Calmodulin-Binding Proteins in Muscle: A Minireview on Nuclear Receptor Interacting Protein, Neurogranin, and Growth-Associated Protein 43



 and
and 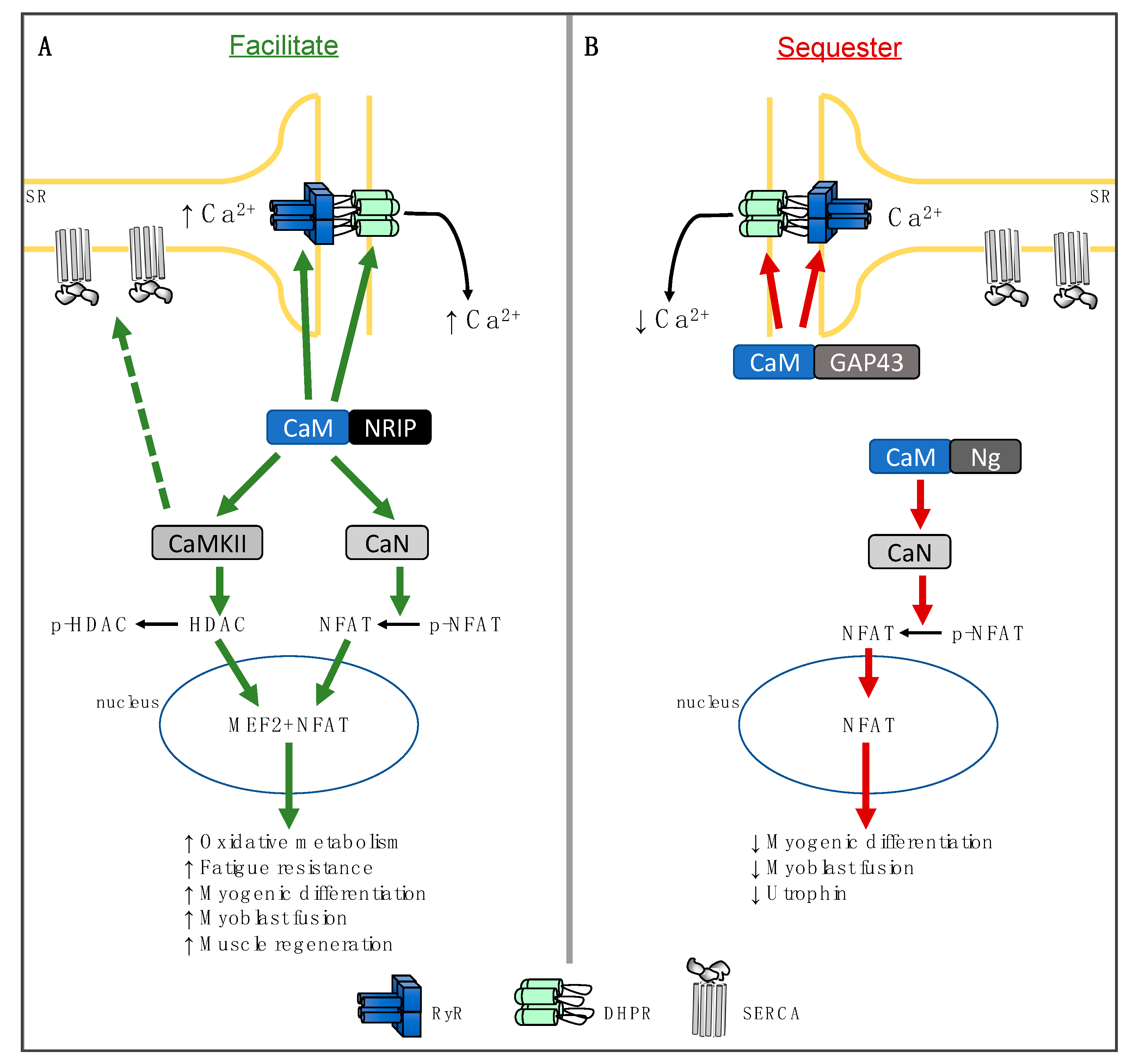{kind=link}
Abstract
1. Introduction
2. Nuclear Receptor Interacting Protein
3. Neurogranin
4. Growth-Associated Protein 43
5. Physiological Significance
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AID | Autoinhibitory domain |
| CaM | Calmodulin |
| CaMKII | Calmodulin dependent kinase II |
| CsA | Cyclosporine A |
| CNM | Centronuclear myopathy |
| CaMBP | Calmodulin-binding protein |
| DHPR | Dihydropyridine receptors |
| DMD | Duchenne muscular dystrophy |
| DM1 | Myotonic dystrophy type 1 |
| GAP43 | Neuron-growth-associated protein 43 |
| MEF2 | Myocyte enhancer factor 2 |
| NFAT | Nuclear factor of activated T-cells |
| NRIP | Nuclear receptor interacting protein |
| Ng | Neurogranin |
| PGC1- | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PLN | Phospholamban |
| RyR | Ryanodine receptor |
| SR | Sarcoplasmic reticulum |
| ROS | Reactive oxygen species |
| RCAN1 | Regulator of calcineurin1 |
| SERCA | Sarco(endo)plasmic reticulum Ca2+ ATPase |
| SLN | Sarcolipin |
| UCP1 | Uncoupling protein 1 |
References
- Ikura, M.; Ames, J.B. Genetic polymorphism and protein conformational plasticity in the calmodulin superfamily: Two ways to promote multifunctionality. Proc. Natl. Acad. Sci. USA 2006, 103, 1159–1164. [Google Scholar] [CrossRef]
- Ojuka, E.O.; Goyaram, V.; Smith, J.A. The role of CaMKII in regulating GLUT4 expression in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E322–E331. [Google Scholar] [CrossRef]
- Tavi, P.; Westerblad, H. The role of in vivo Ca(2)(+) signals acting on Ca(2)(+)-calmodulin-dependent proteins for skeletal muscle plasticity. J. Physiol. 2011, 589, 5021–5031. [Google Scholar] [CrossRef]
- Stull, J.T.; Kamm, K.E.; Vandenboom, R. Myosin light chain kinase and the role of myosin light chain phosphorylation in skeletal muscle. Arch. Biochem. Biophys. 2011, 510, 120–128. [Google Scholar] [CrossRef]
- Olson, E.N.; Williams, R.S. Remodeling muscles with calcineurin. BioEssays 2000, 22, 510–519. [Google Scholar] [CrossRef]
- Michel, R.N.; Chin, E.R.; Chakkalakal, J.V.; Eibl, J.K.; Jasmin, B.J. Ca2+/calmodulin-based signalling in the regulation of the muscle fibre phenotype and its therapeutic potential via modulation of utrophin A and myostatin expression. Appl. Physiol. Nutr. Metab. 2007, 32, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.R. Role of Ca2+/calmodulin-dependent kinases in skeletal muscle plasticity. J. Appl. Physiol. 2005, 99, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Bahler, M.; Rhoads, A. Calmodulin signaling via the IQ motif. FEBS Lett. 2002, 513, 107–113. [Google Scholar] [CrossRef]
- Chen, H.H.; Chen, W.P.; Yan, W.L.; Huang, Y.C.; Chang, S.W.; Fu, W.M.; Su, M.J.; Yu, I.S.; Tsai, T.C.; Yan, Y.T.; et al. NRIP is newly identified as a Z-disc protein, activating calmodulin signaling for skeletal muscle contraction and regeneration. J. Cell Sci. 2015, 128, 4196–4209. [Google Scholar] [CrossRef]
- Chang, S.W.; Tsao, Y.P.; Lin, C.Y.; Chen, S.L. NRIP, a novel calmodulin binding protein, activates calcineurin to dephosphorylate human papillomavirus E2 protein. J. Virol. 2011, 85, 6750–6763. [Google Scholar] [CrossRef]
- Yang, K.C.; Chuang, K.W.; Yen, W.S.; Lin, S.Y.; Chen, H.H.; Chang, S.W.; Lin, Y.S.; Wu, W.L.; Tsao, Y.P.; Chen, W.P.; et al. Deficiency of nuclear receptor interaction protein leads to cardiomyopathy by disrupting sarcomere structure and mitochondrial respiration. J. Mol. Cell Cardiol. 2019, 137, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Liu, Y.; Cseresnyes, Z.; Hawkins, A.; Randall, W.R.; Schneider, M.F. Activity- and calcineurin-independent nuclear shuttling of NFATc1, but not NFATc3, in adult skeletal muscle fibers. Mol. Biol. Cell 2006, 17, 1570–1582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kohlhaas, M.; Backs, J.; Mishra, S.; Phillips, W.; Dybkova, N.; Chang, S.; Ling, H.; Bers, D.M.; Maier, L.S.; et al. CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J. Biol. Chem. 2007, 282, 35078–35087. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Olson, E.N. MEF2: A central regulator of diverse developmental programs. Dev. 2007, 134, 4131–4140. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Investig. 2007, 117, 2459–2467. [Google Scholar] [CrossRef]
- Smith, J.A.; Kohn, T.A.; Chetty, A.K.; Ojuka, E.O. CaMK activation during exercise is required for histone hyperacetylation and MEF2A binding at the MEF2 site on the Glut4 gene. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E698–E704. [Google Scholar] [CrossRef]
- Angus, L.M.; Chakkalakal, J.V.; Mejat, A.; Eibl, J.K.; Belanger, G.; Megeney, L.A.; Chin, E.R.; Schaeffer, L.; Michel, R.N.; Jasmin, B.J. Calcineurin-NFAT signaling, together with GABP and peroxisome PGC-1[1], drives utrophin gene expression at the neuromuscular junction. Am. J. Physiol. Cell Physiol. 2005, 289, C908–C917. [Google Scholar] [CrossRef]
- Jiang, L.Q.; Garcia-Roves, P.M.; de Castro Barbosa, T.; Zierath, J.R. Constitutively active calcineurin in skeletal muscle increases endurance performance and mitochondrial respiratory capacity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E8–E16. [Google Scholar] [CrossRef]
- Pate, P.; Mochca-Morales, J.; Wu, Y.; Zhang, J.Z.; Rodney, G.G.; Serysheva, I.I.; Williams, B.Y.; Anderson, M.E.; Hamilton, S.L. Determinants for calmodulin binding on voltage-dependent Ca2+ channels. J. Biol. Chem. 2000, 275, 39786–39792. [Google Scholar] [CrossRef]
- Stroffekova, K. Ca2+/CaM-dependent inactivation of the skeletal muscle L-type Ca2+ channel (Cav1.1). Pflugers Arch. 2008, 455, 873–884. [Google Scholar] [CrossRef]
- Stroffekova, K. The IQ motif is crucial for Cav1.1 function. J. Biomed. Biotechnol. 2011, 2011, 504649. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Xin, C.; Meissner, G. Identification of apocalmodulin and Ca2+-calmodulin regulatory domain in skeletal muscle Ca2+ release channel, ryanodine receptor. J. Biol. Chem. 2001, 276, 22579–22585. [Google Scholar] [CrossRef] [PubMed]
- Samso, M.; Wagenknecht, T. Apocalmodulin and Ca2+-calmodulin bind to neighboring locations on the ryanodine receptor. J. Biol. Chem. 2002, 277, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine receptors: Structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010, 2, a003996. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef]
- Zhang, Y.; Ye, J.; Chen, D.; Zhao, X.; Xiao, X.; Tai, S.; Yang, W.; Zhu, D. Differential expression profiling between the relative normal and dystrophic muscle tissues from the same LGMD patient. J. Transl. Med. 2006, 4, 53. [Google Scholar] [CrossRef]
- Van der Velden, J.L.; Schols, A.M.; Willems, J.; Kelders, M.C.; Langen, R.C. Glycogen synthase kinase 3 suppresses myogenic differentiation through negative regulation of NFATc3. J. Biol. Chem. 2008, 283, 358–366. [Google Scholar] [CrossRef]
- Fajardo, V.A.; Watson, C.J.F.; Bott, K.N.; Moradi, F.; Maddalena, L.A.; Bellissimo, C.A.; Turner, K.D.; Peters, S.J.; LeBlanc, P.J.; MacNeil, A.J.; et al. Neurogranin is expressed in mammalian skeletal muscle and inhibits calcineurin signaling and myoblast fusion. Am. J. Physiol. Cell Physiol. 2019, 317, C1025–C1033. [Google Scholar] [CrossRef]
- Park, S.Y.; Yun, Y.; Lim, J.S.; Kim, M.J.; Kim, S.Y.; Kim, J.E.; Kim, I.S. Stabilin-2 modulates the efficiency of myoblast fusion during myogenic differentiation and muscle regeneration. Nat. Commun. 2016, 7, 10871. [Google Scholar] [CrossRef]
- Sakuma, K.; Nishikawa, J.; Nakao, R.; Watanabe, K.; Totsuka, T.; Nakano, H.; Sano, M.; Yasuhara, M. Calcineurin is a potent regulator for skeletal muscle regeneration by association with NFATc1 and GATA-2. Acta Neuropathol. 2003, 105, 271–280. [Google Scholar] [CrossRef]
- Stupka, N.; Schertzer, J.D.; Bassel-Duby, R.; Olson, E.N.; Lynch, G.S. Calcineurin-A alpha activation enhances the structure and function of regenerating muscles after myotoxic injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R686–R694. [Google Scholar] [CrossRef][Green Version]
- Horsley, V.; Friday, B.B.; Matteson, S.; Kegley, K.M.; Gephart, J.; Pavlath, G.K. Regulation of the growth of multinucleated muscle cells by an NFATC2-dependent pathway. J. Cell Biol. 2001, 153, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Horsley, V.; Jansen, K.M.; Mills, S.T.; Pavlath, G.K. IL-4 acts as a myoblast recruitment factor during mammalian muscle growth. Cell 2003, 113, 483–494. [Google Scholar] [CrossRef]
- Horsley, V.; Pavlath, G.K. Prostaglandin F2(alpha) stimulates growth of skeletal muscle cells via an NFATC2-dependent pathway. J. Cell Biol. 2003, 161, 111–118. [Google Scholar] [CrossRef]
- Martzen, M.R.; Slemmon, J.R. The dendritic peptide neurogranin can regulate a calmodulin-dependent target. J. Neurochem. 1995, 64, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Krucker, T.; Siggins, G.R.; McNamara, R.K.; Lindsley, K.A.; Dao, A.; Allison, D.W.; De Lecea, L.; Lovenberg, T.W.; Sutcliffe, J.G.; Gerendasy, D.D. Targeted disruption of RC3 reveals a calmodulin-based mechanism for regulating metaplasticity in the hippocampus. J. Neurosci. 2002, 22, 5525–5535. [Google Scholar] [CrossRef]
- Petersen, A.; Gerges, N.Z. Neurogranin regulates CaM dynamics at dendritic spines. Sci. Rep. 2015, 5, 11135. [Google Scholar] [CrossRef]
- Pak, J.H.; Huang, F.L.; Li, J.; Balschun, D.; Reymann, K.G.; Chiang, C.; Westphal, H.; Huang, K.P. Involvement of neurogranin in the modulation of calcium/calmodulin-dependent protein kinase II, synaptic plasticity, and spatial learning: A study with knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 11232–11237. [Google Scholar] [CrossRef]
- Hoffman, L.; Chandrasekar, A.; Wang, X.; Putkey, J.A.; Waxham, M.N. Neurogranin alters the structure and calcium binding properties of calmodulin. J. Biol. Chem. 2014, 289, 14644–14655. [Google Scholar] [CrossRef]
- Gaertner, T.R.; Putkey, J.A.; Waxham, M.N. RC3/Neurogranin and Ca2+/calmodulin-dependent protein kinase II produce opposing effects on the affinity of calmodulin for calcium. J. Biol. Chem. 2004, 279, 39374–39382. [Google Scholar] [CrossRef]
- Chakkalakal, J.V.; Stocksley, M.A.; Harrison, M.A.; Angus, L.M.; Deschenes-Furry, J.; St-Pierre, S.; Megeney, L.A.; Chin, E.R.; Michel, R.N.; Jasmin, B.J. Expression of utrophin A mRNA correlates with the oxidative capacity of skeletal muscle fiber types and is regulated by calcineurin/NFAT signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 7791–7796. [Google Scholar] [CrossRef]
- Zhong, L.; Gerges, N.Z. Neurogranin targets calmodulin and lowers the threshold for the induction of long-term potentiation. PLoS ONE 2012, 7, e41275. [Google Scholar] [CrossRef]
- Guarnieri, S.; Morabito, C.; Paolini, C.; Boncompagni, S.; Pilla, R.; Fano-Illic, G.; Mariggio, M.A. Growth associated protein 43 is expressed in skeletal muscle fibers and is localized in proximity of mitochondria and calcium release units. PLoS ONE 2013, 8, e53267. [Google Scholar] [CrossRef]
- Caprara, G.A.; Perni, S.; Morabito, C.; Mariggio, M.A.; Guarnieri, S. Specific association of growth-associated protein 43 with calcium release units in skeletal muscles of lower vertebrates. Eur. J. Histochem. 2014, 58, 2453. [Google Scholar] [CrossRef]
- Caprara, G.A.; Morabito, C.; Perni, S.; Navarra, R.; Guarnieri, S.; Mariggio, M.A. Evidence for Altered Ca(2+) Handling in Growth Associated Protein 43-Knockout Skeletal Muscle. Front. Physiol. 2016, 7, 493. [Google Scholar] [CrossRef]
- Long, Y.C.; Glund, S.; Garcia-Roves, P.M.; Zierath, J.R. Calcineurin regulates skeletal muscle metabolism via coordinated changes in gene expression. J. Biol. Chem. 2007, 282, 1607–1614. [Google Scholar] [CrossRef]
- Frey, N.; Frank, D.; Lippl, S.; Kuhn, C.; Kogler, H.; Barrientos, T.; Rohr, C.; Will, R.; Muller, O.J.; Weiler, H.; et al. Calsarcin-2 deficiency increases exercise capacity in mice through calcineurin/NFAT activation. J. Clin. Investig. 2008, 118, 3598–3608. [Google Scholar] [CrossRef]
- Oh, M.; Rybkin, I.I.; Copeland, V.; Czubryt, M.P.; Shelton, J.M.; van Rooij, E.; Richardson, J.A.; Hill, J.A.; De Windt, L.J.; Bassel-Duby, R.; et al. Calcineurin is necessary for the maintenance but not embryonic development of slow muscle fibers. Mol. Cell Biol. 2005, 25, 6629–6638. [Google Scholar] [CrossRef][Green Version]
- Pattanakuhar, S.; Pongchaidecha, A.; Chattipakorn, N.; Chattipakorn, S.C. The effect of exercise on skeletal muscle fibre type distribution in obesity: From cellular levels to clinical application. Obes. Res. Clin. Pract. 2017, 11, 112–132. [Google Scholar] [CrossRef]
- Wu, H.; Rothermel, B.; Kanatous, S.; Rosenberg, P.; Naya, F.J.; Shelton, J.M.; Hutcheson, K.A.; DiMaio, J.M.; Olson, E.N.; Bassel-Duby, R.; et al. Activation of MEF2 by muscle activity is mediated through a calcineurin-dependent pathway. EMBO J. 2001, 20, 6414–6423. [Google Scholar] [CrossRef]
- Ryder, J.W.; Bassel-Duby, R.; Olson, E.N.; Zierath, J.R. Skeletal muscle reprogramming by activation of calcineurin improves insulin action on metabolic pathways. J. Biol. Chem. 2003, 278, 44298–44304. [Google Scholar] [CrossRef] [PubMed]
- Rotter, D.; Peiris, H.; Grinsfelder, D.B.; Martin, A.M.; Burchfield, J.; Parra, V.; Hull, C.; Morales, C.R.; Jessup, C.F.; Matusica, D.; et al. Regulator of Calcineurin 1 helps coordinate whole-body metabolism and thermogenesis. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Bombardier, E.; Smith, I.C.; Gamu, D.; Fajardo, V.A.; Vigna, C.; Sayer, R.A.; Gupta, S.C.; Bal, N.C.; Periasamy, M.; Tupling, A.R. Sarcolipin trumps beta-adrenergic receptor signaling as the favored mechanism for muscle-based diet-induced thermogenesis. FASEB J. 2013, 27, 3871–3878. [Google Scholar] [CrossRef] [PubMed]
- Bombardier, E.; Smith, I.C.; Vigna, C.; Fajardo, V.A.; Tupling, A.R. Ablation of sarcolipin decreases the energy requirements for Ca2+ transport by sarco(endo)plasmic reticulum Ca2+-ATPases in resting skeletal muscle. FEBS Lett. 2013, 587, 1687–1692. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Fuller-Jackson, J.P.; Henry, B.A. Adipose and skeletal muscle thermogenesis: Studies from large animals. J. Endocrinol. 2018, 237, R99–R115. [Google Scholar] [CrossRef]
- Consolino, C.M.; Brooks, S.V. Susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of mdx mice. J. Appl. Physiol. 2004, 96, 633–638. [Google Scholar] [CrossRef]
- Webster, C.; Silberstein, L.; Hays, A.P.; Blau, H.M. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 1988, 52, 503–513. [Google Scholar] [CrossRef]
- Wang, J.; Campos, B.; Jamieson, G.A., Jr.; Kaetzel, M.A.; Dedman, J.R. Functional elimination of calmodulin within the nucleus by targeted expression of an inhibitor peptide. J. Biol. Chem. 1995, 270, 30245–30248. [Google Scholar] [CrossRef]
- Chakkalakal, J.V.; Michel, S.A.; Chin, E.R.; Michel, R.N.; Jasmin, B.J. Targeted inhibition of Ca2+ /calmodulin signaling exacerbates the dystrophic phenotype in mdx mouse muscle. Hum. Mol. Genet. 2006, 15, 1423–1435. [Google Scholar] [CrossRef]
- Stupka, N.; Schertzer, J.D.; Bassel-Duby, R.; Olson, E.N.; Lynch, G.S. Stimulation of calcineurin Aalpha activity attenuates muscle pathophysiology in mdx dystrophic mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R983–R992. [Google Scholar] [CrossRef] [PubMed]
- Stupka, N.; Gregorevic, P.; Plant, D.R.; Lynch, G.S. The calcineurin signal transduction pathway is essential for successful muscle regeneration in mdx dystrophic mice. Acta Neuropathol. 2004, 107, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Maurya, S.K.; Bal, N.C.; Sopariwala, D.H.; Pant, M.; Rowland, L.A.; Shaikh, S.A.; Periasamy, M. Sarcolipin Is a Key Determinant of the Basal Metabolic Rate, and Its Overexpression Enhances Energy Expenditure and Resistance against Diet-induced Obesity. J. Biol. Chem. 2015, 290, 10840–10849. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, V.A.; Chambers, P.J.; Juracic, E.S.; Rietze, B.A.; Gamu, D.; Bellissimo, C.; Kwon, F.; Quadrilatero, J.; Russell Tupling, A. Sarcolipin deletion in mdx mice impairs calcineurin signalling and worsens dystrophic pathology. Hum. Mol. Genet. 2018. [Google Scholar] [CrossRef]
- Ravel-Chapuis, A.; Belanger, G.; Cote, J.; Michel, R.N.; Jasmin, B.J. Misregulation of calcium-handling proteins promotes hyperactivation of calcineurin-NFAT signaling in skeletal muscle of DM1 mice. Hum. Mol. Genet. 2017, 26, 2192–2206. [Google Scholar] [CrossRef]
- Fajardo, V.A.; Gamu, D.; Mitchell, A.; Bloemberg, D.; Bombardier, E.; Chambers, P.J.; Bellissimo, C.; Quadrilatero, J.; Tupling, A.R. Sarcolipin deletion exacerbates soleus muscle atrophy and weakness in phospholamban overexpressing mice. PLoS ONE 2017, 12, e0173708. [Google Scholar] [CrossRef]
- Schiaffino, S.; Serrano, A. Calcineurin signaling and neural control of skeletal muscle fiber type and size. Trends Pharmacol. Sci. 2002, 23, 569–575. [Google Scholar] [CrossRef]
- Chin, E.R.; Olson, E.N.; Richardson, J.A.; Yang, Q.; Humphries, C.; Shelton, J.M.; Wu, H.; Zhu, W.; Bassel-Duby, R.; Williams, R.S. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 1998, 12, 2499–2509. [Google Scholar] [CrossRef]
- Dunn, S.E.; Burns, J.L.; Michel, R.N. Calcineurin is required for skeletal muscle hypertrophy. J. Biol. Chem. 1999, 274, 21908–21912. [Google Scholar] [CrossRef]
- Dunn, S.E.; Chin, E.R.; Michel, R.N. Matching of calcineurin activity to upstream effectors is critical for skeletal muscle fiber growth. J. Cell Biol. 2000, 151, 663–672. [Google Scholar] [CrossRef]
- Allen, D.L.; Roy, R.R.; Edgerton, V.R. Myonuclear domains in muscle adaptation and disease. Muscle Nerve 1999, 22, 1350–1360. [Google Scholar] [CrossRef]
- Kurgan, N.; Whitley, K.C.; Maddalena, L.A.; Moradi, F.; Stoikos, J.; Hamstra, S.I.; Rubie, E.A.; Kumar, M.; Roy, B.D.; Woodgett, J.R.; et al. A Low-Therapeutic Dose of Lithium Inhibits GSK3 and Enhances Myoblast Fusion in C2C12 Cells. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, B.J.; Dai, Y.S.; Bueno, O.F.; Parsons, S.A.; Xu, J.; Plank, D.M.; Jones, F.; Kimball, T.R.; Molkentin, J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 2004, 94, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.; Barrientos, T.; Shelton, J.M.; Frank, D.; Rutten, H.; Gehring, D.; Kuhn, C.; Lutz, M.; Rothermel, B.; Bassel-Duby, R.; et al. Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat. Med. 2004, 10, 1336–1343. [Google Scholar] [CrossRef]
- Heineke, J.; Wollert, K.C.; Osinska, H.; Sargent, M.A.; York, A.J.; Robbins, J.; Molkentin, J.D. Calcineurin protects the heart in a murine model of dilated cardiomyopathy. J. Mol. Cell Cardiol. 2010, 48, 1080–1087. [Google Scholar] [CrossRef][Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moradi, F.; Copeland, E.N.; Baranowski, R.W.; Scholey, A.E.; Stuart, J.A.; Fajardo, V.A. Calmodulin-Binding Proteins in Muscle: A Minireview on Nuclear Receptor Interacting Protein, Neurogranin, and Growth-Associated Protein 43. Int. J. Mol. Sci. 2020, 21, 1016. https://doi.org/10.3390/ijms21031016
Moradi F, Copeland EN, Baranowski RW, Scholey AE, Stuart JA, Fajardo VA. Calmodulin-Binding Proteins in Muscle: A Minireview on Nuclear Receptor Interacting Protein, Neurogranin, and Growth-Associated Protein 43. International Journal of Molecular Sciences. 2020; 21(3):1016. https://doi.org/10.3390/ijms21031016
Chicago/Turabian StyleMoradi, Fereshteh, Emily N. Copeland, Ryan W. Baranowski, Aiden E. Scholey, Jeffrey A. Stuart, and Val A. Fajardo. 2020. "Calmodulin-Binding Proteins in Muscle: A Minireview on Nuclear Receptor Interacting Protein, Neurogranin, and Growth-Associated Protein 43" International Journal of Molecular Sciences 21, no. 3: 1016. https://doi.org/10.3390/ijms21031016
APA StyleMoradi, F., Copeland, E. N., Baranowski, R. W., Scholey, A. E., Stuart, J. A., & Fajardo, V. A. (2020). Calmodulin-Binding Proteins in Muscle: A Minireview on Nuclear Receptor Interacting Protein, Neurogranin, and Growth-Associated Protein 43. International Journal of Molecular Sciences, 21(3), 1016. https://doi.org/10.3390/ijms21031016