The Dual Role of Myeloperoxidase in Immune Response

Abstract
1. Immune Response and Tissue Destruction
2. Short Overview about Myeloperoxidase Properties
2.1. Selected Structural Properties
2.2. Heme States and Redox Properties of Myeloperoxidase
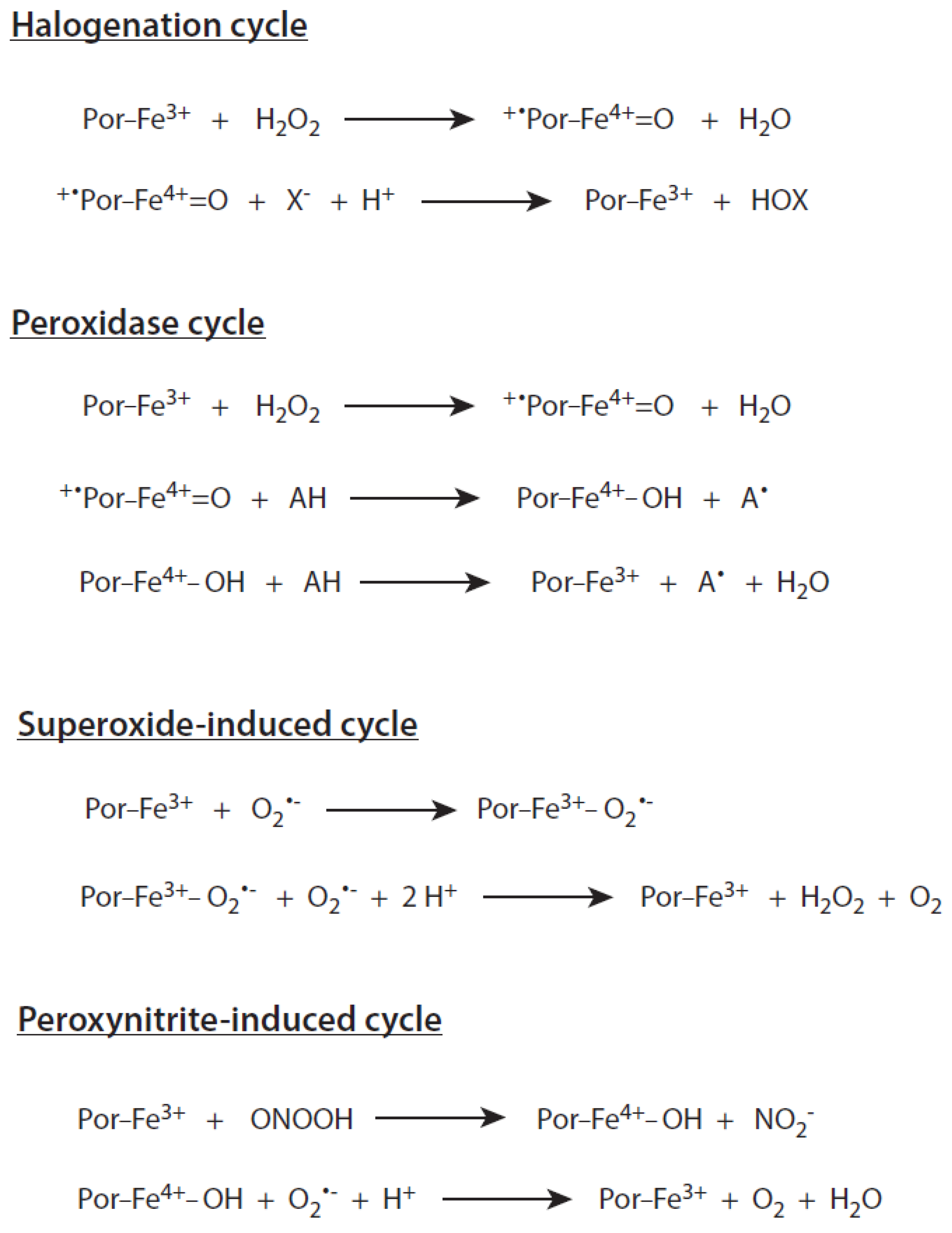
2.3. Reaction Cycles of Myeloperoxidase
3. Neutrophils and Myeloperoxidase at Inflammatory Sites
3.1. Recruitment of PMNs to Inflamed Sites
3.2. Important Components of Azurophilic and Specific Granules of Neutrophils
3.3. Conditions of Phagosomal Digestion
3.4. Potential Role of Myeloperoxidase in Phagosomes
3.5. Redundancy in Deactivation and Killing Pathways and MPO Deficiency
3.6. Cell Death of Neutrophils and Formation of Extracellular Traps
3.7. Frustrated Phagocytosis
3.8. Degradation of Ingested Material by Macrophages
4. Involvement of Myeloperoxidase in Disease Progression
4.1. The Fate of Myeloperoxidase at Inflammatory Sites
4.2. Important Binding Sites for Myeloperoxidase
4.3. The Protective Role of Ceruloplasmin
4.4. Myeloperoxidase in Atherosclerotic Plaques
4.5. Myeloperoxidase and Cardiovascular Diseases
4.6. Vasculitis Induced by Antineutrophil Cytoplasmic Antibodies
4.7. Involvement of Myeloperoxidase in Other Disease Scenarios
5. Chronicity of Inflammatory States
5.1. Chronic Inflammatory Processes
5.2. Protection of Surrounding Media Against Damaging Agents
5.3. Immunosuppression
5.4. Sepsis
6. Conclusions
Funding
Conflicts of Interest
References
- Arnhold, J. Immune response and tissue damage. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 155–204. [Google Scholar]
- Arnhold, J. Acute-phase proteins and additional protective systems. In Cell and Tissue Destruction. mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 205–228. [Google Scholar]
- Muller, W.A. Leukocyte-endothelial interactions in the inflammatory response. Lab. Investig. 2002, 82, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.R.; Gallo, R.I. Glycosaminoglycans and their proteoglycans: Host-associated molecular pattern for initiation and modulation of inflammation. FASEB J. 2002, 20, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.C.; Stevens, D.L. The circulating phagocyte reflects the in vivo state of immune response. Curr. Opin. Infect. Dis. 2006, 5, 389–398. [Google Scholar] [CrossRef]
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef]
- Arnhold, J.; Flemmig, J. Human myeloperoxidase in innate and acquired immunity. Arch. Biochem. Biophys. 2010, 500, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Bos, A.; Wever, R.; Roos, D. Characterization and quantification of the peroxidase in human monocytes. Biochim. Biophys. Acta 1978, 525, 37–44. [Google Scholar] [CrossRef]
- van der Veen, B.S.; de Winther, M.P.; Heeringa, P. Myeloperoxidase: Molecular mechanisms and their relevance to human health and disease. Antioxid. Redox Signal. 2009, 11, 2899–2937. [Google Scholar] [CrossRef]
- Owen, C.A.; Campbell, M.A.; Boukedes, S.S.; Stockley, R.A.; Campbell, E.J. A discrete subpopulation of human monocytes expresses a neutrophil-like proinflammatory (P) phenotype. Am. J. Physiol. 1994, 267, L775–L785. [Google Scholar] [CrossRef]
- Nagra, R.M.; Becher, B.; Trottellotte, W.W.; Antel, J.P.; Gold, D.; Paladino, T.; Smith, R.A.; Nelson, J.R.; Reynolds, W.F. Immunohistochemical and genetic evidence of myeloperoxidase involvement in multiple sclerosis. J. Neuroimmunol. 1997, 78, 97–107. [Google Scholar] [CrossRef]
- Sugiyama, S.; Okada, Y.; Suhkova, G.K.; Virmani, R.; Heinecke, J.W.; Libby, P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am. J. Pathol. 2001, 158, 879–891. [Google Scholar] [CrossRef]
- Odobasic, D.; Kitching, A.R.; Holdsworth, S.R. Neutrophil-mediated regulation of innate and adaptive immunity: The role of myeloperoxidase. J. Immunol. Res. 2016, 2016, 2349817. [Google Scholar] [CrossRef]
- Shepherd, V.L.; Hoidal, J.R. Clearance of neutrophil-derived myeloperoxidase by the macrophage mannose receptor. Am. J. Respir. Cell Mol. Biol. 1990, 2, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Lazzaretto, B.; Fadeel, B. Intra- and extracellular degradation of neutrophil extracellular traps by macrophages and dendritic cells. J. Immunol. 2019, 203, 2276–2290. [Google Scholar] [CrossRef] [PubMed]
- Furtmüller, P.G.; Zederbauer, M.; Jantschko, W.; Helm, J.; Bogner, M.; Jakopitsch, C.; Obinger, C. Active site structure and catalytic mechanisms of human peroxidases. Arch. Biochem. Biophys. 2006, 445, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J.; Kettle, A.J.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: A front-line defender against phagocytosed microorganisms. J. Leukoc. Biol. 2013, 93, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.C.; Parnes, C.; Krinsky, N.I. Comparison of myeloperoxidase and hemi-myeloperoxidase with respect to catalysis, regulation, and bactericidal activity. Arch. Biochem. Biophys. 1984, 228, 439–442. [Google Scholar] [CrossRef]
- Fiedler, T.J.; Davey, C.A.; Fenna, R.E. X-ray crystal structure and characterization of halide binding sites of human myeloperoxidase at 1.8 Å resolution. J. Biol. Chem. 2000, 275, 11964–11971. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, D.; Forman, A.; Borg, D.C.; Fajer, J.; Felton, R.H. Compound I of catalase and horseradish peroxidase: π-cation radicals. Proc. Natl. Acad. Sci. USA 1971, 68, 614–618. [Google Scholar] [CrossRef]
- Odajima, T.; Yamazaki, I. Myeloperoxidase of the leukocytes of normal blood: III. The reaction of ferric myeloperoxidase with superoxide anion. Biochim. Biophys. Acta 1972, 284, 355–359. [Google Scholar]
- Arnhold, J.; Furtmüller, P.G.; Obinger, C. Redox properties of myeloperoxidase. Redox Rep. 2003, 8, 179–186. [Google Scholar] [CrossRef]
- Arnhold, J.; Monzani, E.; Furtmüller, P.G.; Zederbauer, M.; Casella, L.; Obinger, C. Kinetics and thermodynamics of halide and nitrite oxidation by mammalian heme peroxidases. Eur. J. Inorg. Chem. 2006, 3801–3811. [Google Scholar] [CrossRef]
- Arnhold, J.; Furtmüller, P.G.; Regelsberger, G.; Obinger, C. Redox properties of the couple compound I/native enzyme of myeloperoxidase and eosinophil peroxidase. Eur. J. Biochem. 2001, 268, 5142–5148. [Google Scholar] [CrossRef] [PubMed]
- Furtmüller, P.G.; Arnhold, J.; Jantschko, W.; Pichler, H.; Obinger, C. Redox properties of the couples compound I/compound II and compound II/native enzyme of human myeloperoxidase. Biochem. Biophys. Res. Commun. 2003, 301, 551–557. [Google Scholar] [CrossRef]
- Furtmüller, P.G.; Burner, U.; Obinger, C. Reaction of myeloperoxidase compound I with chloride, bromide, iodide, and thiocyanate. Biochemistry 1998, 37, 17923–17930. [Google Scholar] [CrossRef]
- van Dalen, C.J.; Whitehouse, M.W.; Winterbourn, C.C.; Kettle, A.J. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 1997, 327, 487–492. [Google Scholar] [CrossRef]
- Tenovuo, J.; Makinen, K.K. Concentration of thiocyanate and ionizable iodine in saliva of smokers and nonsmokers. J. Dent. Res. 1976, 55, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C.P.; Ahmed, M.K.; Dawes, C.; Mantsch, H.H. Thiocyanate levels in human saliva: Quantitation by Fourier transform infrared spectroscopy. Anal. Biochem. 1996, 240, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.D.; Day, B.J. Thiocyanate: A potential useful therapeutic agent with host defense and antioxidant properties. Biochem. Pharmacol. 2012, 84, 1381–1387. [Google Scholar] [CrossRef]
- Chandler, J.D.; Day, B.J. Biochemical mechanisms and therapeutic potential of pseudohalide thiocyanate in human health. Free Radic. Res. 2015, 49, 695–710. [Google Scholar] [CrossRef]
- Flemmig, J.; Gau, J.; Schlorke, D.; Arnhold, J. Lactoperoxidase as potential drug target. Expert Opin. Ther. Targets 2016, 20, 447–461. [Google Scholar] [CrossRef]
- Bakkenist, A.R.J.; de Boer, J.E.G.; Plat, H.; Wever, R. The halide complexes of myeloperoxidase and the mechanism of halogenation reactions. Biochim. Biophys. Acta 1980, 613, 337–348. [Google Scholar] [CrossRef]
- Kettle, A.J.; Winterbourn, C.C. Superoxide modulates the activity of myeloperoxidase and optimizes the production of hypochlorous acid. Biochem. J. 1988, 252, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Zuurbier, K.W.M.; Bakkenist, A.R.J.; Wever, R.; Muijsers, A.O. The chlorinating activity of myeloperoxidase: High initial activity at neutral pH and activation by electron donors. Biochim. Biophys. Acta 1990, 1037, 140–146. [Google Scholar] [CrossRef]
- Kettle, A.J.; Winterbourn, C.C. Assays for the chlorination activity of myeloperoxidase. Meth. Enzymol. 1994, 233, 502–512. [Google Scholar]
- Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase: A key regulator of neutrophil oxidant production. Redox Rep. 1997, 3, 3–15. [Google Scholar] [CrossRef]
- Jerlich, A.; Horakova, L.; Fabjan, J.S.; Giessauf, A.; Jürgens, G.; Schaur, J.R. Correlation of low-density lipoprotein modification by myeloperoxidase with hypochlorous acid formation. Int. J. Clin. Lab. Res. 1999, 29, 155–161. [Google Scholar] [CrossRef]
- Panasenko, O.M.; Spalteholz, H.; Schiller, J.; Arnhold, J. Myeloperoxidase-induced formation of chlorohydrins and lysophospholipids from unsaturated phosphatidylcholines. Free Radic. Biol. Med. 2003, 34, 553–562. [Google Scholar] [CrossRef]
- Spalteholz, H.; Panasenko, O.M.; Arnhold, J. Formation of reactive halide species by myeloperoxidase and eosinophil peroxidase. Arch. Biochem. Biophys. 2006, 445, 225–234. [Google Scholar] [CrossRef]
- Furtmüller, P.G.; Obinger, C.; Hsuanyu, Y.; Dunford, H.B. Mechanism of reaction of myeloperoxidase with hydrogen peroxide and chloride ion. Eur. J. Biochem. 2000, 267, 5858–5864. [Google Scholar] [CrossRef]
- Marquez, L.A.; Dunford, H.B. Chlorination of taurine by myeloperoxidase: Kinetic evidence for an enzyme-bound intermediate. J. Biol. Chem. 1994, 269, 7950–7956. [Google Scholar]
- Ramos, D.R.; Victoria Garcia, M.; Canle, L.M.; Santaballa, J.; Furtmüller, P.G.; Obinger, C. Myeloperoxidase-catalyzed taurine chlorination: Initial versus equilibrium rate. Arch. Biochem. Biophys. 2007, 466, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.L.; Grisham, M.B.; Melton, D.F.; Jefferson, M.M. Evidence for the role of taurine in the in vitro oxidative toxicity of neutrophils toward erythrocytes. J. Biol. Chem. 1985, 260, 3321–3329. [Google Scholar]
- Learn, D.B.; Fried, V.A.; Thomas, E.L. Taurine and hypotaurine content of human leukocytes. J. Leukoc. Biol. 1990, 48, 174–182. [Google Scholar] [CrossRef]
- Blomgran, R.; Zheng, L.; Stendahl, O. Cathepsin-cleaved Bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization. J. Leukoc. Biol. 2007, 81, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, A.; Miyamoto, Y. Apoptosis triggered by phagocytosis-related oxidative stress through FLIPs down-regulation and JNK activation. J. Leukoc. Biol. 2007, 82, 1344–1352. [Google Scholar] [CrossRef]
- Pattison, D.I.; Davies, M.J. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem. Res. Toxicol. 2001, 14, 453–464. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Pattison, D.I.; Davies, M.J. Hypochlorite-induced oxidation of amino acids, peptides, and proteins. Amino Acids 2003, 25, 259–274. [Google Scholar] [CrossRef]
- Pattison, D.I.; Davies, M.J. Kinetic analysis of the reaction of hypobromous acid with protein components: Implication for cellular damage and the use of 3-bromotyrosine as a marker of oxidative stress. Biochemistry 2004, 43, 4799–4809. [Google Scholar] [CrossRef]
- Skaff, O.; Pattison, D.I.; Davies, M.J. Hypothiocyanous acid reactivity with low-molecular-mass and protein thiols: Absolute rate constants and assessment of biological relevance. Biochem. J. 2009, 442, 111–117. [Google Scholar] [CrossRef]
- Hawkins, C.L. The role of hypothiocyanous acid (HOSCN) in biological systems. Free Radic. Res. 2009, 43, 1147–1158. [Google Scholar] [CrossRef]
- Barrett, T.J.; Hawkins, C.L. Hypothiocyanous acid: Benign or deadly? Chem. Res. Toxicol. 2012, 25, 263–273. [Google Scholar] [CrossRef]
- Salavej, P.; Spalteholz, H.; Arnhold, J. Modification of amino acid residues in human serum albumin by myeloperoxidase. Free Radic. Biol. Med. 2006, 40, 516–525. [Google Scholar] [CrossRef]
- Senthilmohan, R.; Kettle, A.J. Bromination and chlorination reactions of myeloperoxidase at physiological concentrations of bromide and chloride. Arch. Biochem. Biophys. 2006, 445, 235–244. [Google Scholar] [CrossRef]
- Marquez, L.A.; Dunford, H.B. Kinetic of oxidation of tyrosine and dityrosine by myeloperoxidase compounds I and II. J. Biol. Chem. 1995, 270, 30434–30440. [Google Scholar] [CrossRef]
- Burner, U.; Jantschko, W.; Obinger, C. Kinetic of oxidation of aliphatic and aromatic thiols by myeloperoxidase compounds I and II. FEBS Lett. 1999, 43, 290–296. [Google Scholar] [CrossRef]
- Burner, U.; Furtmüller, P.G.; Kettle, A.J.; Koppenol, W.H.; Obinger, C. Mechanism of reaction of myeloperoxidase with nitrite. J. Biol. Chem. 2000, 275, 20597–20601. [Google Scholar] [CrossRef]
- Jantschko, W.; Furtmüller, P.G.; Allegra, M.; Livrea, M.A.; Jakopitsch, C.; Regelsberger, G.; Obinger, C. Redox intermediates of plant and mammalian peroxidases: A comparative transient-kinetic study of their reactivity toward indole derivatives. Arch. Biochem. Biophys. 2002, 398, 12–22. [Google Scholar] [CrossRef]
- Meotti, F.C.; Jameson, G.N.L.; Turner, R.; Harwood, T.D.; Stockwell, S.; Rees, M.D.; Thomas, S.R.; Kettle, A.J. Urate as a physiological substrate for myeloperoxidase. Implications for hyperuricemia and inflammation. J. Biol. Chem. 2011, 286, 12901–12911. [Google Scholar] [CrossRef]
- Spalteholz, H.; Furtmüller, P.G.; Jakopitsch, C.; Obinger, C.; Schewe, T.; Sies, H.; Arnhold, J. Kinetic evidence for rapid oxidation of (−)-epicatechin by human myeloperoxidase. Biochem. Biophys. Res. Commun. 2008, 371, 810–813. [Google Scholar] [CrossRef]
- Kirchner, T.; Flemmig, J.; Furtmüller, P.G.; Obinger, C.; Arnhold, J. (−)-Epicatechin enhances the chlorinating activity of human myeloperoxidase. Arch. Biochem. Biophys. 2010, 495, 21–27. [Google Scholar] [CrossRef]
- Gau, J.; Furtmüller, P.G.; Obinger, C.; Prévost, M.; van Antwerpen, P.; Arnhold, J.; Flemmig, J. Flavonoids as promoters of the (pseudo)halogenating activity of lactoperoxidase and myeloperoxidase. Free Radic. Biol. Med. 2016, 97, 307–319. [Google Scholar] [CrossRef]
- Bolscher, B.G.J.M.; Zoutberg, G.R.; Cuperus, R.A.; Wever, R. Vitamin C stimulates the chlorinating activity of human myeloperoxidase. Biochim. Biophys. Acta 1984, 784, 189–191. [Google Scholar] [CrossRef]
- Dunford, H.B.; Hsuanyu, Y. Kinetics of oxidation of serotonin by myeloperoxidase compounds I and II. Biochem. Cell Biol. 1999, 77, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Kettle, A.J.; Anderson, R.F.; Hampton, M.B.; Winterbourn, C.C. Reactions of superoxide with myeloperoxidase. Biochemistry 2007, 46, 4888–4897. [Google Scholar] [CrossRef] [PubMed]
- Kettle, A.J.; Winterbourn, C.C. Mechanism of inhibition of myeloperoxidase by anti-inflammatory drugs. Biochem. Pharmacol. 1991, 41, 1485–1492. [Google Scholar] [CrossRef]
- Nève, J.; Parij, N.; Moguilevsky, N. Inhibition of the myeloperoxidase chlorinating activity by non-steroidal anti-inflammatory drugs investigated with a human recombinant enzyme. Eur. J. Pharmacol. 2001, 417, 37–43. [Google Scholar] [CrossRef]
- Kettle, A.J.; Sangster, D.F.; Gebicki, J.M.; Winterbourn, C.C. A pulse radiolysis investigation of the reactions of myeloperoxidase with superoxide and hydrogen peroxide. Biochim. Biophys. Acta 1988, 956, 58–62. [Google Scholar] [CrossRef]
- Bielski, B.H.J.; Cabelli, D.E.; Arudi, R.L. Reactivity of HO2/O2− radicals in aqueous solution. J. Phys. Chem. Ref. Data 1985, 14, 1041–1100. [Google Scholar] [CrossRef]
- Huie, R.E.; Padmaja, S. Reactions of NO and O2−. Free Radic. Res. Commun. 1993, 18, 195–199. [Google Scholar] [CrossRef]
- Kissner, R.; Nauser, T.; Bugnon, P.; Lye, P.G.; Koppenol, W.H. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem. Res. Toxicol. 1997, 10, 1285–1292. [Google Scholar] [CrossRef]
- Furtmüller, P.G.; Jantschko, W.; Zederbauer, M.; Schwanninger, M.; Jakopitsch, C.; Herold, S.; Koppenol, W.H.; Obinger, C. Peroxynitrite efficiently mediates the interconversions of redox intermediates of myeloperoxidase. Biochem. Biophys. Res. Commun. 2005, 337, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Koyani, C.N.; Flemmig, J.; Malle, E.; Arnhold, J. Myeloperoxidase scavenges peroxynitrite: A novel anti-inflammatory action of the heme enzyme. Arch. Biochem. Biophys. 2015, 571, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, H.N.; Henson, P.M.; Otani, A.; Hugli, T.E. Chemotactic response of human C3a and C5a anaphylatoxins. I. Evaluation of C3a and C5a leukotaxis in vitro and under simulated in vivo conditions. J. Immunol. 1978, 120, 109–115. [Google Scholar]
- Witko-Sarsat, V.; Rieu, P.; Descamps-Latscha, B.; Lesavre, P.; Halbwachs-Mecarelli, L. Neutrophils: Molecules, functions, and pathophysiological aspects. Lab. Investig. 2000, 80, 617–653. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N. Pathophysiological roles of interleukin 8/CXCL8 in pulmonary diseases. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L566–L577. [Google Scholar] [CrossRef]
- Phillipson, M.; Kubes, P. The neutrophil in vascular inflammation. Nat. Med. 2011, 17, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Schlorke, D.; Thomas, L.; Samsonov, S.A.; Huster, D.; Arnhold, J.; Pichert, A. The influence of glycosaminoglycans on IL-8-mediated functions of neutrophils. Carbohydr. Res. 2012, 356, 196–203. [Google Scholar] [CrossRef]
- Sengeløv, H.; Kjeldson, L.; Borregaard, N. Control of exocytosis in early neutrophil activation. J. Immunol. 1993, 150, 1535–1543. [Google Scholar]
- Sengeløv, H.; Follin, P.; Kjeldson, L.; Lollike, K.; Dahlgren, C.; Borregaard, N. Mobilization of granules and secretory vesicles during in vivo exudation of human neutrophils. J. Immunol. 1995, 154, 4157–4165. [Google Scholar]
- Edwards, S.W. Biochemistry and Physiology of the Neutrophil; Cambridge University Press: New York, NY, USA, 1994. [Google Scholar]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef]
- Elsbach, P. The bactericidal/permeability-increasing protein (BPI) in antibacterial host defense. J. Leukoc. Biol. 1998, 64, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Lindbom, L. Neutrophil-derived azurocidin alarms the immune system. J. Leukoc. Biol. 2009, 85, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Lu, W. α-Defensins in human innate immunity. Immunol. Rev. 2012, 245, 84–112. [Google Scholar] [CrossRef]
- Odell, E.W.; Sarra, R.; Foxworthy, M.; Chapple, D.S.; Evans, R.W. Antibacterial activities of peptides homologous to a loop region in lactoferrin. FEBS Lett. 1996, 382, 175–178. [Google Scholar] [CrossRef]
- Famaud, S.; Evans, R.W. Lactoferrin—A multifunctional protein with antimicrobial properties. Mol. Immunol. 2003, 40, 395–405. [Google Scholar]
- Wardlaw, A.C. The complement-dependent bacteriolytic activity of normal human serum. I. The effect of pH and ionic strength and the role of lysozyme. J. Exp. Med. 1962, 115, 1231–1249. [Google Scholar] [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef]
- Fasciglione, G.F.; Marini, S.; D’Alessio, S.; Politi, V.; Coletta, M. pH- and temperature-dependence of functional modulation in metalloproteinases. A comparison between neutrophil collagenase and gelatinases A and B. Biophys. J. 2000, 79, 2138–2149. [Google Scholar] [CrossRef]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef]
- Foote, J.R.; Behe, P.; Frampton, M.; Lewine, A.P.; Segal, A.W. An exploration of charge compensating ion channels across the phagocytic vacuole of neutrophils. Front. Pharmacol. 2017, 8, 94. [Google Scholar] [CrossRef]
- Segal, A.W.; Geisow, M.; Garcia, R.; Harper, A.; Miller, R. The respiratory burst of phagocytic cells is associated with a rise in vacuolar pH. Nature 1981, 290, 406–409. [Google Scholar] [CrossRef]
- Cech, P.; Lehrer, R.I. Phagolysosomal pH of human neutrophils. Blood 1984, 63, 88–95. [Google Scholar] [CrossRef]
- Levine, A.P.; Duchen, M.R.; de Villiers, S.; Rich, P.R.; Segal, A.W. Alkalinity of neutrophil phagocytic vacuoles is modulated by HVCN1 and has consequences for myeloperoxidase activity. PLoS ONE 2015, 10, e0125906. [Google Scholar] [CrossRef]
- Foote, J.R.; Patel, A.A.; Yona, S.; Segal, A.W. Variations in the phagosomal environment of human neutrophils and mononuclear subsets. Front. Immunol. 2019, 10, 00188. [Google Scholar] [CrossRef] [PubMed]
- Murata, K. Acidic glycosaminoglycans in human platelets and leukocytes: The isolation and enzymatic characterization of chondroitin-4-sulfate. Clin. Chim. Acta 1974, 57, 115–124. [Google Scholar] [PubMed]
- Kolset, S.O.; Gallaher, J.T. Proteoglycans in haemopoietic cells. Biochim. Biophys. Acta 1990, 1032, 191–211. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathway in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [PubMed]
- Flint, D.H.; Tominello, J.F.; Emptage, M.H. The investigation of Fe-S cluster containing hydrolyases by superoxide. J. Biol. Chem. 1993, 268, 22369–22376. [Google Scholar]
- Gardner, P.R. Aconitase: Sensitive target and measure of superoxide. Meth. Enzymol. 2002, 349, 9–23. [Google Scholar]
- Jantschko, W.; Furtmüller, P.G.; Zederbauer, M.; Lanz, M.; Jakopitsch, C.; Obinger, C. Direct conversion of ferrous myeloperoxidase to compound II by hydrogen peroxide: An anaerobic stopped-flow study. Biochem. Biophys. Res. Commun. 2003, 312, 292–298. [Google Scholar] [CrossRef]
- Hoogland, H.; Dekker, H.L.; van Riel, C.; van Kuilenberg, A.; Muijsers, A.O.; Wever, R. A steady-state study on the formation of compounds II and III of myeloperoxidase. Biochim. Biophys. Acta 1988, 955, 337–345. [Google Scholar] [CrossRef]
- Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Involvement of superoxide and myeloperoxidase in oxygen-dependent killing of Staphylococcus aureus by neutrophils. Infect. Immun. 1996, 64, 3512–3517. [Google Scholar] [CrossRef] [PubMed]
- Kettle, A.J.; Geyde, C.A.; Winterbourn, C.C. Mechanism of inactivation of myeloperoxidase by 4-aminobenzoic acid hydrazide. Biochem. J. 1997, 321, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Bolscher, B.G.J.M.; Wever, R. A kinetic study of the reaction between human myeloperoxidase, hydroperoxides and cyanide. Inhibition by chloride and thiocyanate. Biochim. Biophys. Acta 1984, 788, 1–10. [Google Scholar] [CrossRef]
- Lehrer, R.I. Inhibition by sulfonamides of the candidacidal activity of human neutrophils. J. Clin. Investig. 1971, 50, 2498–2505. [Google Scholar] [CrossRef]
- Koch, C. Effect of sodium azide upon normal and pathological granulocyte function. Acta Pathol. Microbiol. Scand. B 1974, 82, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Miyoshi-Koshio, T.; Utsuki, Y.; Mizuno, S.; Suzuki, K. Virucidal activity and viral protein modification by myeloperoxidase: A candidate for defense factor of human polymorphonuclear leukocytes against influenza virus infection. J. Infect. Dis. 1991, 164, 8–14. [Google Scholar] [CrossRef]
- Aratani, Y.; Koyama, H.; Nyui, S.-I.; Suzuki, K.; Kura, F.; Maeda, N. Severe impairment in early host defense against Candida albicans in mice deficient in myeloperoxidase. Infect. Immun. 1999, 67, 1828–1836. [Google Scholar] [CrossRef]
- Chapman, A.L.; Hampton, M.B.; Senthilmohan, R.; Winterbourn, C.C.; Kettle, A.J. Chlorination of bacterial and neutrophil proteins during phagocytosis and killing of Staphylococcus aureus. J. Biol. Chem. 2002, 277, 9757–9762. [Google Scholar] [CrossRef]
- Reeves, E.P.; Nagl, M.; Godovac-Zimmermann, J.; Segal, A.W. Reassessment of the microbicidal activity of reactive oxygen species and hypochlorous acid with reference to the phagocytic vacuole of the neutrophil granulocyte. J. Med. Microbiol. 2003, 53, 643–651. [Google Scholar] [CrossRef]
- Hirche, T.O.; Gaut, J.P.; Heinecke, J.W.; Belaaouaj, A. Myeloperoxidase plays critical roles in killing Klebsiella pneumoniae and inactivating elastase: Effects of host defense. J. Immunol. 2005, 174, 1557–1565. [Google Scholar] [CrossRef]
- Parry, M.F.; Root, R.K.; Metcalf, J.A.; Delaney, K.K.; Kaplow, L.S.; Richar, W.J. Myeloperoxidase deficiency: Prevalence and clinical significance. Ann. Intern. Med. 1981, 85, 293–301. [Google Scholar] [CrossRef]
- Kutter, D. Prevalence of myeloperoxidase deficiency: Population studies using Bayer-Technicon automated hematology. J. Mol. Med. 1998, 76, 669–675. [Google Scholar] [CrossRef]
- Trasher, A.J.; Keep, N.H.; Wientjes, F.; Segal, A.W. Chronic granulomatous disease. Biochim. Biophys. Acta 1994, 1227, 1–24. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Cline, M.J. Leukocyte myeloperoxidase deficiency and disseminated candidiasis: The role of myeloperoxidase in resistance to Candida infection. J. Clin. Investig. 1969, 48, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Cech, P.; Stadler, H.; Widmann, J.J.; Rohner, A.; Miescher, P.A. Leukocyte myeloperoxidase deficiency and diabetes mellitus associated with Candida albicans liver abscess. Am. J. Med. 1979, 66, 149–153. [Google Scholar] [CrossRef]
- Nguyen, C.; Katner, H.P. Myeloperoxidase deficiency manifesting as pustular candida dermatitis. Clin. Infect. Dis. 1997, 24, 258–260. [Google Scholar] [CrossRef] [PubMed]
- Gaut, J.P.; Yeh, G.C.; Tran, H.D.; Byun, J.; Henderson, J.P.; Richter, G.M.; Brennan, M.-L.; Lusis, A.J.; Belaaouiaj, A.; Hotchkiss, R.S.; et al. Neutrophils employ the myeloperoxidase system to generate antimicrobial brominating and chlorinating oxidants during sepsis. Proc. Natl. Acad. Sci. USA 2001, 98, 11961–11966. [Google Scholar] [CrossRef]
- Brennan, M.-L.; Gaur, A.; Pahuja, A.; Lusis, A.J.; Reynolds, W.F. Mice lacking myeloperoxidase are more susceptible to experimental encephalomyelitis. J. Neuroimmunol. 2001, 112, 97–105. [Google Scholar] [CrossRef]
- Aratani, Y.; Kura, F.; Watanabe, H.; Akagawa, H.; Takano, Y.; Ishida-Okawara, A.; Suzuki, K.; Maeda, N.; Koyama, H. Contribution of the myeloperoxidase-dependent oxidative systems to host defence against Cryptococcus neoformans. J. Med. Microbiol. 2006, 55, 1291–1299. [Google Scholar] [CrossRef]
- Zschaler, J.; Schlorke, D.; Arnhold, J. Differences in innate immune response between man and mouse. Crit. Rev. Immunol. 2014, 34, 433–454. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, P.B.; Lehrer, R.I. Mouse neutrophils lack defensins. Infect. Immun. 1992, 60, 3446–3447. [Google Scholar] [CrossRef]
- Lehrer, R.I.; Lichtenstein, A.K.; Ganz, T. Defensins: Antimicrobial and cytotoxic peptides of mammalian cells. Annu. Rev. Immunol. 1993, 11, 105–128. [Google Scholar] [CrossRef]
- Rausch, P.G.; Moore, T.G. Granule enzymes of polymorphonuclear neutrophils: A phylogentic comparison. Blood 1975, 46, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, N.; Nakano, K.; Aratani, Y.; Koyama, H.; Kodama, T.; Niki, E. Role of myeloperoxidase in the neutrophil-induced oxidation of low density lipoprotein as studied by myeloperoxidase-knockout mouse. J. Biochem. 2000, 127, 971–976. [Google Scholar] [CrossRef]
- Lennartsson, A.; Pieters, K.; Vidovic, K.; Gullberg, U. A murine antibacterial ortholog to human bactericidal/permeability-increasing protein (BPI) is expressed in testis, epididymis, and bone marrow. J. Leukoc. Biol. 2005, 77, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.-U. Neutrophil apoptosis pathways and their modification in inflammation. Immunol. Rev. 2003, 193, 101–110. [Google Scholar] [CrossRef]
- Walker, A.; Ward, C.; Taylor, E.I.; Dransfield, I.; Hart, S.P.; Haslett, C.; Rossi, A.G. Regulation of neutrophil apoptosis and removal of apoptotic cells. Curr. Drug Targets—Inflamm. Allergy 2005, 4, 447–454. [Google Scholar] [CrossRef]
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef]
- Barton, G.M. A calculated response: Control of inflammation by the innate immune system. J. Clin. Investig. 2008, 118, 413–420. [Google Scholar] [CrossRef]
- Flemmig, J.; Leßig, J.; Reibetanz, U.; Dautel, P.; Arnhold, J. Non-vital polymorphonuclear leukocytes express myeloperoxidase on their surface. Cell. Physiol. Biochem. 2008, 21, 287–296. [Google Scholar] [CrossRef]
- Vandivier, R.W.; Fadok, V.A.; Hoffmann, P.R.; Bratton, D.L.; Penvari, C.; Brown, K.K.; Brain, J.D.; Accurso, F.J.; Henson, P.M. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J. Clin. Investig. 2002, 109, 661–670. [Google Scholar] [CrossRef]
- Wang, R.; Town, T.; Gokarn, V.; Flavell, R.A.; Chandawarkar, R.Y. HSP70 enhances macrophage phagocytosis by interaction with lipid raft-associated TLR-7 and upregulating p38 MAPK and PI3K pathways. J. Surg. Res. 2006, 136, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Vandivier, R.W.; Henson, P.M.; Douglas, I.S. Burying the death: The impact of failed apoptosis removal (efferocytosis) on chronic inflammatory lung disease. Chest 2006, 129, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production hrough autocrine/paracrine mechanisms involving TGF-beta, PGE2 and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B. Programmed cell clearance. Cell. Mol. Life Sci. 2003, 60, 2575–2585. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; D’Herde, K.; Vandenabeele, P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis 2006, 11, 1673–1682. [Google Scholar] [CrossRef]
- Erwig, L.-P.; Henson, P.M. Immunological consequences of apoptotic cell phagocytosis. Am. J. Pathol. 2007, 171, 2–8. [Google Scholar] [CrossRef]
- Keel, M.; Ungethüm, U.; Steckholzer, U.; Niederer, E.; Hartung, T.; Trentz, O.; Ertel, W. Interleukin-10 counterregulates proinflammatory cytokine-induced inhibition of neutrophil apoptosis during severe sepsis. Blood 1997, 90, 3356–3363. [Google Scholar] [CrossRef]
- Haslett, C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am. J. Respir. Crit. Care Med. 1999, 160, S5–S11. [Google Scholar] [CrossRef]
- Hirahashi, J.; Mekala, D.; van Ziffie, J.; Xiao, L.; Saffaripour, S.; Wagner, D.D.; Shapiro, S.D.; Lowell, C.; Mayadas, T.N. Mac-1 signalling via SRc-family and Syk kinases results in elastase-dependent vasculopathy. Immunity 2006, 25, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Bratton, D.L.; Guthrie, L.; Henson, P.M. Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: Role of proteases. J. Immunol. 2001, 166, 8647–8654. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Emert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, 1000639. [Google Scholar] [CrossRef]
- Papayannopoulos, Y.; Zychlinsky, A. NETs: A new strategy for using old weapons. Trends Immunol. 2009, 30, 513–521. [Google Scholar] [CrossRef]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Uhlemann, Y.; Myers, D.D., Jr.; Wroblewski, S.K.; Wakefield, T.W.; Hartwig, J.H.; et al. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef]
- Martinod, K.; Demers, M.; Fuchs, T.A.; Wong, S.L.; Brill, A.; Gallant, M.; Hu, J.; Wang, Y.; Wagner, D.D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8674–8679. [Google Scholar] [CrossRef]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papyannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef]
- Parker, H.; Albrett, A.M.; Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J. Leukoc. Biol. 2012, 91, 369–376. [Google Scholar] [CrossRef]
- Sheppard, F.R.; Kelher, M.R.; Moore, E.E.; McLaughlin, N.J.; Banerjee, A.; Silliman, C.C. Structural organization of the neutrophil NADPH oxidase: Phosphorylation and translocation during priming and activation. J. Leukoc. Biol. 2005, 78, 1025–1042. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Khezri, R.; Glogauer, M.; Grinstein, S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol. Biol. Cell 2014, 25, 3330–3341. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Schneemann, M.; Schoeden, G. Macrophage biology and immunology: Man is not a mouse. J. Leukoc. Biol. 2007, 81, 579–580. [Google Scholar] [CrossRef]
- Yates, R.M.; Hermetter, A.; Taylor, G.A.; Russell, D.G. Macrophage activation downregulates the degradative capacity of the phagosome. Traffic 2007, 8, 241–250. [Google Scholar] [CrossRef]
- Balce, D.R.; Li, B.; Allan, E.R.; Rybicka, J.M.; Krohn, R.M.; Yates, R.M. Alternative activation of macrophages by IL-4 enhances the proteolytic capacity of their phagosomes through synergistic mechanisms. Blood 2011, 118, 4199–4208. [Google Scholar] [CrossRef]
- Lefkowitz, D.L.; Mone, J.; Lefkowitz, S.S. Myeloperoxidase: The good, the bad, and the ugly. Crit. Immunol. Rev. 2010, 6, 123–129. [Google Scholar] [CrossRef]
- Khan, A.A.; Alsahli, M.A.; Rahmani, A.H. Myeloperoxidase as an active biomarker: Recent biochemical and pathological perspectives. Med. Sci. 2018, 6, 33. [Google Scholar] [CrossRef]
- Willard, B.B.; Ruse, C.I.; Keightley, J.A.; Bond, M.; Kinter, M. Site-specific quantitation of protein nitration using liquid chromatography/tandem mass spectrometry. Anal. Chem. 2003, 75, 2370–2376. [Google Scholar] [CrossRef]
- Tiruppathi, C.; Naqvi, T.; Wu, Y.; Vogel, S.M.; Minshall, R.D.; Malik, A.B. Albumin mediates the transcystosis of myeloperoxidase by means of caveolae in endothelial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 7699–7704. [Google Scholar] [CrossRef]
- Capeillere-Blandin, C.; Gausson, V.; Descamps-Latscha, B.; Witko-Sarsat, V. Biochemical and spectrophotometric significance of advanced oxidized protein products. Biochim. Biophys. Acta 2004, 1689, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Matheson, M.R.; Wong, P.S.; Travis, J. Enzymatic inactivation of human alpha-1 proteinase inhibitor by neutrophil myeloperoxidase. Biochem. Biophys. Res. Commun. 1979, 88, 402–409. [Google Scholar] [CrossRef]
- Patterson, S.D. Mammalian α1-antitrypsins: Comparative biochemistry and genetics of the major plasma serpins. Comp. Biochem. Physiol. B 1991, 100, 439–454. [Google Scholar] [CrossRef]
- Hiemstra, P.S.; van Wetering, S.; Stolk, J. Neutrophil serine proteinases and defensins in chronic obstructive pulmonary disease: Effects on pulmonary epithelium. Eur. Respir. J. 1988, 12, 1200–1208. [Google Scholar] [CrossRef]
- Bouriche, H.; Salavei, P.; Lessig, J.; Arnhold, J. Differential effects of flavonols on inactivation of α1-antitrypsin induced by hypohalous acids and the myeloperoxidase-hydrogen peroxide-halide systems. Arch. Biochem. Biophys. 2007, 459, 137–143. [Google Scholar] [CrossRef]
- Zheng, L.; Nukuna, B.; Brennan, M.-L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, X.; Fu, L.; Thomson, P.L.; Ischiripoulos, H.; et al. Apolipoprotein A-1 is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Malle, E.; Marsche, G.; Panzenboeck, U.; Sattler, W. Myeloperoxidase-mediated oxidation of high-density lipoproteins: Fingerprints of newly recognized potential proatherogenic lipoproteins. Arch. Biochem. Biophys. 2006, 45, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Nybo, T.; Cai, H.; Chuang, C.Y.; Gamon, L.F.; Rogowska-Wrzesinska, A.; Davies, M.J. Chlorination and oxidation of human plasma fibronectin by myeloperoxidase-derived oxidants, and its consequences for smooth muscle cell function. Redox Biol. 2018, 19, 388–400. [Google Scholar] [CrossRef]
- Vanichkitrungruang, S.; Chuang, C.Y.; Hawkins, C.L. Myeloperoxidase-derived damage to human plasma fibronectin: Modulation by protein binding and thiocyanate ions (SCN−). Redox Biol. 2020, 36, 101641. [Google Scholar] [CrossRef]
- Daphna, E.M.; Michaela, S.; Eynat, P.; Irit, A.; Rimon, S. Association of myeloperoxidase with heparin: Oxidative inactivation of proteins on the surface of endothelial cells by the bound enzyme. Mol. Cell. Biochem. 1998, 183, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Baldus, S.; Eiserich, J.P.; Mani, A.; Castrom, L.; Figueroa, M.; Chumley, P.; Ma, W.; Tousson, A.; White, R.; Bullard, D.C.; et al. Endothelial transcytosis of myeloperoxidase confers specificity to vascular ECM proteins as targets for tyrosine nitration. J. Clin. Investig. 2001, 108, 1759–1770. [Google Scholar] [CrossRef] [PubMed]
- Eiserich, J.P.; Baldus, S.; Brennan, M.-L.; Ma, W.; Zhang, C.; Tousson, A.; Castro, L.; Lusis, A.J.; Nauseef, W.M.; White, C.R.; et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science 2002, 196, 2391–2394. [Google Scholar] [CrossRef]
- Kubala, L.; Kolářová, H.; Vitećek, J.; Kremserová, S.; Klinke, A.; Lau, D.; Chapman, A.L.P.; Baldus, S.; Eiserich, J.P. The potentiation of myeloperoxidase activity by the glycosaminoglycan-dependent binding of myeloperoxidase to proteins of the extracellular matrix. Biochim. Biophys. Acta 2013, 1830, 4524–4536. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Chuang, C.Y.; Hawkins, C.L.; Davies, M.J. Binding of myeloperoxidase to the extracellular matrix of smooth muscle cells and subsequent matrix modification. Sci. Rep. 2020, 10, 666. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.D.; Whitelock, J.M.; Malle, E.; Chuang, C.Y.; Iozzo, R.V.; Nilasaroya, A.; Davies, M.J. Myeloperoxidase-derived oxidants selectively disrupt the protein core of the heparan sulfate proteoglycan perlecan. Matrix Biol. 2010, 29, 63–73. [Google Scholar] [CrossRef]
- Nybo, T.; Dieterich, S.; Gamon, L.F.; Chuang, C.Y.; Hammer, A.; Hoefler, G.; Malle, E.; Rogowska-Wrzesinska, A.; Davies, M.J. Chlorination and oxidation of the extracellular matrix protein laminin and basement membrane extracts by hypochlorous acid and myeloperoxidase. Redox Biol. 2019, 20, 496–513. [Google Scholar] [CrossRef]
- Manchanda, K.; Kolářová, H.; Kerkenpaß, C.; Mollenhauer, M.; Vitećek, J.; Rudolph, V.; Kubala, L.; Baldus, S.; Adam, M.; Klinke, A. MPO (myeloperoxidase) reduces endothelial glycocalyx thickness dependent on its cationic charge. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1859–1867. [Google Scholar] [CrossRef]
- Sokolov, A.V.; Ageeva, K.V.; Cherkalina, O.S.; Pulina, M.O.; Zakharova, E.T.; Prozorovskii, V.N.; Aksenov, D.V.; Vasilyev, V.B.; Panasenko, O.M. Identification and properties of complexes formed by myeloperoxidase with lipoproteins and ceruloplasmin. Chem. Phys. Lipids 2010, 163, 347–353. [Google Scholar] [CrossRef]
- Chapman, A.L.P.; Mocatta, T.J.; Shiva, S.; Seidel, A.; Chen, B.; Khalilova, I.; Paumann-Page, M.E.; Jameson, G.N.L.; Winterbourn, C.C.; Kettle, A.J. Ceruloplasmin is an endogenous inhibitor of myeloperoxidase. J. Biol. Chem. 2013, 288, 6464–6477. [Google Scholar] [CrossRef]
- Stoj, C.; Kosman, D.J. Cuprous oxidase activity of yeast Fet3p and human ceruloplasmin: Implication for function. FEBS Lett. 2003, 554, 422–426. [Google Scholar] [CrossRef]
- Sokolov, A.V.; Pulina, M.O.; Ageeva, K.V.; Ayrapetov, M.I.; Berlov, M.I.; Volgin, G.N.; Markov, A.G.; Yablonsky, P.K.; Kolodkin, N.I.; Zakharova, E.T.; et al. Interaction of ceruloplasmin, lactoferrin, and myeloperoxidase. Biochemistry (Moscow) 2007, 72, 409–415. [Google Scholar] [CrossRef]
- Sokolov, A.V.; Pulina, M.O.; Ageeva, K.V.; Runova, O.I.; Zakharova, E.T.; Vasilyev, V.B. Identification of leukocyte cationic proteins that interact with ceruloplasmin. Biochemistry (Moscow) 2007, 72, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Vasilyev, V.B.; Kachurin, A.M.; Soronka, N.V. Dismutation of superoxide anion radicals by ceruloplasmin. Details of the mechanism. Biokhimija 1988, 83, 2051–2058. [Google Scholar]
- Segelmark, M.; Persson, B.; Hellmark, T.; Wieslander, J. Binding and inhibition of myeloperoxidase (MPO): A major function of ceruloplasmin? Clin. Exp. Immunol. 1997, 108, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.S.; Suzuki, K.; Mumby, S.; Taniguchi, N.; Gutteridge, J.M.C. Antioxidant binding of caeruloplasmin to myeloperoxidase: Myeloperoxidase is inhibited, but oxidase, peroxidase, and immunoreactive properties of caeruloplasmin remain intact. Free Radic. Res. 2000, 33, 261–265. [Google Scholar] [CrossRef]
- Sokolov, A.V.; Ageeva, K.V.; Pulina, M.O.; Cherkalina, O.S.; Samygina, V.R.; Vlasova, I.I.; Panasenko, O.M.; Zakharova, E.T.; Vasilyev, V.B. Ceruloplasmin and myeloperoxidase in complex affect the enzymatic properties of each other. Free Radic. Res. 2008, 42, 989–998. [Google Scholar] [CrossRef]
- Samygina, V.R.; Sokolov, A.V.; Bourenkov, G.; Petoukhov, M.V.; Pulina, M.O.; Zakharova, E.T.; Vasilyev, V.B.; Bartunik, H.; Svergun, D.I. Ceruloplasmin: Macromolecular assemblies with iron-containing acute phase proteins. PLoS ONE 2013, 8, e67145. [Google Scholar] [CrossRef]
- Sokolov, A.V.; Kostevich, V.A.; Zakharova, E.T.; Samygina, V.R.; Panasenko, O.M.; Vasilyev, V.B. Interaction of ceruloplasmin with eosinophil peroxidase as compared to its interplay with myeloperoxidase: Reciprocal effect on enzymatic properties. Free Radic. Res. 2015, 49, 800–811. [Google Scholar] [CrossRef]
- Griffin, S.V.; Chapman, P.T.; Lianos, E.A.; Lockwood, C.M. The inhibition of myeloperoxidase by ceruloplasmin can be reversed by anti-myeloperoxidase antibodies. Kidney Int. 1999, 55, 917–925. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Wick, G.; Knoflach, M.; Xu, Q. Autoimmune and inflammatory mechanisms in atherosclerosis. Annu. Rev. Atheroscl. 2004, 22, 361–403. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Garcia-Gardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef]
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J. W: Pathophysiology and treatment of atherosclerosis. Current view and future perspective on lipoprotein modification treatment. Neth. Heart J. 2017, 25, 231–242. [Google Scholar] [CrossRef]
- Gisterå, A.; Hansson, G. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond cholesterol: Modification of low-density lipoprotein that increases its atherogenicity. N. Engl. J. Med. 1989, 320, 915–924. [Google Scholar]
- Teng, N.; Maghzal, G.; Talib, J.; Rashid, I.; Lau, A.K.; Stocker, R. The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox Rep. 2017, 22, 51–73. [Google Scholar] [CrossRef]
- Daugherty, A.; Rateri, D.L.; Dunn, J.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef]
- Malle, E.; Waeg, G.; Schreiber, R.; Gröne, E.F.; Sattler, W.; Gröne, H.J. Immunological evidence for the myeloperoxidase/H2O2/halide system in human atherosclerotic lesions. Eur. J. Biochem. 2000, 267, 4495–4503. [Google Scholar] [CrossRef] [PubMed]
- Hazen, S.L.; Heinecke, J.W. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Investig. 1997, 99, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, J.; Byun, J.; Nhan, T.Q.; Pritchard, D.K.; Pennathur, S.; Schwartz, S.M.; Chait, A.; Heinecke, J.W. Myeloperoxidase generates 5-chlorouracil in human atherosclerotic tissue: A potential pathway for somatic mutagenesis by macrophages. J. Biol. Chem. 2005, 281, 3096–3104. [Google Scholar] [CrossRef] [PubMed]
- Hazell, L.J.; Stocker, R. Oxidation of low-density lipoprotein with hypochlorite causes transformation of the lipoprotein into a high-uptake form for macrophages. Biochem. J. 1993, 290, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Gu, Z.W.; Yang, M.; Lin, S.N.; Garcia-Prats, A.J.; Rogers, L.K.; Welty, S.E.; Smith, C.V. Selective modification of apoB-100 in the oxidation of low density lipoprotein by myeloperoxidase in vitro. J. Lipid Res. 1999, 40, 686–698. [Google Scholar]
- Smith, J.D. Dysfunctional HDL as a diagnostic and therapeutic target. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.A.; Feig, J.E.; Hewing, B.; Hazen, S.L.; Smith, J.D. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2813–2820. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.; Reynolds, W.F.; Topol, E.; DiDonato, J.A.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 113, 1176–1184. [Google Scholar] [CrossRef]
- Delporte, C.; Boudjeltia, K.Z.; Furtmüller, P.G.; Maki, R.A.; Dieu, M.; Noyon, C.; Soudi, M.; Dufour, D.; Coremans, C.; Nuyens, V.; et al. Myeloperoxidase-catalyzed oxidation of cyanide to cyanate: A potential carbamylation route involved in the formation of atherosclerotic plaques? J. Biol. Chem. 2018, 293, 6374–6386. [Google Scholar] [CrossRef]
- Zhang, C.; Reiter, C.; Eiserich, J.P.; Boersma, B.; Parks, D.A.; Beckman, J.S.; Barnes, S.; Kirk, M.; Baldus, S.; Darley-Usmar, V.M.; et al. L-Arginine chlorination products inhibit endothelial nitric oxide production. J. Biol. Chem. 2001, 276, 27159–27165. [Google Scholar] [CrossRef]
- Koeth, R.A.; Haselden, V.; Tang, W.H. Myeloperoxidase in cardiovascular disease. Adv. Clin. Chem. 2013, 62, 1–32. [Google Scholar]
- Wang, Y.; Rosen, H.; Madtes, D.K.; Shao, B.; Martin, T.R.; Heinecke, J.W.; Fu, X. Myeloperoxidase inactivates TIMP-1 by oxidizing its N-terminal cysteine residue: An oxidative mechanism for regulating proteolysis during inflammation. J. Biol. Chem. 2007, 282, 31826–31834. [Google Scholar] [CrossRef]
- Ehrenfeld, P.; Matus, C.E.; Pavicic, F.; Toledo, C.; Nualart, F.; Gonzalez, C.B.; Burgos, R.A.; Bhoola, K.D.; Figueroa, C.D. Kinin B1 receptor activation turns on exocytosis of matrixmetalloprotease-0 and myeloperoxidase in human neutrophils: Involvement of mitogen-activated protein kinase family. J. Leukoc. Biol. 2009, 86, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Baldus, S.; Heeschen, C.; Meinertz, T.; Zeiher, A.M.; Eiserich, J.P.; Münzel, T.; Simoons, M.L.; Hamm, C.W. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation 2003, 108, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Katz, R.; Brennan, M.-L.; Tracy, R.P.; Aviles, R.J.; Psaty, B.M.; Hazen, S.L. Usefulness of myeloperoxidase levels in healthy elderly subjects to predict risk of developing heart failure. Am. J. Cardiol. 2009, 103, 1269–1274. [Google Scholar] [CrossRef]
- Nussbaum, C.; Klinke, A.; Adam, M.; Baldus, S.; Sperandio, M. Myeloperoxidase: A leukocyte-derived protagonist of inflammation and cardiovascular disease. Antioxid. Redox Signal. 2013, 18, 692–713. [Google Scholar] [CrossRef]
- Narula, J.; Nakano, M.; Virmani, R.; Kolodgie, F.D.; Petersen, R.; Newcomb, R.; Malik, S.; Fuster, V.; Finn, A.V. Histopathologic characteristics of atherosclerotic coronary disease and implications of the findings for the invasive and noninvasive detection of vulnerable plaques. J. Am. Cell Cardiol. 2013, 61, 1041–1051. [Google Scholar] [CrossRef]
- Jennette, J.C.; Falk, R.J.; Hu, P.; Xiao, H. Pathogenesis of anti-neutrophil cytoplasmic autoantibodies associated small vessel vasculitis. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 139–160. [Google Scholar]
- Crisford, H.; Sapey, E.; Stockley, R.A. Proteinase 3; a potential target in chronic obstructive pulmonary disease and other chronic inflammatory diseases. Respir. Res. 2018, 19, 180. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Puerta, J.A.; Bosch, X. Anti-neutrophil cytoplasmic antibody pathogenesis in small-vessel vasculitis. Am. J. Pathol. 2009, 175, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Tuttle, R.H.; Hogan, S.L.; Taylor, J.G.; Philipps, B.D.; Falk, R.J.; Jennette, J.C. Target antigens for anti-neutrophil cytoplasmic autoantibodies (ANCA) are on the surface of primed and apoptotic but not unstimulated neutrophils. Clin. Exp. Immunol. 2000, 121, 165–172. [Google Scholar] [CrossRef]
- Kain, R.; Exner, M.; Brandes, R.; Ziebermayr, R.; Cunningham, D.; Alderson, D.A.; Davidovits, A.; Raab, I.; Jahn, R.; Ashour, O.; et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat. Med. 2008, 14, 1088–1096. [Google Scholar] [CrossRef]
- Jennette, J.C.; Nachman, P.H. ANCA glomerulonephritis and vasculitis. Clin. J. Am. Soc. Nephrol. 2017, 12, 1680–1691. [Google Scholar] [CrossRef] [PubMed]
- Greenan, K.; Vassallo, D.; Chinnadurai, R.; Ritchie, J.; Shepard, K.; Green, D.; Ponnusamy, A.; Sinha, S. Respiratory manifestations of ANCA-associated vasculitis. Clin. Respir. J. 2018, 12, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.; Al-Shabrawey, M.; Caldwell, R.W.; Caldwell, R.B. Inflammation and diabetic retinal microvascular complications. J. Cardiovasc. Dis. Res. 2011, 2, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Baskin, E.; Bakkaloglu, A.; Besbas, N.; Hascelik, G.; Saatci, U.; Gök, F.; Ozen, S. Ceruloplasmin levels in antineutrophil cytoplasmic antibody-positive patients. Pediatr. Nephrol. 2002, 17, 917–919. [Google Scholar] [CrossRef]
- Chooklin, S.; Pereyaslov, A.; Bihalskyy, I. Pathogenic role of myeloperoxidase in acute pancreatitis. Hepatobiliary Pancreat. Dis. Int. 2009, 8, 627–631. [Google Scholar] [PubMed]
- Klangprapan, S.; Chaiyarit, P.; Hormdee, D.; Kampichai, A.; Khampitak, T.; Daduang, J.; Tavichakorntrakool, R.; Panijpan, B.; Boonsiri, P. Salivary myeloperoxidase, assessed by 3,3’-diaminobenzidine colorimetry, can differentiate periodontal patients from nonperiodontal subjects. Enzyme Res. 2016, 2016, 7517928. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.W.; Hallett, M.B. Seeing the wood for the trees: The forgotten role of neutrophils in rheumatoid arthritis. Immunol. Today 1997, 18, 320–324. [Google Scholar] [CrossRef]
- Green, P.S.; Mendez, A.J.; Jacob, J.S.; Crowley, J.R.; Growdon, W.; Hyman, B.T.; Heinecke, J.W. Neuronal expression of myeloperoxidase is increased in Alzheimer’s disease. J. Neurochem. 2004, 90, 724–733. [Google Scholar] [CrossRef]
- Maki, R.A.; Tyurin, V.A.; Lyon, R.C.; Hamilton, R.L.; DeKosky, S.T.; Kagan, V.E.; Reynolds, W.F. Aberrant expression of myeloperoxidase in astrocytes promotes phospholipid oxidation and memory deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2009, 284, 3158–3169. [Google Scholar] [CrossRef]
- Choi, D.-K.; Pennathur, S.; Perier, C.; Tieu, K.; Teismann, P.; Wu, D.-C.; Jackson-Lewis, V.; Vila, M.; Vonsattel, J.-P.; Heinecke, J.W.; et al. Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson’s disease in mice. J. Neurosci. 2005, 25, 6594–6600. [Google Scholar] [CrossRef]
- Teismann, P. Myeloperoxidase in the neurodegenerative process of Parkinson’s disease. Dtsch. Med. Wochenschr. 2014, 139, 99–102. [Google Scholar] [PubMed]
- Gray, E.; Thomas, T.L.; Betmouni, S.; Scolding, N.; Love, S. Elevated myeloperoxidase activity in white matter in multiple sclerosis. Neurosci. Lett. 2008, 444, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Olza, J.; Aguilera, C.M.; Gil-Campos, M.; Leis, R.; Bueno, G.; Martinez-Jiménez, M.D.; Valle, M.; Cañete, R.; Tojo, R.; Moreno, L.A.; et al. Myeloperoxidase is an early biomarker of inflammation and cardiovascular risk in prepubertal obese children. Diabetes Care 2012, 35, 2373–2376. [Google Scholar] [CrossRef] [PubMed]
- Demoly, P.; Crampette, L.; Mondain, M.; Enander, I.; Jones, I.; Bousquet, J. Myeloperoxidase and interleukin-8 levels in chronic sinusitis. Clin. Exp. Allergy 1997, 27, 672–675. [Google Scholar] [CrossRef]
- Sagel, S.D.; Wagner, B.D.; Anthony, M.M.; Emmett, P.; Zemanick, E.T. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 857–865. [Google Scholar] [CrossRef]
- Hansberry, D.R.; Shah, K.; Agarwal, P.; Agarwal, N. Fecal myeloperoxidase as a biomarker for inflammatory bowel disease. Cureus 2017, 9, e1004. [Google Scholar] [CrossRef]
- Charin, B.; Martin, N.J.J.; Dennis, J.H.; Witting, P.K. Myeloperoxidase in the inflamed colon: A novel target for treating inflammatory bowel disease. Arch. Biochem. Biophys. 2018, 645, 61–71. [Google Scholar]
- Malle, E.; Woenckhaus, C.; Waeg, G.; Esterbauer, H.; Gröne, E.F.; Gröne, H.-J. Immunological evidence for hypochlorite-modified proteins in human kidney. Am. J. Pathol. 1997, 150, 603–615. [Google Scholar]
- Malle, E.; Buch, T.; Gröne, H.-J. Myeloperoxidase in kidney disease. Kidney Int. 2003, 64, 1956–1967. [Google Scholar] [CrossRef]
- Hanumegowda, U.M.; Copple, B.I.; Shibuya, M.; Malle, E.; Ganey, P.E.; Roth, R.A. Basement membrane and matrix metalloproteinases in monocrotaline-induced liver injury. Toxicol. Sci. 2003, 76, 237–246. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Li, M.O.; Flavell, R.A. Contextual regulation of inflammation: A duet by transforming growth factor-beta and interleukin-10. Immunity 2008, 28, 468–476. [Google Scholar] [CrossRef]
- Yoshimura, A.; Wakabayashi, Y.; Mori, T. Cellular and molecular basis for the regulation of inflammation by TGF-β. J. Biochem. 2010, 147, 781–792. [Google Scholar] [CrossRef]
- Chandrasekharan, J.A.; Sharma-Walia, N. Lipoxins: Nature’s way to resolve inflammation. J. Inflamm. Res. 2015, 8, 181–192. [Google Scholar]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Rother, R.P.; Bell, L.; Hillman, P.; Gladwin, M.T. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin. J. Am. Med. Assoc. 2005, 293, 1653–1662. [Google Scholar] [CrossRef]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Sauret, J.M.; Marinides, G.; Wang, G.K. Rhabdomyolysis. Am. Fam. Phys. 2002, 65, 907–912. [Google Scholar]
- Hunter, J.D.; Gregg, K.; Damani, Z. Rhabdomyolysis. Cont. Ed. Anaesth. Crit. Care Pain 2006, 6, 141–143. [Google Scholar] [CrossRef]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercelotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef]
- Lin, T.; Sammy, F.; Yang, H.; Thundivalappil, S.; Hellman, J.; Tracey, K.C.; Warren, H.S. Identification of hemopexin as an anti-inflammatory factor that inhibits synergy of hemoglobin with HMGB1 in sterile and infectious inflammation. J. Immunol. 2012, 189, 2017–2022. [Google Scholar] [CrossRef]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercelotti, G.M. Hemolysis and free heme revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef]
- Flemmig, J.; Schlorke, D.; Kühne, F.-W.; Arnhold, J. Inhibition of the heme-induced hemolysis of red blood cells by the chlorite-based drug WF10. Free Radic. Res. 2016, 50, 1386–1395. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef]
- Antonyuk, S.V.; Strange, R.W.; Marklund, S.L.; Hasnain, S.S. The structure of human extracellular copper-zinc superoxide dismutase at 1.7 Å resolution: Insight into heparin and collagen binding. J. Mol. Biol. 2009, 388, 310–326. [Google Scholar] [CrossRef]
- Low, F.M.; Hampton, M.B.; Winterbourn, C.C. Prx2 and peroxide metabolism in the erythrocyte. Antioxidants Redox Signal. 2008, 10, 1621–1630. [Google Scholar] [CrossRef]
- Goyal, M.M.; Basak, A. Human catalase: Looking for complete identity. Prot. Cell 2010, 1, 888–897. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1840, 3289–3303. [Google Scholar] [CrossRef]
- Ponka, P. Cellular iron metabolism. Kidney Int. 1999, 55, S2–S11. [Google Scholar] [CrossRef]
- Massover, W.H. Ultrastructure of ferritin and apoferritin: A review. Micron 1993, 24, 389–437. [Google Scholar] [CrossRef]
- Chiabrando, D.; Vinchi, F.; Florito, V.; Tolosano, E. Haptoglobin and hemopexin in heme detoxification and iron recycling. In Acute Phase Proteins—Regulation and Functions of Acute Phase Proteins; Veas, F., Ed.; Intech: Rijeka, Croatia, 2011; pp. 261–288. [Google Scholar]
- Niki, E. Antioxidants in relation to lipid peroxidation. Chem. Phys. Lipids 1987, 44, 227–253. [Google Scholar] [CrossRef]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, α-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Hochstein, P.; Hatch, L.; Sevanian, A. Uric acid: Functions and determinations. Meth. Enzymol. 1984, 105, 162–166. [Google Scholar]
- Nyyssönen, K.; Porkkala-Sarataho, E.; Kaikkonen, J.; Salonen, J.T. Ascorbate and urate are the strongest determinants of plasma antioxidative capacity and serum lipid resistance to oxidation in Finnish men. Atherosclerosis 1997, 130, 223–233. [Google Scholar] [CrossRef]
- Koj, A. Biological functions of acute-phase proteins. In The Acute Phase Response to Injury and Infection; Gordon, A.H., Koj, A., Eds.; Elsevier: Amsterdam, The Netherlands, 1985; pp. 145–160. [Google Scholar]
- Baumann, H.; Gauldie, J. The acute phase response. Immunol. Today 1994, 15, 74–80. [Google Scholar] [CrossRef]
- Musci, G. Structure/function relationships in ceruloplasmin. Adv. Exp. Med. Biol. 1999, 448, 175–182. [Google Scholar] [PubMed]
- Taggart, C.C.; Greene, C.M.; Carroll, T.P.; O’Neill, S.J.; McElvaney, N.G. Elastolytic proteases. Inflammation regulation and dysregulation in chronic infective lung disease. Am. J. Respir. Crit. Care Med. 2005, 171, 1070–1076. [Google Scholar] [CrossRef]
- Brode, S.K.; Ling, S.C.; Chapman, K.R. Alpha-1 antitrypsin deficiency: A commonly overlooked cause of lung disease. Can. Med. Assoc. J. 2012, 184, 1365–1371. [Google Scholar] [CrossRef]
- Silverman, E.K.; Sandhaus, R.A. Alpha1-antitrypsin deficiency. N. Engl. J. Med. 2009, 360, 2749–2757. [Google Scholar] [CrossRef]
- Sandhaus, R.A.; Turino, G.; Brantly, M.L.; Campos, M.; Cross, C.E.; Goodman, K.; Hogart, D.K.; Knight, S.L.; Stocks, J.M. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chron. Obstr. Pulm. Dis. 2016, 3, 668–682. [Google Scholar] [CrossRef]
- Townsend, S.A.; Edgar, R.G.; Kantas, P.R.; Newsome, P.N.; Turner, A.M. Systematic review: The natural history of alpha-1 antitrypsin deficiency, and associated liver disease. Alim. Pharmacol. Ther. 2018, 47, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune response. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Reber, A.J.; Chirkova, T.; Kim, J.H.; Cao, W.; Biber, R.; Shay, D.K.; Sambhara, S. Immunosenescence and challenges of vaccination against influenza in the aging population. Aging Dis. 2012, 3, 68–90. [Google Scholar] [PubMed]
- Sepkowitz, K.A. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin. Infect. Dis. 2002, 34, 1098–1107. [Google Scholar] [CrossRef]
- Kampitak, T.; Suwanpimolkul, G.; Browne, S.; Suankratay, C. Anti-interferon-γ autoantibody and opportunistic infections: Case series and review of the literature. Infection 2011, 39, 65–71. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellamo, R.; Bernard, G.R.; Chiche, J.-D.; Coppersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). J. Am. Med. Assoc. 2016, 23, 801–810. [Google Scholar] [CrossRef]
- Podnos, Y.D.; Jiminez, J.C.; Wilson, S.E. Intraabdominal sepsis in elderly persons. Clin. Infect. Dis. 2002, 35, 62–68. [Google Scholar] [CrossRef]
- Williams, M.D.; Braun, L.A.; Cooper, I.M.; Johnston, J.; Weiss, R.V.; Qualy, R.I.; Linde-Zwirble, W. Hospitalized cancer patients with severe sepsis: Analysis of incidence, mortality, and associated costs of care. Crit. Care 2004, 8, R291–R298. [Google Scholar] [CrossRef]
- Jiminez, M.F.; Watson, R.W.; Parodo, J.; Evans, D.; Foster, D.; Steinberg, M.; Rotstein, O.D.; Marshall, J.C. Dysregulated expression of neutrophil apoptosis in the systemic inflammatory response syndrome. Arch. Surg. 1997, 132, 1263–1270. [Google Scholar] [CrossRef]
- Demaret, J.; Venet, F.; Friggeri, A.; Cauzalis, M.-A.; Plassais, J.; Jallades, L.; Malcus, C.; Poitevin-Later, F.; Textoris, J.; Lepape, A.; et al. Marked alterations in neutrophil functions during sepsis-induced immunosuppression. J. Leukoc. Biol. 2015, 98, 1081–1090. [Google Scholar] [CrossRef]
- Brown, K.A.; Brain, S.D.; Pearson, J.D.; Edgeworth, J.D.; Lewis, S.M.; Treacher, D.F. Neutrophils in development of multiorgan failure in sepsis. Lancet 2006, 368, 157–169. [Google Scholar] [CrossRef]
- Kovach, M.A.; Standiford, T.J. The functions of neutrophils in sepsis. Curr. Opin. Infect. Dis. 2012, 25, 321–327. [Google Scholar] [CrossRef]
- Crouser, E.; Exline, M.; Wewers, M.D. Sepsis: Links between pathogen sensing and organ damage. Curr. Pharmaceut. Des. 2008, 14, 1840–1852. [Google Scholar] [CrossRef] [PubMed]
- Rittirsch, D.; Flierl, M.A.; Ward, P.A. Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 2008, 8, 776–787. [Google Scholar] [CrossRef]
- Maruchi, Y.; Tsuda, M.; Mori, H.; Takenaka, N.; Gocho, T.; Huq, M.A.; Takeyama, N. Plasma myeloperoxidase-conjugated DNA level predicts outcomes and organ dysfunction in patients with septic shock. Crit. Care 2018, 22, 176. [Google Scholar] [CrossRef]
- Schrijver, I.T.; Kempermann, H.; Roest, M.; Kesecioglu, J.; de Lange, D.W. Myeloperoxidase can differentiate between sepsis and non-infectious SIRS and predicts mortality in intensive care patients with SIRS. Intens. Care Med. Exp. 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Kothari, N.; Keshari, R.S.; Bogra, J.; Kohli, M.; Abbas, H.; Malik, A.; Dikshit, M.; Barthwal, M.K. Increased myeloperoxidase enzyme activity in plasma is an indicator of inflammation and onset of sepsis. J. Crit. Care 2011, 26, 435.e1–453.e7. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.S.; Yoon, J.M.; Jung, W.J.; Kim, Y.W.; Kim, T.H.; Kim, O.H.; Cha, K.C.; Kim, H.; Hwang, S.O.; Lee, K.H. Evaluation of usefulness of myeloperoxidase index (MPXI) for differential diagnosis of systemic inflammatory response syndrome (SIRS) in the emergency department. Emerg. Med. J. 2015, 32, 304–307. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Heme State of MPO | Short Denomination | Formal Oxidation State Versus Resting MPO |
|---|---|---|
| resting MPO | Por−Fe3+ a | |
| Compound I | •+Por−Fe4+=O | +2 |
| Compound II | Por−Fe4+−OH | +1 |
| Compound III | Por−Fe3+−O2•− | 0 |
| Redox Couple | E’° (at pH 7) | Number of Transferred Electrons | References |
|---|---|---|---|
| Compound I/resting MPO | 1.16 V | 2 | [24] |
| Compound I/Compound II | 1.35 V | 1 | [25] |
| Compound II/resting MPO | 0.97 V | 1 | [25] |
| Potentially Damaging Agents | Antagonizing Principle | References |
|---|---|---|
| Myeloperoxidase | Ceruloplasmin | [182,183] |
| Proteases from PMNs, mast cells and others | Anti-proteinases such as α1-antiproteinase, α1-antichymotrypsin, secretory leukocyte protease inhibitor, elafin, α2-macroglobulin | [90] |
| Superoxide anion radicals | Superoxide dismutases, cytochrome c, ceruloplasmin | [187,257,258,259] |
| Hydrogen peroxide | Glutathione peroxidases, peroxiredoxins, catalase | [260,261,262] |
| Free metal ions | Ceruloplasmin, chelators, lactoferrin, ferritin | [88,184,263,264] |
| Free hemoglobin, free myoglobin | Haptoglobin | [265] |
| Free heme | Hemopexin | [265] |
| Lipid-based oxidants | Lipid-soluble antioxidants such as tocopherols, carotenoids, ubiquinol | [266,267] |
| Water-based oxidants | Ascorbic acid, urate | [268,269] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnhold, J. The Dual Role of Myeloperoxidase in Immune Response. Int. J. Mol. Sci. 2020, 21, 8057. https://doi.org/10.3390/ijms21218057
Arnhold J. The Dual Role of Myeloperoxidase in Immune Response. International Journal of Molecular Sciences. 2020; 21(21):8057. https://doi.org/10.3390/ijms21218057
Chicago/Turabian StyleArnhold, Jürgen. 2020. "The Dual Role of Myeloperoxidase in Immune Response" International Journal of Molecular Sciences 21, no. 21: 8057. https://doi.org/10.3390/ijms21218057
APA StyleArnhold, J. (2020). The Dual Role of Myeloperoxidase in Immune Response. International Journal of Molecular Sciences, 21(21), 8057. https://doi.org/10.3390/ijms21218057