Myeloperoxidase-Derived 2-Chlorohexadecanal Is Generated in Mouse Heart during Endotoxemia and Induces Modification of Distinct Cardiomyocyte Protein Subsets In Vitro

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
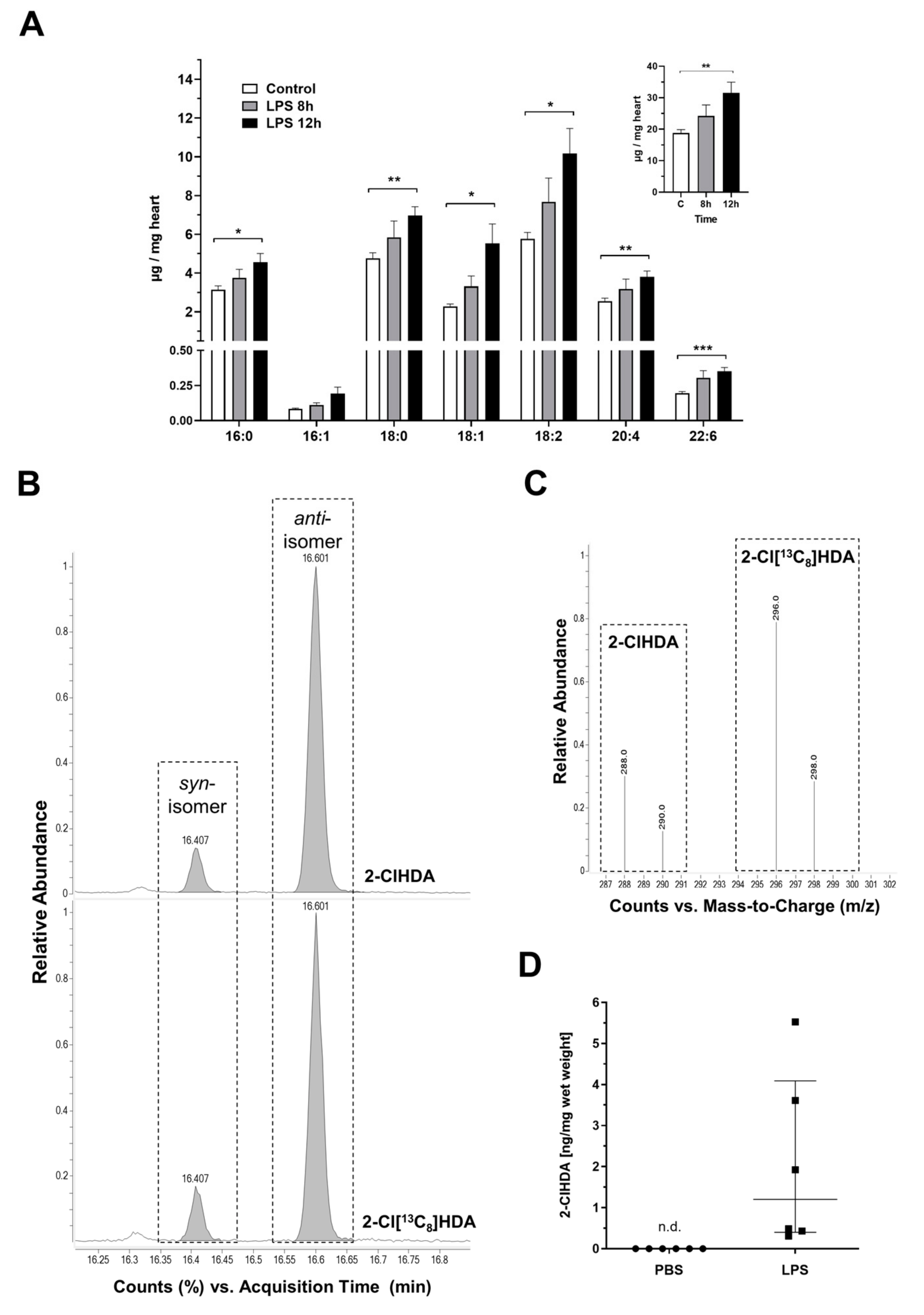
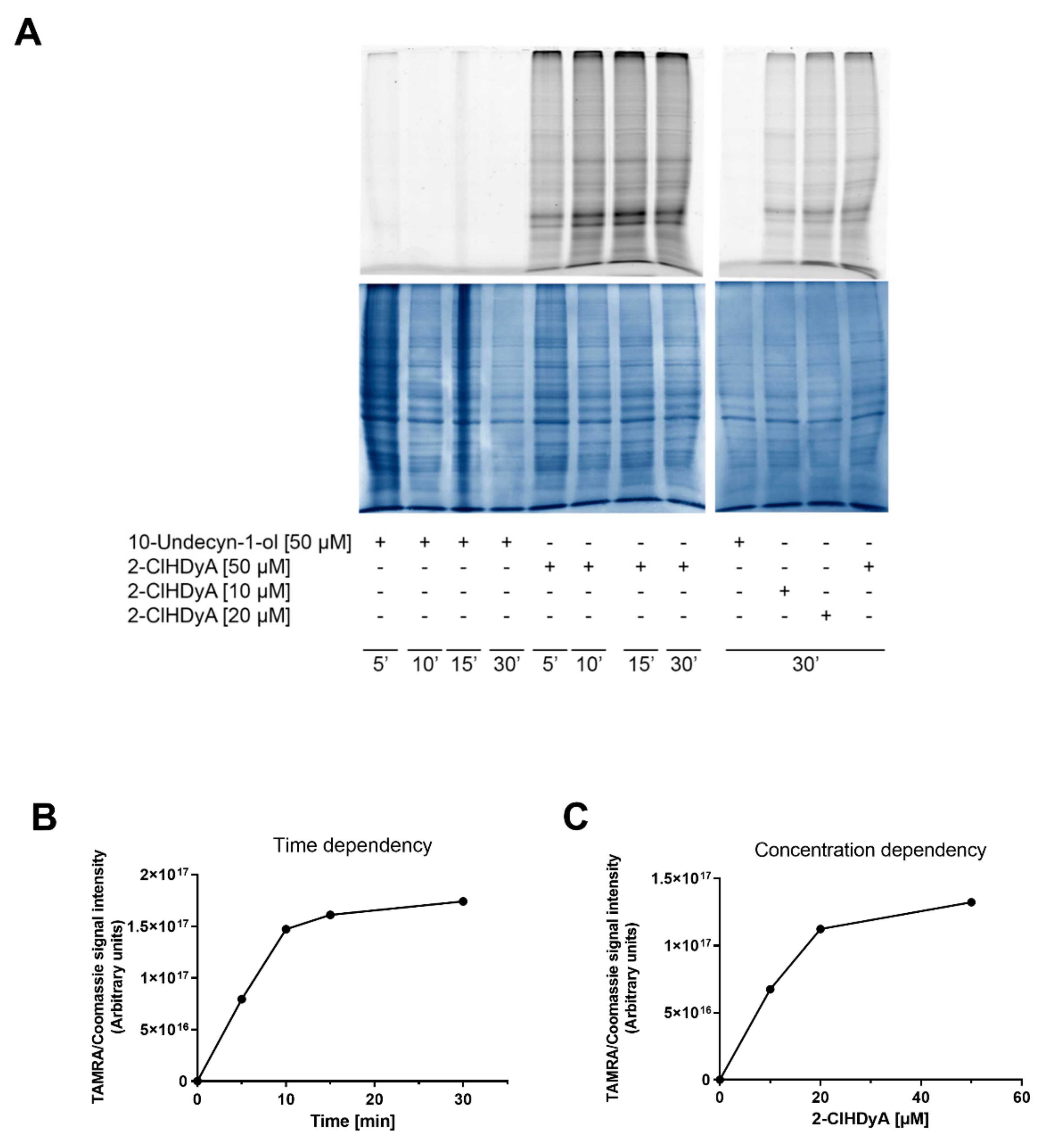
2. Results
- (i).
- Cytoskeletal proteins that belong to microfilaments (Actbl2), intermediate filaments (e.g., Des and Vim), or microtubules (members of the tubulin beta chain family);
- (ii).
- Chaperones and stress response including heat shock proteins (HSPs), Cct subunits of the chaperonin-containing T-complex (CCT), protein disulfide isomerases (Pdia3 and 6), and eukaryotic translation initiation factors;
- (iii).
- Proteins involved in cellular energy metabolism, primarily cytosolic glycolytic enzymes but also mitochondrial proteins like Idh3a, Ndufs2, Uacrc1 and 2; and
- (iv).
- Miscellaneous proteins that were not included in the groups listed above.
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Immunohistochemistry
4.3. Cell Culture
4.4. FA Analysis
4.5. Analysis of 2-ClHDA in Murine Cardiac Tissue
4.6. Metabolic Conversion of 2-ClHDA to 2-ClHA and 2-ClHOH
4.7. Derivatization Procedures
4.8. NICI-GC-MS Analysis
4.9. HOCl-Induced Formation of 2-ClHDA and 2-ClHA in HL-1 Cardiomyocytes
4.10. Synthesis and Analysis of Chlorinated Lipids
4.11. CellTiter-Glo Assay
4.12. Identification of 2-ClHDyA-Modified Proteins in HL-1 Cardiomyocytes
4.13. Click Chemistry
4.14. 1D-Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE)
4.15. 2D-GE
4.16. Coomassie-Blue Staining of 2D-gels
4.17. Liquid Chromatography (LC)-MS/MS Analysis
4.18. STRING Network Analysis
4.19. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-ClHA | 2-chlorohexadecanoic acid |
| 2-ClHDA | 2-chlorohexadecanal |
| 2-ClHDyA | 2-chlorohexadec-15-yn-1-al |
| 2-ClHOH | 2-chlorohexadecanol |
| 2D-GE | Two dimensional-gel electrophoresis |
| CCT | Chaperonin-containing T-complex |
| DMSO | Dimethyl sulfoxide |
| FA | Fatty acid |
| FALDH | Fatty aldehyde dehydrogenase |
| FBS | Fetal bovine serum |
| GO | Gene ontology |
| GSH | Glutathione |
| HNE | 4-Hydroxynonenal |
| HOCl | Hypochlorous acid |
| HRP | Horseradish peroxidase |
| HSP | Heat shock protein |
| HSR | Heat shock response |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
| LPS | Lipopolysaccharide |
| MI | Myocardial infarction |
| MPO | Myeloperoxidase |
| N3-TAMRA | 5-tetramethylrhodamin azide |
| NETs | Neutrophil extracellular traps |
| NICI-GC-MS | Negative ion chemical ionization-gas chromatography-mass spectrometry |
| NO | Nitric oxide |
| PBS | Phosphate-buffered saline |
| PFB | Pentafluorobenzyl |
| RES | Reactive electrophile species |
| ROS | Reactive oxygen species |
| RT | Room temperature |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SIM | Selected ion monitoring |
| TLR4 | Toll-like receptor 4 |
| TNFα | Tumor necrosis factor α |
References
- Lagu, T.; Rothberg, M.B.; Shieh, M.S.; Pekow, P.S.; Steingrub, J.S.; Lindenauer, P.K. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit. Care Med. 2012, 40, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Bellissant, E.; Cavaillon, J.M. Septic shock. Lancet 2005, 365, 63–78. [Google Scholar] [CrossRef]
- Drosatos, K.; Lymperopoulos, A.; Kennel, P.J.; Pollak, N.; Schulze, P.C.; Goldberg, I.J. Pathophysiology of sepsis-related cardiac dysfunction: Driven by inflammation, energy mismanagement, or both? Curr. Heart Fail. Rep. 2015, 12, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D.; Doddi, M. Sepsis and the heart. Br. J. Anaesth. 2010, 104, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Dhainaut, J.F.; Huyghebaert, M.F.; Monsallier, J.F.; Lefevre, G.; Dall’Ava-Santucci, J.; Brunet, F.; Villemant, D.; Carli, A.; Raichvarg, D. Coronary hemodynamics and myocardial metabolism of lactate, free fatty acids, glucose, and ketones in patients with septic shock. Circulation 1987, 75, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Arnhold, J. The Dual Role of Myeloperoxidase in Immune Response. Int. J. Mol. Sci. 2020, 21, 8057. [Google Scholar] [CrossRef]
- Klebanoff, S.J.; Kettle, A.J.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: A front-line defender against phagocytosed microorganisms. J. Leukoc. Biol. 2013, 93, 185–198. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef]
- Liew, P.X.; Kubes, P. The Neutrophil’s Role During Health and Disease. Physiol. Rev. 2019, 99, 1223–1248. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Casas, A.I.; Dao, V.T.; Daiber, A.; Maghzal, G.J.; Di Lisa, F.; Kaludercic, N.; Leach, S.; Cuadrado, A.; Jaquet, V.; Seredenina, T.; et al. Reactive Oxygen-Related Diseases: Therapeutic Targets and Emerging Clinical Indications. Antioxid. Redox Signal. 2015, 23, 1171–1185. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Sohnle, P.G. Generation of free radical intermediates from foreign compounds by neutrophil-derived oxidants. J. Clin. Investig. 1985, 75, 1618–1622. [Google Scholar] [CrossRef] [PubMed]
- Katrantzis, M.; Baker, M.S.; Handley, C.J.; Lowther, D.A. The oxidant hypochlorite (OCl-), a product of the myeloperoxidase system, degrades articular cartilage proteoglycan aggregate. Free Radic. Biol. Med. 1991, 10, 101–109. [Google Scholar] [CrossRef]
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef]
- Palladino, E.N.D.; Hartman, C.L.; Albert, C.J.; Ford, D.A. The chlorinated lipidome originating from myeloperoxidase-derived HOCl targeting plasmalogens: Metabolism, clearance, and biological properties. Arch. Biochem. Biophys. 2018, 641, 31–38. [Google Scholar] [CrossRef]
- Teng, N.; Maghzal, G.J.; Talib, J.; Rashid, I.; Lau, A.K.; Stocker, R. The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox Rep. Commun. Free Radic. Res. 2017, 22, 51–73. [Google Scholar] [CrossRef]
- Thukkani, A.K.; McHowat, J.; Hsu, F.F.; Brennan, M.L.; Hazen, S.L.; Ford, D.A. Identification of alpha-chloro fatty aldehydes and unsaturated lysophosphatidylcholine molecular species in human atherosclerotic lesions. Circulation 2003, 108, 3128–3133. [Google Scholar] [CrossRef]
- Marsche, G.; Hammer, A.; Oskolkova, O.; Kozarsky, K.F.; Sattler, W.; Malle, E. Hypochlorite-modified high density lipoprotein, a high affinity ligand to scavenger receptor class B, type I, impairs high density lipoprotein-dependent selective lipid uptake and reverse cholesterol transport. J. Biol. Chem. 2002, 277, 32172–32179. [Google Scholar] [CrossRef]
- Ray, R.S.; Katyal, A. Myeloperoxidase: Bridging the gap in neurodegeneration. Neurosci. Biobehav. Rev. 2016, 68, 611–620. [Google Scholar] [CrossRef]
- Thukkani, A.K.; Martinson, B.D.; Albert, C.J.; Vogler, G.A.; Ford, D.A. Neutrophil-mediated accumulation of 2-ClHDA during myocardial infarction: 2-ClHDA-mediated myocardial injury. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2955–H2964. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cao, Z.; Moore, D.R.; Jackson, P.L.; Barnes, S.; Lambeth, J.D.; Thannickal, V.J.; Cheng, G. Microbicidal activity of vascular peroxidase 1 in human plasma via generation of hypochlorous acid. Infect. Immun. 2012, 80, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- Paumann-Page, M.; Katz, R.S.; Bellei, M.; Schwartz, I.; Edenhofer, E.; Sevcnikar, B.; Soudi, M.; Hofbauer, S.; Battistuzzi, G.; Furtmuller, P.G.; et al. Pre-steady-state Kinetics Reveal the Substrate Specificity and Mechanism of Halide Oxidation of Truncated Human Peroxidasin 1. J. Biol. Chem. 2017, 292, 4583–4592. [Google Scholar] [CrossRef] [PubMed]
- Albert, C.J.; Crowley, J.R.; Hsu, F.F.; Thukkani, A.K.; Ford, D.A. Reactive chlorinating species produced by myeloperoxidase target the vinyl ether bond of plasmalogens: Identification of 2-chlorohexadecanal. J. Biol. Chem. 2001, 276, 23733–23741. [Google Scholar] [CrossRef]
- Marsche, G.; Heller, R.; Fauler, G.; Kovacevic, A.; Nuszkowski, A.; Graier, W.; Sattler, W.; Malle, E. 2-chlorohexadecanal derived from hypochlorite-modified high-density lipoprotein-associated plasmalogen is a natural inhibitor of endothelial nitric oxide biosynthesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2302–2306. [Google Scholar] [CrossRef]
- Thukkani, A.K.; Hsu, F.F.; Crowley, J.R.; Wysolmerski, R.B.; Albert, C.J.; Ford, D.A. Reactive chlorinating species produced during neutrophil activation target tissue plasmalogens: Production of the chemoattractant, 2-chlorohexadecanal. J. Biol. Chem. 2002, 277, 3842–3849. [Google Scholar] [CrossRef]
- Thukkani, A.K.; Albert, C.J.; Wildsmith, K.R.; Messner, M.C.; Martinson, B.D.; Hsu, F.F.; Ford, D.A. Myeloperoxidase-derived reactive chlorinating species from human monocytes target plasmalogens in low density lipoprotein. J. Biol. Chem. 2003, 278, 36365–36372. [Google Scholar] [CrossRef]
- Nusshold, C.; Ullen, A.; Kogelnik, N.; Bernhart, E.; Reicher, H.; Plastira, I.; Glasnov, T.; Zangger, K.; Rechberger, G.; Kollroser, M.; et al. Assessment of electrophile damage in a human brain endothelial cell line utilizing a clickable alkyne analog of 2-chlorohexadecanal. Free Radic. Biol. Med. 2016, 90, 59–74. [Google Scholar] [CrossRef]
- Rizzo, W.B. Fatty aldehyde and fatty alcohol metabolism: Review and importance for epidermal structure and function. Biochim. Biophys. Acta 2014, 1841, 377–389. [Google Scholar] [CrossRef]
- Anbukumar, D.S.; Shornick, L.P.; Albert, C.J.; Steward, M.M.; Zoeller, R.A.; Neumann, W.L.; Ford, D.A. Chlorinated lipid species in activated human neutrophils: Lipid metabolites of 2-chlorohexadecanal. J. Lipid Res. 2010, 51, 1085–1092. [Google Scholar] [CrossRef]
- Wang, W.Y.; Albert, C.J.; Ford, D.A. Alpha-chlorofatty Acid accumulates in activated monocytes and causes apoptosis through reactive oxygen species production and endoplasmic reticulum stress. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, E.; Kogelnik, N.; Prasch, J.; Gottschalk, B.; Goeritzer, M.; Depaoli, M.R.; Reicher, H.; Nusshold, C.; Plastira, I.; Hammer, A.; et al. 2-Chlorohexadecanoic acid induces ER stress and mitochondrial dysfunction in brain microvascular endothelial cells. Redox Biol. 2018, 15, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wang, M.; Wang, D.; Kalogeris, T.J.; McHowat, J.; Ford, D.A.; Korthuis, R.J. Chlorinated Lipids Elicit Inflammatory Responses in vitro and in vivo. Shock 2019, 51, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Palladino, E.N.D.; Katunga, L.A.; Kolar, G.R.; Ford, D.A. 2-Chlorofatty acids: Lipid mediators of neutrophil extracellular trap formation. J. Lipid Res. 2018, 59, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.J.; Reilly, J.P.; Feng, R.; Christie, J.D.; Hazen, S.L.; Albert, C.J.; Franke, J.D.; Hartman, C.L.; McHowat, J.; Ford, D.A. Myeloperoxidase-derived 2-chlorofatty acids contribute to human sepsis mortality via acute respiratory distress syndrome. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.J.; Seymour, C.W.; Rosengart, M.R. Current Murine Models of Sepsis. Surg. Infect. 2016, 17, 385–393. [Google Scholar] [CrossRef]
- Balija, T.M.; Lowry, S.F. Lipopolysaccharide and sepsis-associated myocardial dysfunction. Curr. Opin. Infect. Dis. 2011, 24, 248–253. [Google Scholar] [CrossRef]
- Duerr, M.A.; Aurora, R.; Ford, D.A. Identification of glutathione adducts of alpha-chlorofatty aldehydes produced in activated neutrophils. J. Lipid Res. 2015, 56, 1014–1024. [Google Scholar] [CrossRef]
- Wildsmith, K.R.; Albert, C.J.; Hsu, F.F.; Kao, J.L.; Ford, D.A. Myeloperoxidase-derived 2-chlorohexadecanal forms Schiff bases with primary amines of ethanolamine glycerophospholipids and lysine. Chem. Phys. Lipids 2006, 139, 157–170. [Google Scholar] [CrossRef]
- Wilkie-Grantham, R.P.; Magon, N.J.; Harwood, D.T.; Kettle, A.J.; Vissers, M.C.; Winterbourn, C.C.; Hampton, M.B. Myeloperoxidase-dependent lipid peroxidation promotes the oxidative modification of cytosolic proteins in phagocytic neutrophils. J. Biol. Chem. 2015, 290, 9896–9905. [Google Scholar] [CrossRef]
- Codreanu, S.G.; Ullery, J.C.; Zhu, J.; Tallman, K.A.; Beavers, W.N.; Porter, N.A.; Marnett, L.J.; Zhang, B.; Liebler, D.C. Alkylation damage by lipid electrophiles targets functional protein systems. Mol. Cell. Proteom. 2014, 13, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.A. The harmful role of c5a on innate immunity in sepsis. J. Innate Immun. 2010, 2, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Bosmann, M.; Ward, P.A. The inflammatory response in sepsis. Trends Immunol. 2013, 34, 129–136. [Google Scholar] [CrossRef] [PubMed]
- El Kazzi, M.; Rayner, B.S.; Chami, B.; Dennis, J.M.; Thomas, S.R.; Witting, P.K. Neutrophil-Mediated Cardiac Damage After Acute Myocardial Infarction: Significance of Defining a New Target Cell Type for Developing Cardioprotective Drugs. Antioxid. Redox Signal. 2020, 33, 689–712. [Google Scholar] [CrossRef] [PubMed]
- Vasilyev, N.; Williams, T.; Brennan, M.L.; Unzek, S.; Zhou, X.; Heinecke, J.W.; Spitz, D.R.; Topol, E.J.; Hazen, S.L.; Penn, M.S. Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation 2005, 112, 2812–2820. [Google Scholar] [CrossRef] [PubMed]
- Askari, A.T.; Brennan, M.L.; Zhou, X.; Drinko, J.; Morehead, A.; Thomas, J.D.; Topol, E.J.; Hazen, S.L.; Penn, M.S. Myeloperoxidase and plasminogen activator inhibitor 1 play a central role in ventricular remodeling after myocardial infarction. J. Exp. Med. 2003, 197, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, V.; Andrie, R.P.; Rudolph, T.K.; Friedrichs, K.; Klinke, A.; Hirsch-Hoffmann, B.; Schwoerer, A.P.; Lau, D.; Fu, X.; Klingel, K.; et al. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat. Med. 2010, 16, 470–474. [Google Scholar] [CrossRef]
- Mollenhauer, M.; Friedrichs, K.; Lange, M.; Gesenberg, J.; Remane, L.; Kerkenpass, C.; Krause, J.; Schneider, J.; Ravekes, T.; Maass, M.; et al. Myeloperoxidase Mediates Postischemic Arrhythmogenic Ventricular Remodeling. Circ. Res. 2017, 121, 56–70. [Google Scholar] [CrossRef]
- Zhong, S.; Li, L.; Shen, X.; Li, Q.; Xu, W.; Wang, X.; Tao, Y.; Yin, H. An update on lipid oxidation and inflammation in cardiovascular diseases. Free Radic. Biol. Med. 2019, 144, 266–278. [Google Scholar] [CrossRef]
- Schilling, J.; Lai, L.; Sambandam, N.; Dey, C.E.; Leone, T.C.; Kelly, D.P. Toll-like receptor-mediated inflammatory signaling reprograms cardiac energy metabolism by repressing peroxisome proliferator-activated receptor gamma coactivator-1 signaling. Circ. Heart Fail. 2011, 4, 474–482. [Google Scholar] [CrossRef]
- Brahmbhatt, V.V.; Albert, C.J.; Anbukumar, D.S.; Cunningham, B.A.; Neumann, W.L.; Ford, D.A. {omega}-Oxidation of {alpha}-chlorinated fatty acids: Identification of {alpha}-chlorinated dicarboxylic acids. J. Biol. Chem. 2010, 285, 41255–41269. [Google Scholar] [CrossRef] [PubMed]
- Pike, D.P.; Vogel, M.J.; McHowat, J.; Mikuzis, P.A.; Schulte, K.A.; Ford, D.A. 2-Chlorofatty acids are biomarkers of sepsis mortality and mediators of barrier dysfunction in rats. J. Lipid Res. 2020, 61, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Long, M.J.C.; Aye, Y. Proteomics and Beyond: Cell Decision-Making Shaped by Reactive Electrophiles. Trends Biochem. Sci. 2019, 44, 75–89. [Google Scholar] [CrossRef] [PubMed]
- West, J.D.; Wang, Y.; Morano, K.A. Small molecule activators of the heat shock response: Chemical properties, molecular targets, and therapeutic promise. Chem. Res. Toxicol. 2012, 25, 2036–2053. [Google Scholar] [CrossRef]
- Parvez, S.; Long, M.J.C.; Poganik, J.R.; Aye, Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem. Rev. 2018, 118, 8798–8888. [Google Scholar] [CrossRef] [PubMed]
- Schopfer, F.J.; Cipollina, C.; Freeman, B.A. Formation and signaling actions of electrophilic lipids. Chem. Rev. 2011, 111, 5997–6021. [Google Scholar] [CrossRef] [PubMed]
- Wildsmith, K.R.; Albert, C.J.; Anbukumar, D.S.; Ford, D.A. Metabolism of myeloperoxidase-derived 2-chlorohexadecanal. J. Biol. Chem. 2006, 281, 16849–16860. [Google Scholar] [CrossRef]
- Ullen, A.; Fauler, G.; Bernhart, E.; Nusshold, C.; Reicher, H.; Leis, H.J.; Malle, E.; Sattler, W. Phloretin ameliorates 2-chlorohexadecanal-mediated brain microvascular endothelial cell dysfunction in vitro. Free Radic. Biol. Med. 2012, 53, 1770–1781. [Google Scholar] [CrossRef]
- Chen, C.H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef]
- Chen, C.H.; Ferreira, J.C.; Gross, E.R.; Mochly-Rosen, D. Targeting aldehyde dehydrogenase 2: New therapeutic opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef]
- Poganik, J.R.; Long, M.J.C.; Aye, Y. Getting the Message? Native Reactive Electrophiles Pass Two Out of Three Thresholds to be Bona Fide Signaling Mediators. Bioessays 2018, 40, e1700240. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Christians, E.S.; Ishiwata, T.; Benjamin, I.J. Small heat shock proteins in redox metabolism: Implications for cardiovascular diseases. Int. J. Biochem. Cell Biol. 2012, 44, 1632–1645. [Google Scholar] [CrossRef] [PubMed]
- Krief, S.; Faivre, J.F.; Robert, P.; Le Douarin, B.; Brument-Larignon, N.; Lefrère, I.; Bouzyk, M.M.; Anderson, K.M.; Greller, L.D.; Tobin, F.L.; et al. Identification and characterization of cvHsp. A novel human small stress protein selectively expressed in cardiovascular and insulin-sensitive tissues. J. Biol. Chem. 1999, 274, 36592–36600. [Google Scholar] [CrossRef]
- Surya, S.L.; Long, M.J.C.; Urul, D.A.; Zhao, Y.; Mercer, E.J.; IM, E.I.; Evans, T.; Aye, Y. Cardiovascular Small Heat Shock Protein HSPB7 Is a Kinetically Privileged Reactive Electrophilic Species (RES) Sensor. ACS Chem. Biol. 2018, 13, 1824–1831. [Google Scholar] [CrossRef]
- Neef, D.W.; Jaeger, A.M.; Gomez-Pastor, R.; Willmund, F.; Frydman, J.; Thiele, D.J. A direct regulatory interaction between chaperonin TRiC and stress-responsive transcription factor HSF1. Cell Rep. 2014, 9, 955–966. [Google Scholar] [CrossRef]
- Monge, C.; Beraud, N.; Tepp, K.; Pelloux, S.; Chahboun, S.; Kaambre, T.; Kadaja, L.; Roosimaa, M.; Piirsoo, A.; Tourneur, Y.; et al. Comparative analysis of the bioenergetics of adult cardiomyocytes and nonbeating HL-1 cells: Respiratory chain activities, glycolytic enzyme profiles, and metabolic fluxes. Can. J. Physiol. Pharmacol. 2009, 87, 318–326. [Google Scholar] [CrossRef]
- Wen, J.J.; Garg, N. Oxidative modification of mitochondrial respiratory complexes in response to the stress of Trypanosoma cruzi infection. Free Radic. Biol. Med. 2004, 37, 2072–2081. [Google Scholar] [CrossRef]
- Puurand, M.; Tepp, K.; Timohhina, N.; Aid, J.; Shevchuk, I.; Chekulayev, V.; Kaambre, T. Tubulin betaII and betaIII Isoforms as the Regulators of VDAC Channel Permeability in Health and Disease. Cells 2019, 8, 239. [Google Scholar] [CrossRef]
- Mado, K.; Chekulayev, V.; Shevchuk, I.; Puurand, M.; Tepp, K.; Kaambre, T. On the role of tubulin, plectin, desmin, and vimentin in the regulation of mitochondrial energy fluxes in muscle cells. Am. J. Physiol. Cell Physiol. 2019, 316, C657–C667. [Google Scholar] [CrossRef]
- Tang, H.L.; Lung, H.L.; Wu, K.C.; Le, A.H.; Tang, H.M.; Fung, M.C. Vimentin supports mitochondrial morphology and organization. Biochem. J. 2008, 410, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Guzun, R.; Karu-Varikmaa, M.; Gonzalez-Granillo, M.; Kuznetsov, A.V.; Michel, L.; Cottet-Rousselle, C.; Saaremae, M.; Kaambre, T.; Metsis, M.; Grimm, M.; et al. Mitochondria-cytoskeleton interaction: Distribution of beta-tubulins in cardiomyocytes and HL-1 cells. Biochim. Biophys. Acta 2011, 1807, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Sattler, W.; Puhl, H.; Hayn, M.; Kostner, G.M.; Esterbauer, H. Determination of fatty acids in the main lipoprotein classes by capillary gas chromatography: BF3/methanol transesterification of lyophilized samples instead of Folch extraction gives higher yields. Anal. Biochem. 1991, 198, 184–190. [Google Scholar] [CrossRef]
- Ullen, A.; Fauler, G.; Kofeler, H.; Waltl, S.; Nusshold, C.; Bernhart, E.; Reicher, H.; Leis, H.J.; Wintersperger, A.; Malle, E.; et al. Mouse brain plasmalogens are targets for hypochlorous acid-mediated modification in vitro and in vivo. Free Radic. Biol. Med. 2010, 49, 1655–1665. [Google Scholar] [CrossRef]
- Bhat, V.K.; Bernhart, E.; Plastira, I.; Fan, K.; Ghaffari-Tabrizi-Wizsy, N.; Wadsack, C.; Rechberger, G.; Eichmann, T.; Asslaber, M.; Spassova, I.; et al. Pharmacological Inhibition of Serine Palmitoyl Transferase and Sphingosine Kinase-1/-2 Inhibits Merkel Cell Carcinoma Cell Proliferation. J. Investig. Dermatol. 2019, 139, 807–817. [Google Scholar] [CrossRef]
- Nusshold, C.; Kollroser, M.; Kofeler, H.; Rechberger, G.; Reicher, H.; Ullen, A.; Bernhart, E.; Waltl, S.; Kratzer, I.; Hermetter, A.; et al. Hypochlorite modification of sphingomyelin generates chlorinated lipid species that induce apoptosis and proteome alterations in dopaminergic PC12 neurons in vitro. Free Radic. Biol. Med. 2010, 48, 1588–1600. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot ID | Short Name | Group | Full Name | Molecular Mass (Da) | Isoelectric Point (pI) |
|---|---|---|---|---|---|
| Q8BFZ3 | Actbl2 | Beta-actin-like protein 2 | 42,345.8 | 5.31 | |
| Q99KJ8 | Dctn2 | Dynactin subunit 2 | 44,230.2 | 5.14 | |
| P31001 | Des | Desmin | 53,553.9 | 5.21 | |
| P48678 | Lmna | Prelamin-A/C | 74,521.5 | 6.54 | |
| P15331 | Prph | Peripherin | 54,380.8 | 5.40 | |
| P14206 | Rpsa | 40S ribosomal protein SA | 32,951.6 | 4.80 | |
| P05214 | Tuba3b | Tubulin alpha-3 chain | 50,643.3 | 4.98 | |
| Q3UX10 | Tubal3 | Tubulin alpha chain-like 3 | 50,728.9 | 5.37 | |
| Q9ERD7 | Tubb3 | Tubulin beta-3 chain | 50,874.2 | 4.82 | |
| Q922F4 | Tubb6 | Tubulin beta-6 chain | 50,545.9 | 4.80 | |
| P20152 | Vim | Vimentin | 53,743.8 | 5.06 | |
| Q9CQV8 | Ywhab | 14-3-3 protein beta/alpha | 28,200.0 | 4.77 | |
| P80318 | Cct3 | T-complex protein 1 subunit gamma | 61,199.5 | 6.28 | |
| P80316 | Cct5 | T-complex protein 1 subunit epsilon | 60,079.5 | 5.72 | |
| P42932 | Cct8 | T-complex protein 1 subunit theta | 60,125.0 | 5.44 | |
| P62737 | Eif3f | Eukaryotic translation initiation factor 3 subunit F | 38,097.7 | 5.33 | |
| P10630 | Eif4a2 | Eukaryotic initiation factor 4A-II | 46,629.7 | 5.33 | |
| P30416 | Fkbp4 | Peptidyl-prolyl cis-trans isomerase FKBP4 | 51,970.8 | 5.54 | |
| P07901 | Hsp90aa1 | Heat shock protein HSP 90-alpha | 85,185.7 | 4.93 | |
| P16627 | Hspa1l | Heat shock 70 kDa protein 1-like | 71,035.4 | 5.91 | |
| P14602 | Hspb1 | Heat shock protein beta-1 | 23,070.5 | 6.12 | |
| P27773 | Pdia3 | Protein disulfide-isomerase A3 | 57,133.7 | 5.88 | |
| Q922R8 | Pdia6 | Protein disulfide-isomerase A6 | 38,097.7 | 5.33 | |
| Q9WVJ2 | Psmd13 | 26S proteasome non-ATPase regulatory subunit 13 | 43,150.9 | 5.46 | |
| Q60864 | Stip1 | Stress-induced-phosphoprotein 1 | 63,208.6 | 6.41 | |
| P11983 | Tcp1 | T-complex protein 1 subunit alpha | 60,904.0 | 5.82 | |
| P10518 | Alad | Delta-aminolevulinic acid dehydratase | 36,479.3 | 6.32 | |
| P47738 | Aldh2 | Aldehyde dehydrogenase, mitochondrial | 57,050.0 | 7.76 | |
| P05064 | Aldoa | Fructose-bisphosphate aldolase A | 39,811.6 | 8.75 | |
| P17182 | Eno1 | Alpha-enolase | 47,482.3 | 6.37 | |
| P16858 | Gapdh | Glyceraldehyde-3-phosphate dehydrogenase | 36,094.6 | 8.76 | |
| Q9D6R2 | Idh3a | Isocitrate dehydrogenase [NAD] subunit alpha, mitochondrial | 40,094.5 | 6.27 | |
| P06151 | Ldha | L-lactate dehydrogenase A chain | 36,840.2 | 7.84 | |
| Q91WD5 | Ndufs2 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 2, mitochondrial | 53,024.1 | 6.52 | |
| Q9DBJ1 | Pgam1 | Phosphoglycerate mutase 1 | 28,945.6 | 6.68 | |
| Q61753 | Phgdh | D-3-phosphoglycerate dehydrogenase | 57,383.3 | 6.12 | |
| Q9CZ13 | Uqcrc1 | Cytochrome b-c1 complex subunit 1, mitochondrial | 53,478.4 | 5.81 | |
| Q9DB77 | Uqcrc2 | Cytochrome b-c1 complex subunit 2, mitochondrial | 48,291.1 | 9.31 | |
| P07724 | Alb | Albumin | 70,745.2 | 5.75 | |
| Q9Z1Q5 | Clic1 | Chloride intracellular channel protein 1 | 27,354.6 | 5.09 | |
| Q8VDW0 | Ddx39 | ATP-dependent RNA helicase DDX39A | 49,580.0 | 5.46 | |
| Q9Z1N5 | Ddx39b | Spliceosome RNA helicase Ddx39b | 49,491.0 | 5.44 | |
| Q9D8N0 | Eef1g | Elongation factor 1-gamma | 50,402.1 | 6.31 | |
| P58252 | Eef2 | Elongation factor 2 | 96,282.3 | 6.41 | |
| Q920E5 | Fdps | Farnesyl pyrophosphate synthase | 40,923.1 | 5.49 | |
| Q8R081 | Hnrnpl | Heterogeneous nuclear ribonucleoprotein L | 64,590.1 | 8.75 | |
| P17897 | Lyz1 | Lysozyme C-1 | 17,250.3 | 10.35 | |
| P17918 | Pcna | Proliferating cell nuclear antigen | 29,126.7 | 4.66 | |
| Q91VI7 | Rnh1 | Ribonuclease inhibitor | 51,527.2 | 4.69 | |
| Q9WTM5 | Ruvbl2 | RuvB-like 2 | 51,282.8 | 5.49 | |
| Q3TW96 | Uap1l1 | UDP-N-acetylhexosamine pyrophosphorylase-like protein 1 | 57,354.4 | 5.27 |
| Analyte (PFB-Derivatives) | m/z | Internal Standard (I.S.) | m/z | I.S. (ng) |
|---|---|---|---|---|
| 2-ClHDA | 288/290 | 2-Cl[13C8]HDA | 296/298 | 1000 |
| 2-ClHA | 289/291 | 2-Cl[13C8]HA | 297/299 | 1000 |
| 2-ClHOH | 470/472 | pentadecanol | 422 | 1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prasch, J.; Bernhart, E.; Reicher, H.; Kollroser, M.; Rechberger, G.N.; Koyani, C.N.; Trummer, C.; Rech, L.; Rainer, P.P.; Hammer, A.; et al. Myeloperoxidase-Derived 2-Chlorohexadecanal Is Generated in Mouse Heart during Endotoxemia and Induces Modification of Distinct Cardiomyocyte Protein Subsets In Vitro. Int. J. Mol. Sci. 2020, 21, 9235. https://doi.org/10.3390/ijms21239235
Prasch J, Bernhart E, Reicher H, Kollroser M, Rechberger GN, Koyani CN, Trummer C, Rech L, Rainer PP, Hammer A, et al. Myeloperoxidase-Derived 2-Chlorohexadecanal Is Generated in Mouse Heart during Endotoxemia and Induces Modification of Distinct Cardiomyocyte Protein Subsets In Vitro. International Journal of Molecular Sciences. 2020; 21(23):9235. https://doi.org/10.3390/ijms21239235
Chicago/Turabian StylePrasch, Jürgen, Eva Bernhart, Helga Reicher, Manfred Kollroser, Gerald N. Rechberger, Chintan N. Koyani, Christopher Trummer, Lavinia Rech, Peter P. Rainer, Astrid Hammer, and et al. 2020. "Myeloperoxidase-Derived 2-Chlorohexadecanal Is Generated in Mouse Heart during Endotoxemia and Induces Modification of Distinct Cardiomyocyte Protein Subsets In Vitro" International Journal of Molecular Sciences 21, no. 23: 9235. https://doi.org/10.3390/ijms21239235
APA StylePrasch, J., Bernhart, E., Reicher, H., Kollroser, M., Rechberger, G. N., Koyani, C. N., Trummer, C., Rech, L., Rainer, P. P., Hammer, A., Malle, E., & Sattler, W. (2020). Myeloperoxidase-Derived 2-Chlorohexadecanal Is Generated in Mouse Heart during Endotoxemia and Induces Modification of Distinct Cardiomyocyte Protein Subsets In Vitro. International Journal of Molecular Sciences, 21(23), 9235. https://doi.org/10.3390/ijms21239235