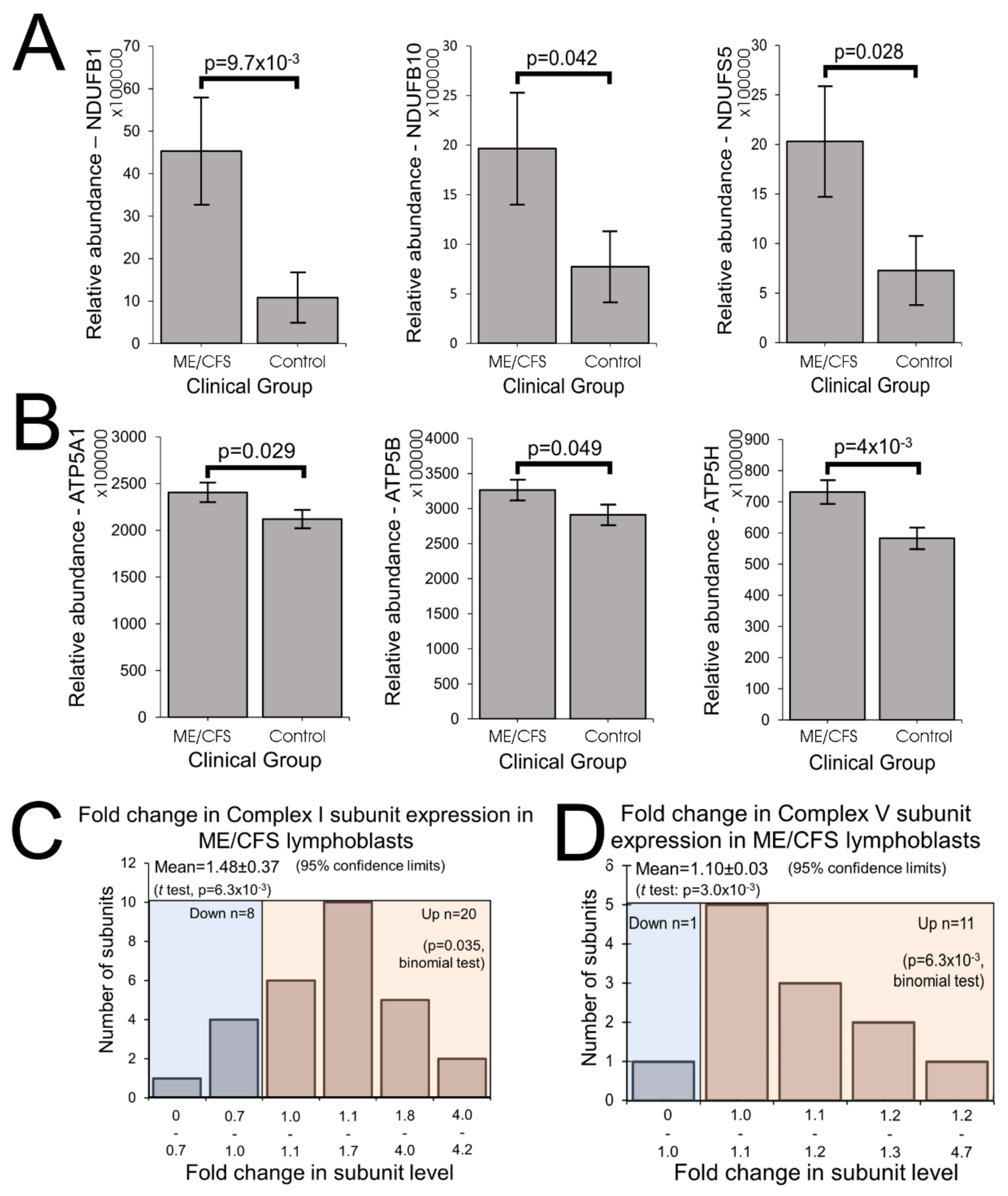
The elevated maximum respiratory capacity, mitochondrial Complex I activity, and proton leak we found in ME/CFS lymphoblasts are consistent with the higher mitochondrial protein expression we observed in these cells, as assayed using both semiquantitative Western blots and whole cell proteomics. Consistent with this, it was reported previously that the expression of genes encoding mitochondrial proteins is upregulated in ME/CFS saliva, lymphocytes, and platelets [
12,
13,
23], while increased translocase activity into the mitochondrial matrix has also been reported [
7]. Together our results suggest a model in which the Complex V defect is a proximal activator of compensatory upregulation of expression of mitochondrial proteins.
The increased expression of diverse mitochondrial proteins in ME/CFS observed by us and others [
11,
12,
13] suggests the possibility that mitochondrial biogenesis more broadly is activated in these cells. However, we found that the mitochondrial membrane “mass” per cell (Mitotracker Green fluorescence) and the copy number of the mitochondrial genome relative to the nuclear genome were unchanged in the ME/CFS lymphoblasts. Accordingly, the mitochondria in these cells appear to have higher concentrations of mitochondrial respiratory proteins and catabolic enzymes.
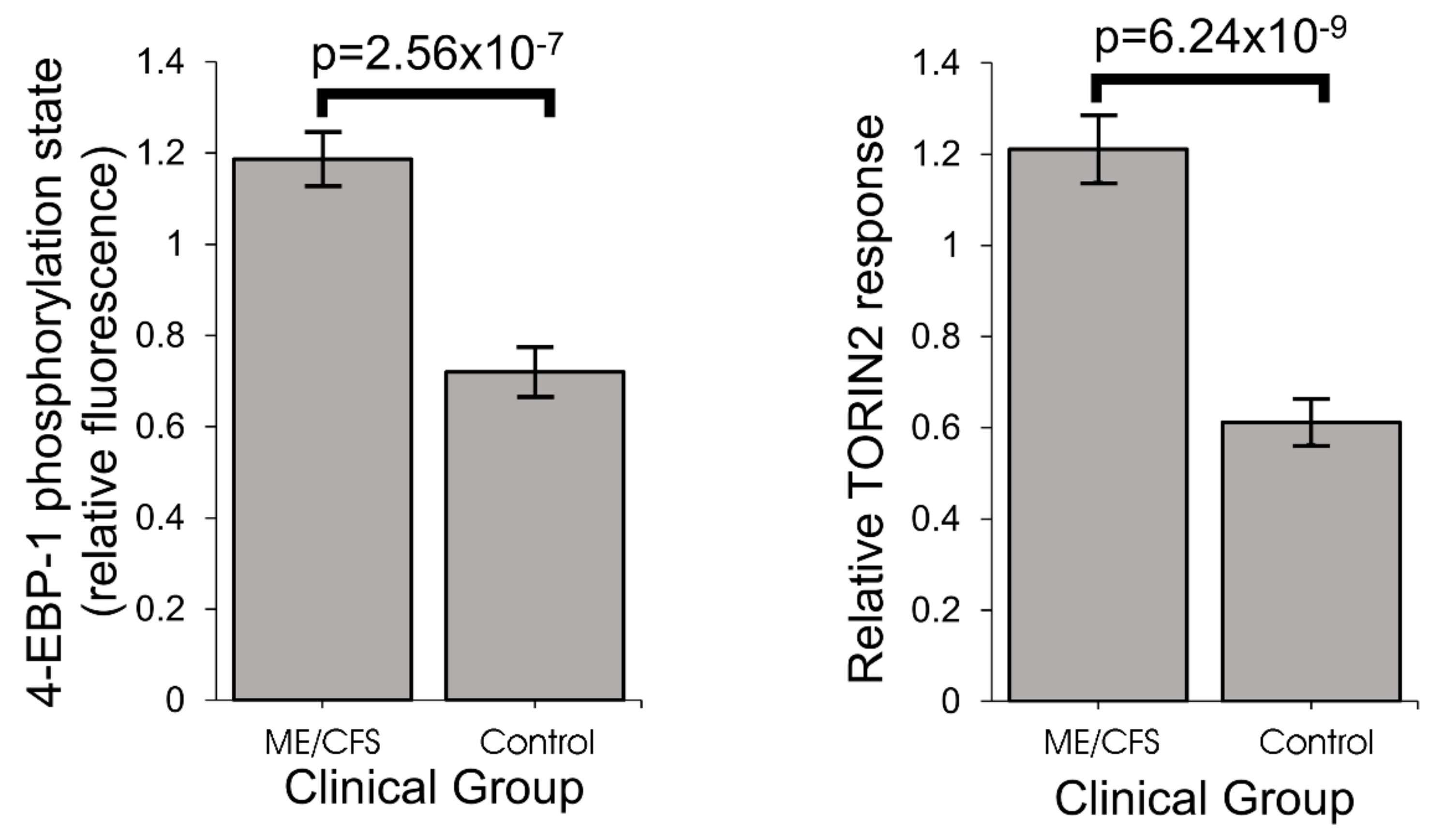
One of the key upstream regulators of mitochondrial protein expression is TOR Complex I (TORC1 whose catalytic subunit is mTOR, the mechanistic Target Of Rapamycin). We found that TORC1 activity is elevated in ME/CFS lymphoblasts. The expression of mitochondrial enzymes involved in electron transport is known to be upregulated by TORC1 via selective activation of translation via inhibitory phosphorylation of the TORC1 target 4E-BP1 [
20]. In addition to its actions on the translation of nuclear-encoded mitochondrial proteins, TORC1 upregulates the expression of transcription factors PGC-1α (transcriptionally via Yin Yang 1) and TFAM (translationally), which respectively induce the transcription of nuclear and mitochondrial genes encoding mitochondrial proteins [
19]. Most notable amongst the mitochondrial proteins whose translation is upregulated by TORC1 are nuclear-encoded subunits of Complexes I and V [
20], the two respiratory complexes whose expression we found to be the most evidently elevated in the whole cell proteomes of ME/CFS lymphoblasts.
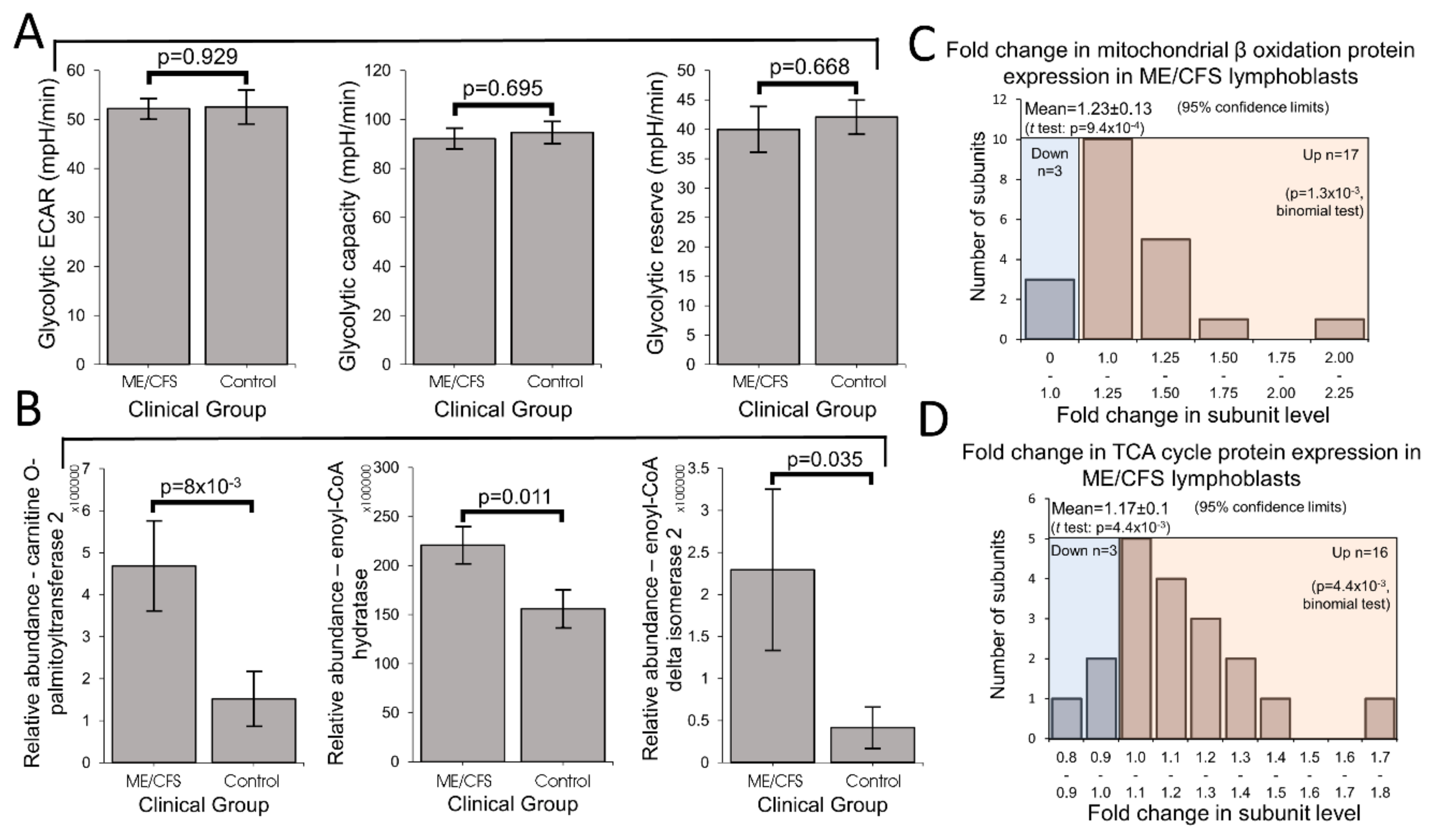
Mitochondrial enzymes involved in the β oxidation of fatty acids are amongst the many proteins whose expression is upregulated by PGC1α and thus indirectly by TORC1. Fatty acid β oxidation provides acetyl CoA to the TCA cycle, as does glycolysis, but it yields more ATP per oxidized carbon than does glycolysis. This also makes thermodynamic sense, given the more highly reduced state of the carbons in fatty acid chains compared to those in sugar molecules. ME/CFS cells could shift their metabolism in favor of fatty acid β oxidation because of a deficiency in glycolysis [
15,
16] or pyruvate dehydrogenase which partially oxidizes pyruvate and supplies acetyl CoA to the TCA cycle [
17]. It has also been suggested that in ME/CFS cells metabolism shifts away from glycolysis in favor of the pentose phosphate pathway which can oxidize sugars in the cytosol to generate reducing power that can be transported into the mitochondria to drive electron transport [
28]. Our results support the existence of a metabolic shift, but we found no deficiency in glycolysis rates, glycolytic reserve or glycolytic capacity. Although there have been reports of reduced rates of glycolysis in ME/CFS lymphocytes [
26], this contrasts with reports by others [
8]. Instead of impaired glycolytic capacity driving the shift in metabolism, our results suggest that the change may be driven by elevated usage of alternatives such as the β oxidation of fatty acids. Fatty acid utilization rates were previously reported to be unchanged in permeabilized ME/CFS lymphocytes [
41]. However, the permeabilization process (with loss of cytoplasmic cellular context, e.g., metabolites) or the metabolic quiescence and greater death rates of ME/CFS lymphocytes may have obscured the result in this previous study. It would be valuable in future experiments to measure fatty acid utilization rates in ME/CFS and control lymphoblasts.
TORC1 is not the only cellular stress sensing protein that regulates expression of proteins involved in cellular metabolism and mitochondrial function. It acts in concert with AMPK as part of a complex stress-sensing network [
31,
35]. AMPK is activated by a variety of cellular stressors including ATP insufficiency, elevated cytosolic Ca
2+ concentrations and oxidative stress [
57,
58]. As the primary ATP sensor [
59], AMPK is implicated in mitochondrial disease [
60], and activates a variety of catabolic pathways that provide alternative oxidizable substrates to the mitochondria—including amino acids or fatty acids [
61]. In ME/CFS lymphoblasts, with their chronically inefficient ATP synthesis, AMPK is expected to be chronically activated and participate in the compensatory upregulation of mitochondrial respiratory capacity that we observe. However, despite lower steady state ATP levels, the AMPK activation state was not significantly different between cultured muscle cells from CFS patients (Fukuda criteria) and healthy controls [
62]. Future work should therefore test the hypothesis that AMPK is chronically activated in ME/CFS lymphoblasts and other cell types.
4. Materials and Methods
4.1. Participant Cohort
All participants were of European descent and belonged to two groups: ME/CFS patients (
n = 51, 86% female, median age 50, age range 26–70) or healthy controls (
n = 22, 68% female, median age 41, age range 21–58) without any family history of ME/CFS or similar myalgias, nor cohabiting with ME/CFS patients. There was no significant difference between the patient and control groups in either the gender proportions (Fisher’s exact test,
p = 0.21) or age distribution (Fisher’s exact test, participants grouped by ages in 5 year increments,
p = 0.19). Neither gender (ANOVA) nor age (multiple regression,
Figure S2) had an effect on any of the parameters of mitochondrial function and TORC1 signaling (
p > 0.05).
Lymphoblastoid cell lines (lymphoblasts) were isolated from all participants. Multiple assays were conducted on both lymphoblasts and the lymphocytes from which they were derived (peripheral blood mononuclear cells, PBMCs). However, because of limited PBMC supplies, subsets of the participant cohort were used for the tests using lymphocytes:
PBMC respirometry assays. The sample selection was determined by the availability of sufficient PBMCs for the experiments. This subcohort included 14 ME/CFS patients (71% female, median age 59, age range 38–71) and nine healthy controls (67% female, median age 41, age range 21–52). The gender proportions were not different (Fisher’s exact test p = 1.0), but the age distributions were (four control but no ME/CFS individuals under 30; seven ME/CFS individuals over 60; Fisher’s exact test p = 0.013 using 15-year bins). Neither age (multiple regression) nor gender (ANOVA) had any influence on the lymphocyte O2 consumption rates in either patients or controls (p > 0.05).
Lymphocyte cell death assays used samples from 35 ME/CFS individuals (89% female, median age 52, age range 26–71) and 14 control individuals (71% female, median age 42, age range 21–58). Neither the gender proportions (Fisher’s exact test p = 0.15) nor the age distributions were significantly different (Fisher’s exact test p = 0.13 using 15-year bins). Neither age (multiple regression) nor gender (ANOVA) had any influence on the lymphocyte death rate in either patients or controls (p > 0.05).
We used a randomly selected subset of lymphoblastoid cell lines to determine if the copy numbers of the EBV genome were different in patient and control groups and if they had any effect on the mitochondrial and cell signaling abnormalities we observed. This subset included 13 MECFS patients (85% female, median age 43, age range 26–62) and 15 controls (53% female, median age 41, age range 21–58). Neither the difference in gender proportions (Fisher’s exact test p = 0.11) nor the distribution of ages (Fisher’s exact test, p = 0.8 using 15 year bins) was statistically significant. Neither age (multiple regression) nor gender (ANOVA) had any influence on the EBV genome copy number in either patients or controls (p > 0.05).
A subset of the lymphoblast cell lines was also used for the proteomics analysis. It included 22 ME/CFS patients (90% female, median age 56, age range 26–71) and 16 controls (56% female, median age 39, age range 21–58). The difference between the patient and control groups in gender proportions (Fisher’s exact test p = 0.02) and age distribution (Fisher’s exact test, p = 0.03 using 10 year bins) was statistically significant. However, there was no significant effect of either age (multiple regression) or gender (ANOVA) on expression of the analyzed mitochondrial proteins in either patients or controls (p > 0.05).
Participants were assessed and samples collected at CFS Discovery Clinic, Melbourne, Australia, who have a long running specialization and interest in ME/CFS. Patients were diagnosed using the Canadian Consensus Criteria [
63], assessed for postorthostatic tachycardia syndrome comorbidity, and asked to complete the Depression, Anxiety, and Stress Scale questionnaire and the Epworth Sleepiness Scale questionnaire. ME/CFS specific severity assessments were also conducted using Richardson and Lidbury’s Weighted Standing Time [
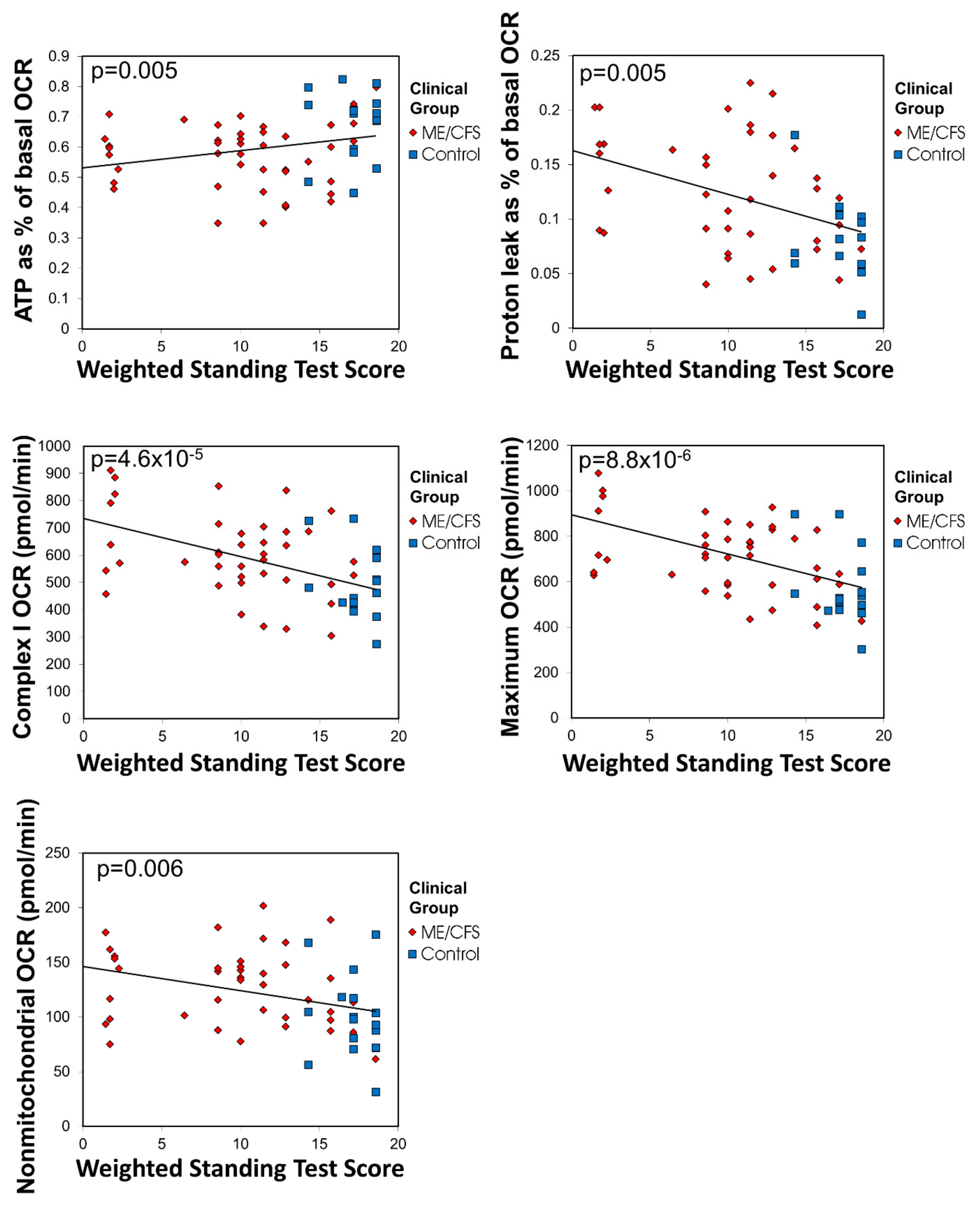
24]. 15 mL of blood was taken per participant in heparin-treated vacutainer tubes (BD). Subjects with other known reasons for fatigue were excluded.
The project was approved by the Australian National University Human Research Ethics Committee (Reference 2015/193) and accepted as an externally approved project by the La Trobe University Human Ethics Committee (26 February 2016).
4.2. PBMC Isolation from Blood Sample and Immortalization
PBMCs were isolated from blood, stored and immortalized to form lymphoblast cultures as previously described [
21]. Briefly, lymphocytes were isolated by Ficoll-Paque density centrifugation and counted. Next, 5 × 10
6 cells were harvested for immortalization and were resuspended in 5 mL RPMI 1640 without L-glutamine (Life Technologies) supplemented with 1× Glutamax (Life Technologies), 10% FBS and 1% Penicillin/Streptomycin. Excess lymphocytes were separated into aliquots of 5 × 10
6 cells, harvested and resuspended in 250 μL of Recovery™ Cell Culture Freezing Medium (Life Technologies) and stored at −80 °C.
For immortalization, 1 mL culture supernatant from B95.8 cells expressing Epstein–Barr virus (EBV) was added, and 150 µL of the mix was seeded per well in a 96-well U-bottom plate, then incubated for one hour within a humidified 5% CO2 incubator at 37 °C. A final concentration of 500 ng/mL Cyclosporin A (Sigma) was then added to each well. Cultures were fed weekly by replacing half of the medium with the same formulation, without disturbing the cells. This process was repeated over a period of approximately three weeks until the cells were confluent and growing rapidly, after which the lymphoblast cultures were processed as described in the following section.
4.3. Lymphoblast Cultures
Confluent lymphoblasts were transferred to T25 flasks in growth medium (Minimum Essential Medium α (Life Technologies), supplemented with 10% FBS and 1% Penicillin/Streptomycin) where they were cultured within a humidified 5% CO2 incubator at 37 °C. Cells were seeded at concentrations of no less than 2 × 105 cells/mL, fed at intervals not exceeding three days by replacing 1/3 of medium with new medium, or split in a 1:3 ratio of cell culture to fresh medium as required. For storage, flasks containing confluent cultures were transferred to tubes, harvested by centrifugation, resuspended by 250 µL aliquots in Recovery™ Cell Culture Freezing Medium and stored at −80 °C. Dozens of lymphoblast aliquots per individual were stored to allow for ongoing access to cultures with low passage numbers for future use. Cells were removed from storage by thawing in a 37 °C water bath, harvested by centrifugation, resuspended in growth medium, and transferred to a fresh T25 flask.
Prior to commencing experiments, lymphoblast lines were cultured over as short a time and as few passages (2–5) as possible. For a set of triplicate, independent experiments, harvest, assay, and conduct of experiments occurred over approximately one week. As previously, two immortalized lymphoblast cell lines created from healthy donor blood were utilized as internal controls to normalize for variation between experiments where appropriate [
19].
4.4. Viable Cell Counts
Lymphoblast or lymphocyte (PBMC) viable counts for all applications were determined by staining with Trypan Blue (Thermo-Fisher Scientific) prior to hemocytometer cell counting. Trypan Blue-stained cells were counted as dead and unstained, intact cells as viable. For the unimmortalized lymphocyte viability measurements over time, frozen aliquots were thawed in a 37 °C water bath, pelleted at 1000× g for 2 min and resuspended in 1 mL RPMI 1640 without L-glutamine supplemented with 1X Glutamax, 10% FBS and 1% Penicillin/Streptomycin. The cells were then washed at 1000× g for 2 min and resuspended in fresh medium of the same formulation. They were then seeded in 96-well U-bottom plate at a density of 1 × 106 cells/mL, and kept in a humidified 5% CO2 incubator at 37 °C over the course of the experiment.
4.5. Mitochondrial Mass and Mitochondrial Membrane Potential (MMP)
Mitochondrial mass and MMP were assayed with modifications as previously using the mitochondrial dyes MitoTracker
® Green FM (Life Technologies) and MitoTracker
® Red CMXRos (Life Technologies) [
21,
64]. Both dyes bind specifically to mitochondrial membranes, MitoTracker
® Red binding being membrane potential (Δψ
m)-dependent, while MitoTracker
® Green binding is not. Mitotracker Green fluorescence thus measures mitochondrial membrane “mass” and Mitotracker Red provides a measurement of Δψm when normalized to the Mitotracker Green signal.
Cells (7 × 105) were harvested at 500× g for 5 min, and 1 × 105 cells were plated per well into six wells of a 96 well black, clear flat bottom plate (Corning). The plate was incubated for 1 h at 37 °C with 5% CO2. To duplicate wells for each dye treatment, MitoTracker® Green and Red were added to final concentrations of 200 nM, PBS added to background wells and Hoechst 33342 Nuclear Stain (Enzo Life Sciences) was included in every well at a final dilution of 1/2000, for normalizing under each treatment condition (excitation 355 nm, emission 455 nm). The plate was then incubated for 1 h at 37 °C and 5% CO2, the supernatant removed via aspiration and replaced with PBS. Fluorescences were read using the BMG Labtech Clariostar microplate reader. Relative mitochondrial mass was determined by background-subtracted MitoTracker® Green FM fluorescence at excitation 470 nm and emission 515 nm, normalized to the background subtracted signal from the same number of cells of the internal control cell line. The MMP was determined from the background-subtracted MitoTracker® Red CMXRos fluorescence (excitation 570 nm, emission 620 nm) divided by the background-subtracted fluorescence of MitoTracker® Green FM.
4.6. Mitochondrial Stress Test (Seahorse Respirometry)
Oxygen consumption rates (OCR) of 8 × 10
5 viable PBMCs or lymphoblasts per well were measured using the Seahorse XFe24 Extracellular Flux Analyzer with Seahorse XF24 FluxPaks (Agilent Technologies, Chicopee, Canada). Immortalized lymphoblasts were cultured in 3 mL growth medium per well in 6-well Costar plates prior to Seahorse experiments while PBMCs were recovered from storage and inoculated immediately. Seahorse assays were carried out as previously described in detail [
21] and illustrated in
Figure S3. Oxygen consumption rates (OCR in pmol/min) were measured (basal OCR) prior to and after successive injection of 1 µM oligomycin (ATP synthase inhibitor), 1 µM CCCP (carbonyl cyanide m-chlorophenyl hydrazone, an uncoupling protonophore), 1 µM rotenone (Complex I inhibitor) and 5 µM antimycin A (Complex III inhibitor). From the resulting data we determined the OCR associated with respiratory ATP synthesis (oligomycin-sensitive), the maximum OCR in CCCP-uncoupled mitochondria and the rotenone-sensitive OCR attributable to uncoupled Complex I activity, the antimycin-sensitive Complex II/III activity, the OCR by mitochondrial functions (e.g., protein import) other than ATP synthesis that are Δψm-driven (so-called ‘proton leak’), non-respiratory oxygen consumption (e.g., by cellular and mitochondrial oxygenases and oxidases), and the respiratory ‘spare-capacity’ (excess capacity of the respiratory electron transport chain that is not being used in basal respiration).
4.7. Glycolytic Stress Test
The extracellular acidification rate (ECAR) of live, intact lymphoblasts was measured using a modified glycolytic stress test in the Seahorse XFe24 Extracellular Flux Analyzer with Seahorse XF24 FluxPaks (Agilent Technologies, Chicopee, Canada). Immortalized lymphoblasts were cultured in growth medium in 6-well plates prior to Seahorse experiments.
In order to measure the ECAR of cells using this method, they must be firmly adhered to and evenly spread across the bottom of the assay plate wells. To achieve this, the Cell Culture Microplate was prepared as previously described with a Matrigel coating in the bottom of each well [
21]. The plate was then left to dry at room temperature under laminar flow, and 8 × 10
5 cells/well were later plated out in XF base medium (Agilent Technologies, Chicopee, Canada) containing 200 mM L-glutamine and 5 mM HEPES, as recommended by the manufacturer for glycolytic assays.
The sensor cartridge apparatus was rehydrated one day in advance by adding 1 mL XF Calibrant to each well and incubating at 37 °C until needed. The injection ports of the sensor cartridge apparatus were loaded with the following drugs, in chronological order of four injections to give the indicated final concentrations in the wells: Glucose-10 mM, Oligomycin-2 µM, Rotenone-1 µM and Antimycin A-5 µM (combined injection), 2-Deoxyglucose-50 mM. The treatment with the rotenone/antimycin combination allowed assessment of the impact of electron transport on ECAR by respiratory acidification coupled to passage of some glycolytic pyruvate through the TCA cycle to supply respiration.
Before and after each successive drug addition, the ECAR was measured over three time points, consisting of a 3 min mix, 2 min wait, and 3 min measurement time. These measurements were subsequently analyzed to determine the magnitudes of various parameters of glycolysis based on the targets of each successive drug injection.
4.8. Steady-State ATP Analysis
Steady-state intracellular ATP concentration was determined by firefly luciferin bioluminescence using the ATP Determination Kit (Invitrogen) as previously described [
21].
4.9. Intracellular Reactive Oxygen Species (ROS) Levels
Intracellular ROS levels were determined using the Fluorometric Intracellular ROS Kit (MAK145-1KT, Sigma). Then 5 × 105 cells were harvested per cell line at 500× g for 5 min, resuspended in 360 µL PBS, and 90 µL loaded into triplicate wells on a 96 well black, clear flat bottom plate. One hundred microliters of reaction mix prepared according to manufacturer’s instructions was added to each well, the plate covered from light and incubated for 1 h at 37 °C with 5% CO2. The fluorescence was then read at excitation 520 nm emission 605 nm in the Clariostar microplate reader as a measure of intracellular ROS. C105 was arbitrarily included as an internal normalization control for between-experiment variation. The fluorescence is proportional to the amount of ROS present.
4.10. Mitochondrial or EBV Genome Copy Number
Relative mitochondrial genome copy number was determined by the amplification and detection of two indicative mitochondrially encoded genes (
mtND1 and
mtND4) as previously described [
21].
Relative EBV genome copy number was determined by the amplification and detection of two indicative EBV genes (
BHRF and
EBNA-1) [
65] following the same qPCR method as previously described [
21].
4.11. 4E-BP1 Phosphorylation Levels (TORC1 Activity)
TORC1 activity in ME/CFS lymphoblast lysates was measured using a Time-resolved FRET-based multiwell plate assay of the phosphorylation state of 4E-BP1, a major TORC-1 substrate (Cisbio Bioassays). Cells were harvested, resuspended in growth medium at 2.75 × 105 cells/mL and plated in four replicates at 5 × 104 cells/well in a 96-well plate. Cells were incubated at 5% CO2/37 °C for 2 h, with two of the replicates subjected to TOR inhibition by 0.5 µM TORIN2. Lysis buffer was added to each well as per manufacturer instructions and the plate mixed on an orbital shaker for 40 min before plating each sample into a 384 well white plate (Corning)—incorporating various controls and antibody mix (anti- 4E-BP1 antibody labelled with d2 acceptor, and anti-phospho-4E-BP1 antibody labelled with Eu3+-cryptate donor) according to manufacturer instructions. After a 2 h incubation at room temperature the plate was scanned by the Clariostar plate reader (BMG) and the ratio of the FRET signal from anti-phospho-4E-BP1 antibody to the donor fluorescence signal from anti-4E-BP1 antibody was measured according to instructions. C105 cells were included as an internal normalization control for between-experiment variation.
4.12. Western Blotting
Cells were lysed in SDS loading buffer (63 mM Tris hydrochloride, 10% glycerol, 2% SDS, 10% mercaptoethanol and 0.0001% Bromophenol Blue) with a protease inhibitor cocktail (complete EDTA-free, Roche). A small aliquot of each sample was briefly sonicated and analyzed for total protein concentration using a Qubit Protein Assay Kit and Qubit 2.0 Fluorometer (Thermo-Fisher Scientific) according to the manufacturer’s instructions.
The samples were then heated to 90 °C for 10 min and 30 µg of total protein was loaded into each well in 12% SDS polyacrylamide gels. After electrophoresis, proteins were transferred onto PVDF membranes (Amersham Hybond-P, GE Healthcare) using a Trans Blot Turbo Blotting apparatus (Bio-Rad) for 30 min at 180 V, 1.0 A, blocked for 1h with blocking buffer (5% skim milk, TBS) and incubated overnight with primary antibodies (Total OXPHOS human WB antibody cocktail, Abcam, ab110411) diluted 1:1000 in blocking buffer. This cocktail is directed against five OXPHOS proteins. Stain-free gel scans were utilized as the internal loading control in combination with an Alexa Fluor 800-labelled secondary antibody for detection (Alexa Fluor 800 goat anti-mouse IgG diluted 1:1000 in TBS) (Thermo-Fisher Scientific). Following incubation with antibodies, the membranes were washed three times with TBS buffer containing 0.5% Tween 20, scanned with a ChemiDoc (Bio-Rad) and analyzed using the Image Lab software (Bio-Rad). Two arbitrarily selected control cell lines (C105 and C0002, two of the study controls) were included in every blot as internal normalization controls for between-experiment variation.
4.13. Whole Cell Proteomics
Each sample (3 × 106 cells in 100 µL PBS) was dried using a SpeedVac Concentrator and Savant Refrigerated Vapor trap (Thermo-Fisher Scientific). Samples were resuspended in 8 M Urea, 100 mM Tris pH = 8.3. 1 µL of TCEP (tris [2-carboxyethyl] phosphine hydrochloride, 200 mM solution in water) was then added to the samples and incubated overnight at 21 °C in a ThermoMixer (Eppendorf AG). Four microliters of 1 M IAA (iodoacetamide in water) was added the following day and incubated in the dark at 21 °C. Next, 500 µL of 50 mM Tris (pH 8.3) and 1 μg trypsin was added to samples and left for 6 h at 37 °C in an incubator. Another 1 μg trypsin was added for double digestion and incubated overnight at 37 °C. The digested samples were purified for mass spectrometry analysis prior to peptide reconstitution and separation using Sep-Pak light C18 cartridges (Waters) according to manufacturer standard procedures. Data were collected on a Q Exactive HF (Thermo-Fisher Scientific) in Data Dependent Acquisition mode using m/z 350–1500 as MS scan range at 60,000 resolution, HCD MS/MS spectra were collected for the 15 most intense ions per MS scan at 15,000 resolution with a normalized collision energy of 28% and an isolation window of 1.4 m/z. Dynamic exclusion parameters were set as follows: exclude isotope on, duration 30 s and peptide match preferred. Other instrument parameters for the Orbitrap were MS maximum injection time 30 ms with AGC target 3 × 106, for a maximum injection time of 25 ms with AGT target of 1 × 105. Raw files consisting of high-resolution MS/MS spectra were processed with MaxQuant version 1.6.1.0 to detect features and identify proteins using the search engine Andromeda. UniProtKB/Swiss-Prot Homo sapiens sequence data was used as the database for the search engine. To assess the false discovery rate (FDR) a decoy data set was generated by MaxQuant after reversing the sequence database. Theoretical spectra were generated using the enzyme as trypsin allowing two missed cleavages. The minimum required peptide length used was seven amino acids. Carbamidomethylation of Cys was set as a fixed modification, while n-acetylation of proteins and oxidation of Met were set as variable modifications. Precursor mass tolerance was set to 5 ppm and MS/MS tolerance to 0.05 Da. The “match between runs” option was enabled in MaxQuant to transfer identifications made between runs on the basis of matching precursors with high mass accuracy. PSM and protein identifications were filtered using a target-decoy approach at a false discovery rate (FDR) of 1%.
4.14. Quantification and Statistical Analysis
Data was analyzed using Microsoft Excel with the Winstat add-in (
http://www.winstat.com) or R using the packages R Commander [
66], REzy [
67] and stats. Proteomics data was analyzed employing the software Scaffold (Proteome Software) and detected proteins were identified as belonging to a single functional group (e.g., TCA cycle) or respiratory complex using the NCBI GO annotation database [
68]. Unless otherwise specified, two-sample tests used the Welch
t-test. ANOVA and Fisher’s exact tests were used as specified and appropriate. The significance of individual coefficients in multiple regression analysis was tested using
t-tests. The binomial test was employed to assess whether all detected proteins in a single functional group or respiratory complex were coordinately up- or down-regulated in the ME/CFS group compared to controls, the null hypothesis being that the levels of each protein had a probability of 0.5 of being above the control average. The single sample
t test was also used to assess whether the average fold change in the levels of all detected proteins in a single functional group or respiratory complex in the ME/CFS cohort was significantly greater than the control value of 1.0.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







