NADPH Oxidase-Mediated Activation of Neutral Sphingomyelinase Is Responsible for Diesel Particulate Extract-Induced Keratinocyte Apoptosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
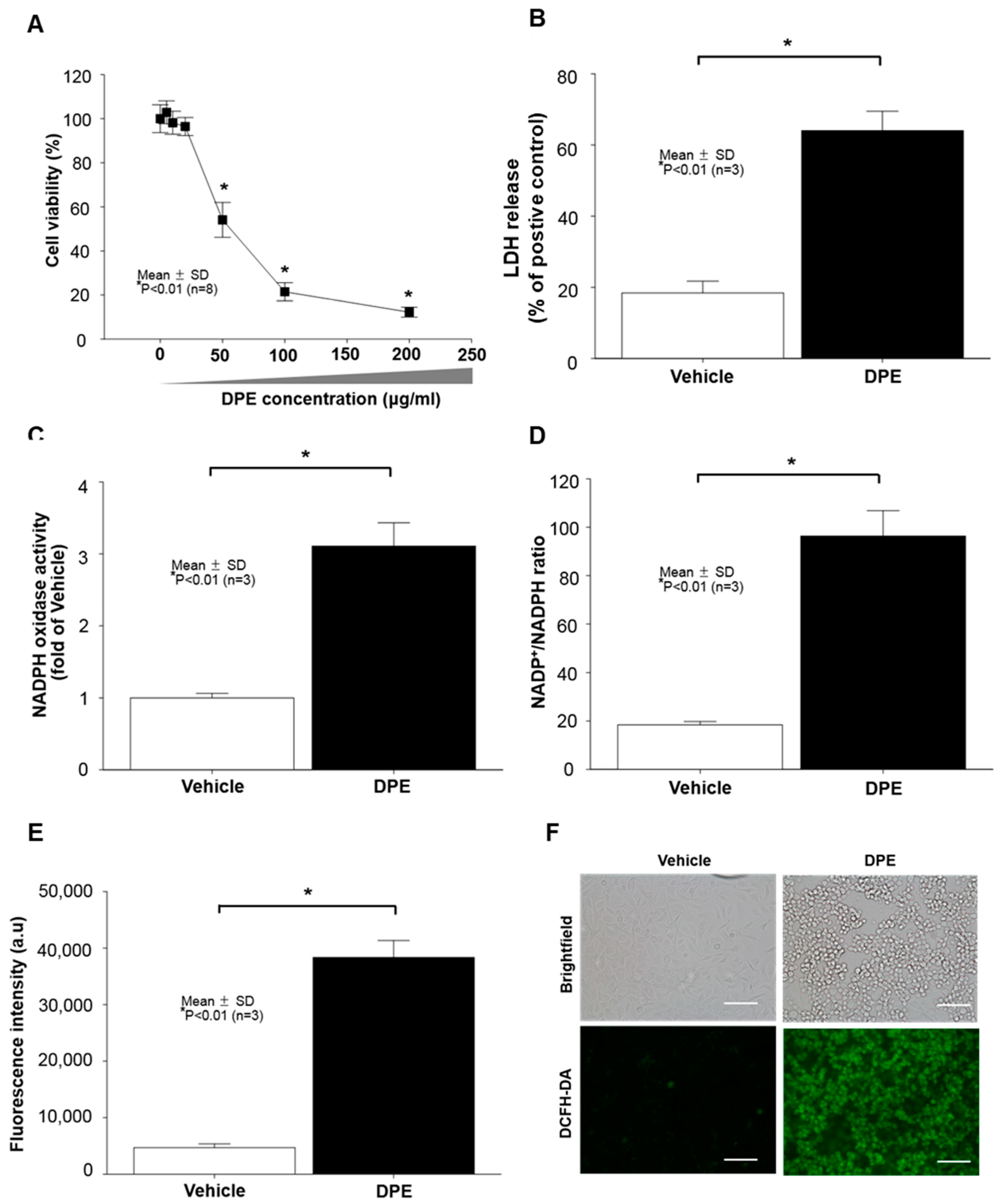
2.1. Diesel Particulate Extract (DPE) Induces Keratinocytes (KC) Apoptosis, Paralleled by Increased NAPDH Oxidation and Reactive Oxygen Species (ROS) Generation
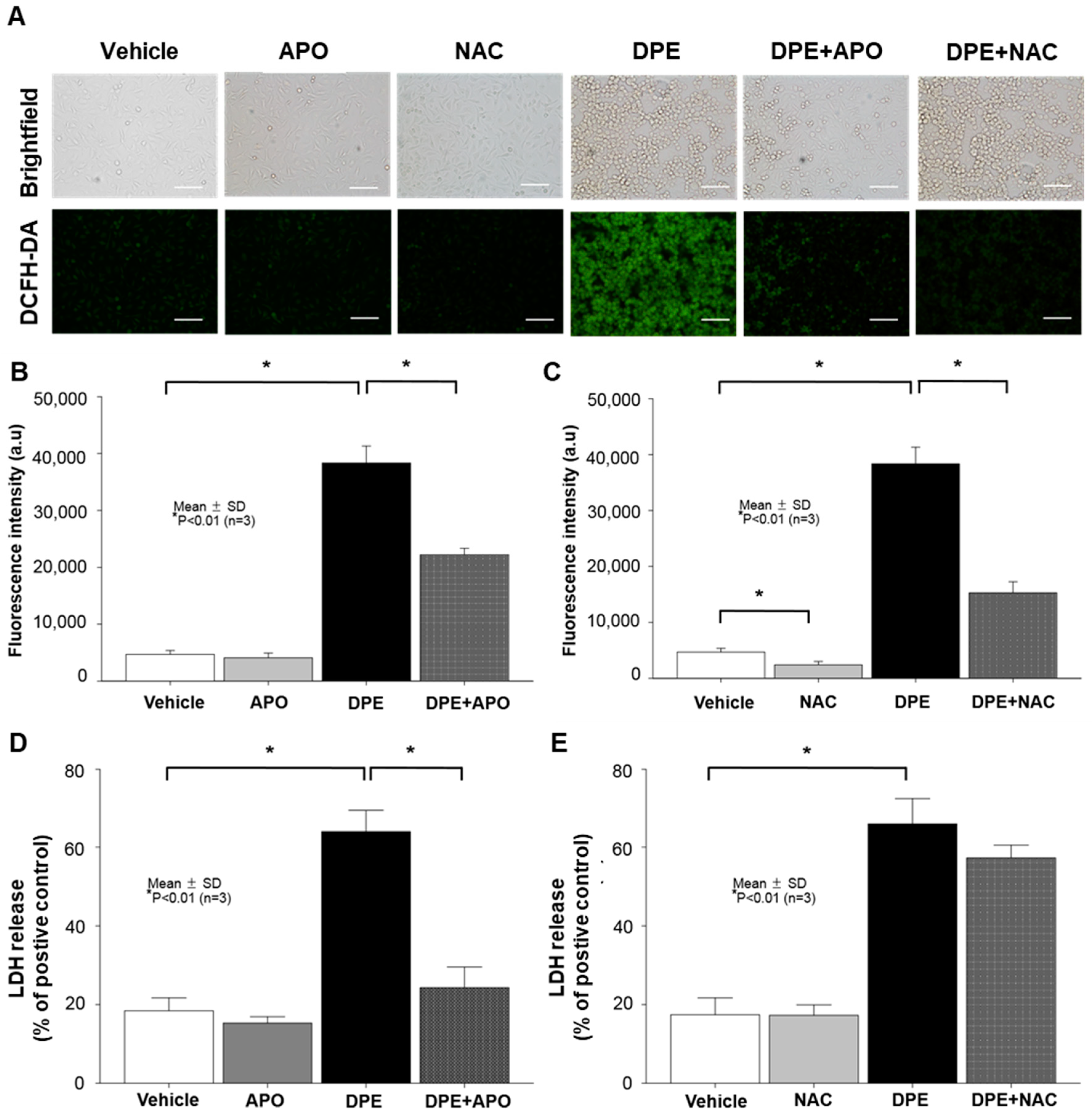
2.2. DPE Induces KC Apoptosis through NOX Activation, but Not ROS-Dependent Mechanism
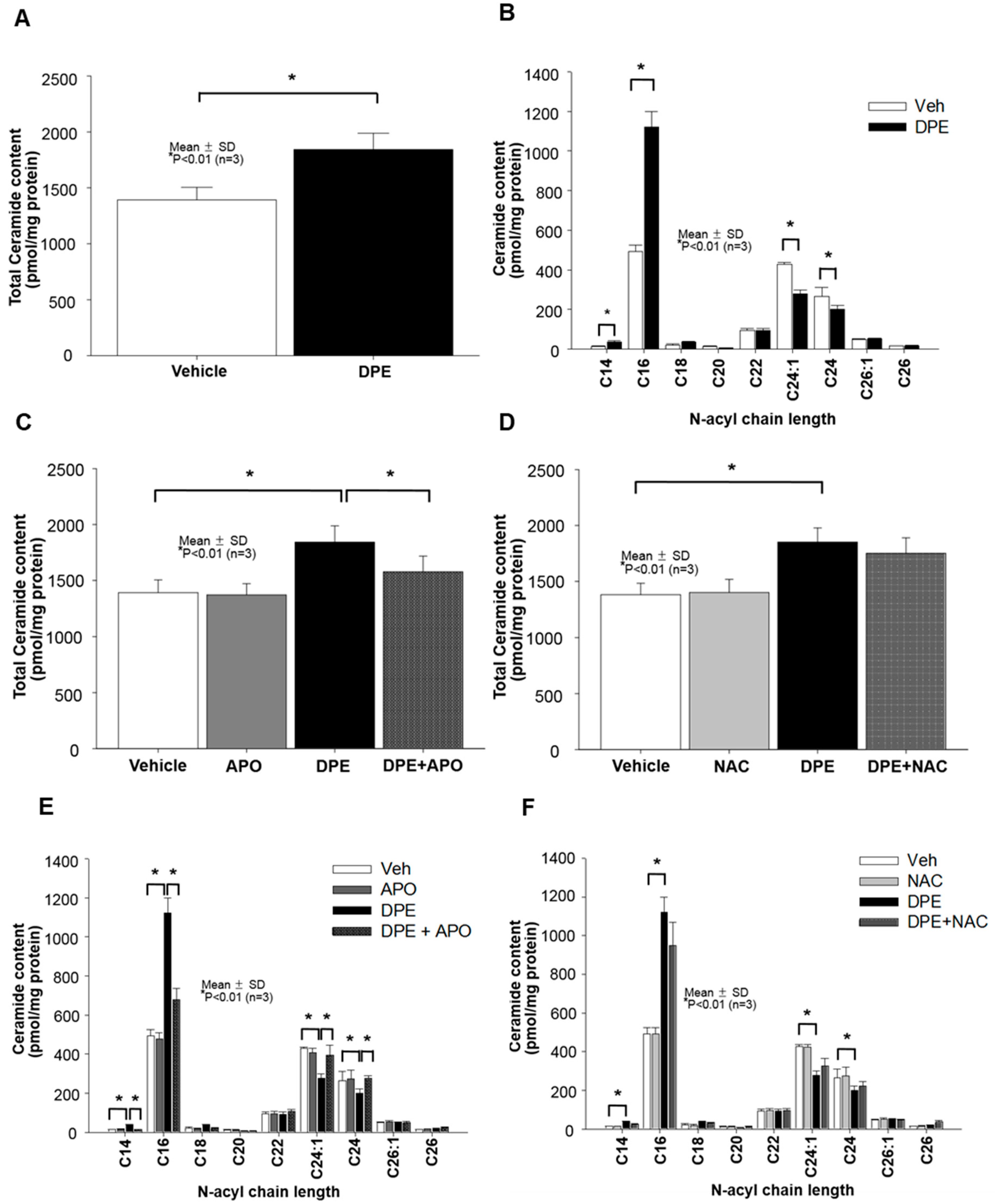
2.3. DPE-Induced Activation of NAPDH Oxidation is Responsible for Increased Overall Ceramide Production in Human KC
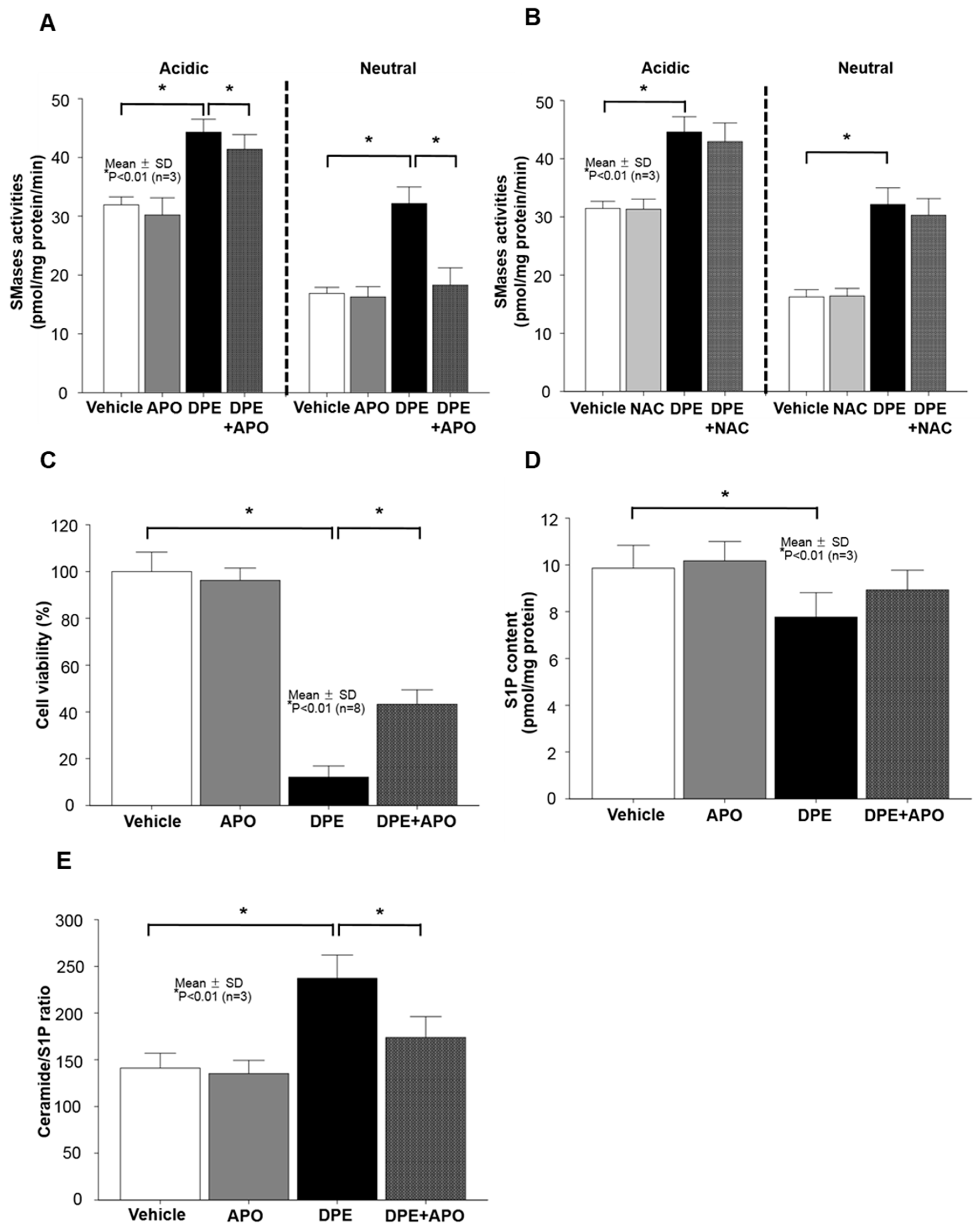
2.4. Neutral Sphingomyelinase (SMase) Is Required for DPE/NOX Activation-Mediated Increase in Ceramide Production and Subsequent KC Apoptosis
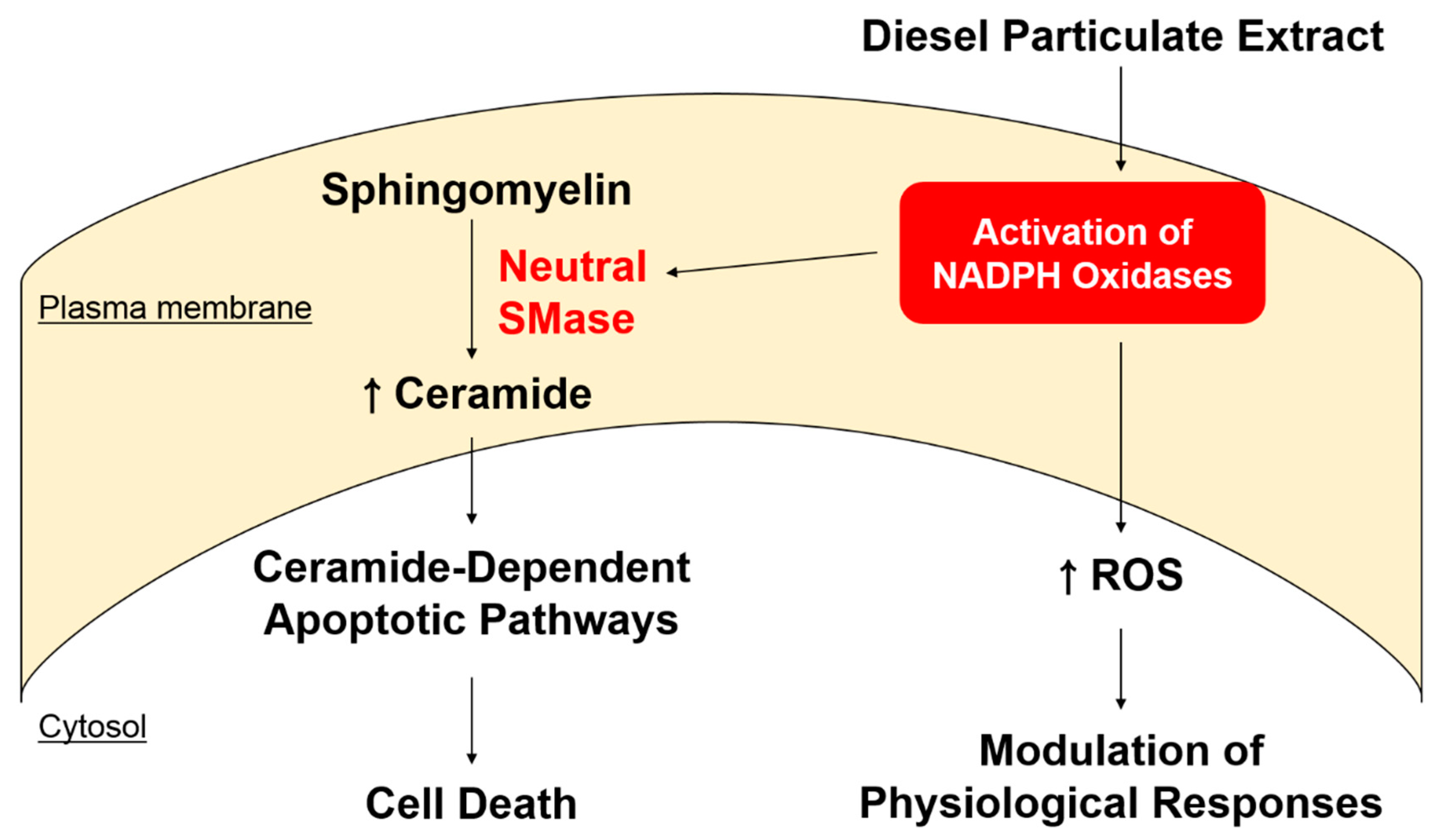
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. DPE Preparation
4.3. Cell Culture
4.4. Cell Viability
4.5. Lactate Dehydrogenase Assay
4.6. Activity of NADPH Oxidases
4.7. 2′,7′-Dichlorodihydrofluorescein Diacetate-Based Detection of Cellular ROS
4.8. Measurement of Ceramide and Sphingosine-1-Phosphate (S1P)
4.9. Enzyme Activity Assay for Sphingomyelinases (SMases)
4.10. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APO | Apocynin |
| DCFH-DA | 2′,7′-dichlorodihydrofluorescein diacetate |
| DPE | Diesel particulate extract |
| FA | Fatty acid |
| JNK | c-jun kinase |
| KC | Keratinocytes |
| LDH | Lactate dehydrogenase |
| MRM | Multiple reaction monitoring mode |
| NAC | N-Acetylcysteine |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NOX | NADPH oxidase |
| PAH | Polyaromatic hydrocarbon |
| PI3K/PKB | Phosphoinositide 3-kinase/ Protein kinase B |
| ROS | Reactive oxygen species |
| SAPK | Stress-activated protein kinase |
| SMase | Sphingomyelinase |
| S1P | Sphingosine-1-phosphate |
| WST | Water-soluble tetrazolium salt |
References
- Elias, P.M.; Menon, G.K. Structural and lipid biochemical correlates of the epidermal permeability barrier. Adv. Lipid. Res. 1991, 24, 1–26. [Google Scholar] [PubMed]
- Lee, C.W.; Lin, Z.C.; Hu, S.C.; Chiang, Y.C.; Hsu, L.F.; Lin, Y.C.; Lee, I.T.; Tsai, M.H.; Fang, J.Y. Urban particulate matter down-regulates filaggrin via COX2 expression/PGE2 production leading to skin barrier dysfunction. Sci. Rep. 2016, 6, 27995. [Google Scholar] [CrossRef] [PubMed]
- Sydbom, A.; Blomberg, A.; Parnia, S.; Stenfors, N.; Sandstrom, T.; Dahlen, S.E. Health effects of diesel exhaust emissions. Eur. Respir. J. 2001, 17, 733–746. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, P.; Jain, A.P.; Nanjappa, V.; Patel, K.; Mangalaparthi, K.K.; Babu, N.; Cavusoglu, N.; Roy, N.; Soeur, J.; Breton, L.; et al. Proteome-wide changes in primary skin keratinocytes exposed to diesel particulate extract-A role for antioxidants in skin health. J. Dermatol. Sci. 2018, 91, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Vattanasit, U.; Navasumrit, P.; Khadka, M.B.; Kanitwithayanun, J.; Promvijit, J.; Autrup, H.; Ruchirawat, M. Oxidative DNA damage and inflammatory responses in cultured human cells and in humans exposed to traffic-related particles. Int. J. Hyg. Env. Health 2014, 217, 23–33. [Google Scholar] [CrossRef]
- Costa, C.; Catania, S.; De Pasquale, R.; Stancanelli, R.; Scribano, G.M.; Melchini, A. Exposure of human skin to benzo[a]pyrene: Role of CYP1A1 and aryl hydrocarbon receptor in oxidative stress generation. Toxicology 2010, 271, 83–86. [Google Scholar] [CrossRef]
- Soeur, J.; Belaidi, J.P.; Chollet, C.; Denat, L.; Dimitrov, A.; Jones, C.; Perez, P.; Zanini, M.; Zobiri, O.; Mezzache, S.; et al. Photo-pollution stress in skin: Traces of pollutants (PAH and particulate matter) impair redox homeostasis in keratinocytes exposed to UVA1. J. Dermatol. Sci. 2017, 86, 162–169. [Google Scholar] [CrossRef]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Chen, J.H.; Chou, F.P.; Lin, H.H.; Wang, C.J. Gaseous nitrogen oxide repressed benzo[a]pyrene-induced human lung fibroblast cell apoptosis via inhibiting JNK1 signals. Arch. Toxicol. 2005, 79, 694–704. [Google Scholar] [CrossRef]
- Lee, F.Y.; Lee, M.S.; Wallace, C.G.; Huang, C.R.; Chu, C.H.; Wen, Z.H.; Huang, J.H.; Chen, X.S.; Wang, C.C.; Yip, H.K. Short-interval exposure to ambient fine particulate matter (PM2.5) exacerbates the susceptibility of pulmonary damage in setting of lung ischemia-reperfusion injury in rodent: Pharmacomodulation of melatonin. Biomed. Pharm. 2019, 113, 108737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Zou, A.P.; Li, P.L. Ceramide-induced activation of NADPH oxidase and endothelial dysfunction in small coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H605–612. [Google Scholar] [CrossRef] [PubMed]
- Von Haefen, C.; Wieder, T.; Gillissen, B.; Starck, L.; Graupner, V.; Dorken, B.; Daniel, P.T. Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene 2002, 21, 4009–4019. [Google Scholar] [CrossRef] [PubMed]
- Haimovitz-Friedman, A.; Kolesnick, R.N.; Fuks, Z. Ceramide signaling in apoptosis. Br. Med. Bull. 1997, 53, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, J. Sphingosine and ceramide signalling in apoptosis. IUBMB Life 2006, 58, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Wattenberg, B.W. The long and the short of ceramides. J. Biol. Chem. 2018, 293, 9922–9923. [Google Scholar] [CrossRef]
- Wanner, R.; Peiser, M.; Wittig, B. Keratinocytes rapidly readjust ceramide-sphingomyelin homeostasis and contain a phosphatidylcholine-sphingomyelin transacylase. J. Investig. Dermatol. 2004, 122, 773–782. [Google Scholar] [CrossRef]
- Shin, K.O.; Kim, K.P.; Cho, Y.; Kang, M.K.; Kang, Y.H.; Lee, Y.M.; Ikushiro, H.; Yokota, M.; Yano, T.; Choe, S.J.; et al. Both Sphingosine Kinase 1 and 2 Coordinately Regulate Cathelicidin Antimicrobial Peptide Production during Keratinocyte Differentiation. J. Investig. Dermatol. 2019, 139, 492–494. [Google Scholar] [CrossRef]
- Uchida, Y. Ceramide signaling in mammalian epidermis. Biochim. Biophys. Acta 2014, 1841, 453–462. [Google Scholar] [CrossRef]
- Alexaki, A.; Clarke, B.A.; Gavrilova, O.; Ma, Y.; Zhu, H.; Ma, X.; Xu, L.; Tuymetova, G.; Larman, B.C.; Allende, M.L.; et al. De Novo Sphingolipid Biosynthesis Is Required for Adipocyte Survival and Metabolic Homeostasis. J. Biol. Chem. 2017, 292, 3929–3939. [Google Scholar] [CrossRef]
- Gegotek, A.; Skrzydlewska, E. The role of transcription factor Nrf2 in skin cells metabolism. Arch. Dermatol. Res. 2015, 307, 385–396. [Google Scholar] [CrossRef]
- Lee, E.Y.; Bae, H.C.; Lee, H.; Jang, Y.; Park, Y.H.; Kim, J.H.; Ryu, W.I.; Choi, B.H.; Kim, J.H.; Jeong, S.H.; et al. Intracellular ROS levels determine the apoptotic potential of keratinocyte by Quantum Dot via blockade of AKT Phosphorylation. Exp. Dermatol. 2017, 26, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Houben, E.; Park, K.; Douangpanya, S.; Lee, Y.M.; Wu, B.X.; Hannun, Y.A.; Radin, N.S.; Elias, P.M.; Holleran, W.M. Hydrolytic pathway protects against ceramide-induced apoptosis in keratinocytes exposed to UVB. J. Investig. Dermatol. 2010, 130, 2472–2480. [Google Scholar] [CrossRef] [PubMed]
- Van Brocklyn, J.R.; Williams, J.B. The control of the balance between ceramide and sphingosine-1-phosphate by sphingosine kinase: Oxidative stress and the seesaw of cell survival and death. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2012, 163, 26–36. [Google Scholar] [CrossRef]
- Hughes, T.J.; Lewtas, J.; Claxton, L.D. Development of a standard reference material for diesel mutagenicity in the Salmonella plate incorporation assay. Mutat. Res. 1997, 391, 243–258. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Park, K.; Elias, P.M.; Oda, Y.; Mackenzie, D.; Mauro, T.; Holleran, W.M.; Uchida, Y. Regulation of cathelicidin antimicrobial peptide expression by an endoplasmic reticulum (ER) stress signaling, vitamin D receptor-independent pathway. J. Biol. Chem. 2011, 286, 34121–34130. [Google Scholar] [CrossRef]
- Lin, Z.C.; Lee, C.W.; Tsai, M.H.; Ko, H.H.; Fang, J.Y.; Chiang, Y.C.; Liang, C.J.; Hsu, L.F.; Hu, S.C.; Yen, F.L. Eupafolin nanoparticles protect HaCaT keratinocytes from particulate matter-induced inflammation and oxidative stress. Int. J. Nanomed. 2016, 11, 3907–3926. [Google Scholar]
- Rosenkranz, A.R.; Schmaldienst, S.; Stuhlmeier, K.M.; Chen, W.; Knapp, W.; Zlabinger, G.J. A microplate assay for the detection of oxidative products using 2,7′-dichlorofluorescin-diacetate. J. Immunol. Methods 1992, 156, 39–45. [Google Scholar] [CrossRef]
- Park, K.; Elias, P.M.; Shin, K.O.; Lee, Y.M.; Hupe, M.; Borkowski, A.W.; Gallo, R.L.; Saba, J.; Holleran, W.M.; Uchida, Y. A novel role of a lipid species, sphingosine-1-phosphate, in epithelial innate immunity. Mol. Cell Biol. 2013, 33, 752–762. [Google Scholar] [CrossRef]
- Park, K.; Ikushiro, H.; Seo, H.S.; Shin, K.O.; Kim, Y.I.; Kim, J.Y.; Lee, Y.M.; Yano, T.; Holleran, W.M.; Elias, P.; et al. ER stress stimulates production of the key antimicrobial peptide, cathelicidin, by forming a previously unidentified intracellular S1P signaling complex. Proc. Natl. Acad. Sci. USA 2016, 113, E1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.P.; Shin, K.O.; Park, K.; Yun, H.J.; Mann, S.; Lee, Y.M.; Cho, Y. Vitamin C Stimulates Epidermal Ceramide Production by Regulating Its Metabolic Enzymes. Biomol. Ther. 2015, 23, 525–530. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-S.; Park, H.Y.; Kwon, S.P.; Kim, B.; Lee, Y.; Kim, S.; Shin, K.-O.; Park, K. NADPH Oxidase-Mediated Activation of Neutral Sphingomyelinase Is Responsible for Diesel Particulate Extract-Induced Keratinocyte Apoptosis. Int. J. Mol. Sci. 2020, 21, 1001. https://doi.org/10.3390/ijms21031001
Lee H-S, Park HY, Kwon SP, Kim B, Lee Y, Kim S, Shin K-O, Park K. NADPH Oxidase-Mediated Activation of Neutral Sphingomyelinase Is Responsible for Diesel Particulate Extract-Induced Keratinocyte Apoptosis. International Journal of Molecular Sciences. 2020; 21(3):1001. https://doi.org/10.3390/ijms21031001
Chicago/Turabian StyleLee, Hyun-Seok, Hye Yoon Park, Sung Pil Kwon, Bogyeong Kim, Yerin Lee, Seongeun Kim, Kyong-Oh Shin, and Kyungho Park. 2020. "NADPH Oxidase-Mediated Activation of Neutral Sphingomyelinase Is Responsible for Diesel Particulate Extract-Induced Keratinocyte Apoptosis" International Journal of Molecular Sciences 21, no. 3: 1001. https://doi.org/10.3390/ijms21031001
APA StyleLee, H.-S., Park, H. Y., Kwon, S. P., Kim, B., Lee, Y., Kim, S., Shin, K.-O., & Park, K. (2020). NADPH Oxidase-Mediated Activation of Neutral Sphingomyelinase Is Responsible for Diesel Particulate Extract-Induced Keratinocyte Apoptosis. International Journal of Molecular Sciences, 21(3), 1001. https://doi.org/10.3390/ijms21031001